
Abstract
Background
Congenital Hyperinsulinism typically occurs with a neonatal hypoglycemia but can appear even in childhood or in adolescence with different types of glucose metabolism derangements. Current diagnostic algorithms don’t take into account cases with a late presentation.
Patients and methods
Clinical and laboratory data of twenty-two subjects diagnosed at Federico II University of Naples have been described: patients have been divided according to the molecular defect into channel defects, metabolic defects and unidentified molecular defects. A particular focus has been made on three cases with a late presentation.
Results and conclusions
Late presentation cases may not be identified by previous diagnostic algorithms. Consequently, it seems appropriate to design a new flow-chart starting from the age of presentation, also considering that late presentation cases can show glucose metabolism derangements other than hypoglycaemic crises such as diabetes, glucose intolerance, postprandial hypoglycaemia and gestational diabetes.
Similar content being viewed by others

Background
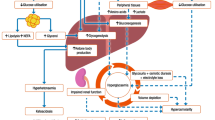
Congenital Hyperinsulinism (CH), first defined by Stanley [1], represents the most common cause of persistent hypoglycemia (HY) in infancy with estimated incidence of 1:40.000–50.000 in general population, up to 1:2500 in case of consanguinity [2]. To date, its prevalence in Italy is not well known [3, 4]. CH is due to alteration of beta-cell membrane channels or intracellular metabolic pathways, involving insulin secretion. In particular, channel defects (ChD), deriving from mutations of the K-ATP channel, represent about 36% of all CH cases [5] and affect ABCC8 and KCJN11 genes, which codify respectively for Sulfonylurea Receptor 1 (SUR1) and Kir6.2 (that are K-ATP channel subunits). Metabolic defects (MeD) are due to defective beta-cell intracellular signaling. To date, 12 genes are known to be responsible for MeD cases [5, 6]; GCK and GLUD1 are the less rarely involved genes. CH is suspected in case of neonatal onset of persistent HY with inappropriately measurable blood insulin levels. A further diagnostic criterion can be an intravenous glucose requirement of more than 8 mg/kg/min [7, 8]. The flow-chart of Maiorana et others [9] proposes to start with metabolic screenings and states that 18F-DOPA PET should be performed in all cases.
It is known that the diagnosis of CH can also occur in adolescents and adults reported for symptoms other than HY, such as mild hyperglycaemia and gestational diabetes mellitus (GDM) [10,11,12,13,14], thus justifying a late diagnosis of CH [15].
In light of our clinical experience, we propose a new flow chart, which represents an extension of the diagnostic range already presented by others in the literature with the advantage of making the diagnosis quicker and more accurate.
Patients and methods
Twenty-two patients, who received the diagnosis of CH and were followed for at least 3 years at the Regional Center for Pediatric Diabetes of the Federico II University of Naples, are reported in our study.
Diagnosis of CH was based on the detection of measurable insulin levels during the hypoglycaemic crises starting in neonatal age/infancy. The molecular diagnosis was performed according to the current diagnostic flow-charts in neonatal cases; an accurate clinical investigation was essential for the diagnosis of the three late presentation cases.
Based on their genetic defects, patients have been classified into three groups: ChD, MeD and unidentified molecular defects.
The data collected are: HY and other glucose metabolism derangements (GMD) in the family, large weight at birth (LGA) and delivery at term or preterm, HY onset before or after 72 h from birth, responsiveness to Diazoxide (Dx), surgical treatment and, in the follow up, presence of diabetes, obesity and neurological impairment. Data from ChD subjects (ABCC8-mutation) has been reported in Table 1, data of MeD and unidentified defects in Table 2.
Results
Of all the 22 patients, 16 have received molecular diagnosis: 12 ABCC8 mutations (54%), 2 GLUD1 (9%), 1 GCK (4%) and 1 HADH (4%). Consequently, in this population the detection rate of genetic defects is 73%, higher than 45–55% reported by Rahman and others [5].
GMD were found in 7/12 families of ChD cases, 2/4 of MeD (both having GLUD1 mutation) and 3/6 of unidentified defects; preterm birth was found in 6/12 ChD cases and in none of other categories; LGA was detected in 6/12 ChD cases and in only one MeD (with GCK mutation); time of presentation of HY in the first 72 h of life was found in 11/12 ChD and in 3/6 unidentified defects; Dx responsiveness was found in 6/12 ChD and in all MeD + unidentified defects subjects; diabetes in the follow-up was found in 4/12 ChD (2 subjects who had undergone pancreatectomy and 2 with late presentation); none in MeD and unidentified defects; obesity in the follow-up was found in 5/12 ChD, in 1/4 MeD and in 3/6 unidentified defects; neurological impairment in the follow-up was found in 4/12 ChD (subject number 5 epilepsy, subject number 7 epilepsy and severe developmental delay, subjects numbers 9 and 10 developmental delay), 1/4 MeD (this is G. V, described extensively below) and 1/4 unidentified defects (epilepsy).
We have performed 18F-DOPA PET-CT to 14/22 of described patients and we have always found diffused forms. A recent newborn with a focal form resolved by surgery (partial pancreatectomy) and four further cases are not included in this study due to the lack of three-year follow-up.
Late-presentation cases
Family B. B.A. (male, 12-years old) has been referred to us because of asymptomatic fasting hyperglycemia occasionally detected. He was born preterm (35 weeks) and LGA (4150 g) from pregnancy characterized by GDM. B.A. and his brother, B. B, 17-years old, had presented neonatal HY and, during childhood, post-meal HY; also B.B. 3 years later, received a diagnosis of diabetes. Their mother, maternal grandmother and aunt presented diabetes treated with metformin. The mother’s first pregnancy was characterized by GDM and hesitated in a LGA newborn dead at birth. After excluding autoimmune diabetes, B.A. has been initially treated with a low glycemic index diet and later with sulfonylurea. Heterozygous mutation c.G4435 > A (p.Gly1479Arg) (already reported in literature as responsible of this phenotypic variability) [12] was detected in B.A., B.B. and their mother; the grandmother and the aunt refused genetic investigations. Data of B.A. and B.B. are reported in Table 1 (Patients number 1 and 2).
G.V. (female, 15 years old) has been referred to us due to an occasionally detected HY. She was born at term (40 weeks), LGA (4800 g) and was adopted at 2 years of age; neonatal neurological damage of unknown origin was reported. During childhood, she presented generalized epilepsy, treated with valproic acid, and moderate psychomotor impairment. Laboratory investigations, executed during HY (glucose value 42 mg/dL), showed hyperinsulinism (20,1 μUI/ml), while ammonium, lactic acid, cortisol, IGF-1, plasmatic aminoacids, acylcarnitines and urinary organic acids profiles were normal. V455L heterozygous mutation of Glucokinase gene (GCK) was detected; this mutation has not been yet described in literature. She started Dx-treatment at the dose of 8 mg/kg/die with a good response. Data of G.V. are reported in Table 2 (patient number 1).
Flow-chart
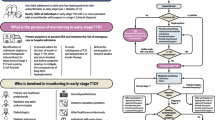
From the observation of the reported cases, particularly in consideration of late presentation cases, we tried to design a new CH diagnostic flow-chart (Fig. 1) expanding the diagnostic part of Maiorana’s algorithm by putting on the first step the age of presentation: section A refers to CH cases with precocious presentation while section B refers to CH cases with late presentation. In section A, the occurrence of post meal HY directs either to GLUD1, HADH or UCP2 [16,17,18,19] defects, depending on the associated metabolic findings. The presence of a non-specific hypoglycemic pattern with positive HY family history can point toward ABCC8/KCJN11 mutations; if they are not altered, firstly GCK and then HNF1α/4α should be tested, as already reported. In section B, the non-specific hypoglycemic pattern with hypo/hyperglycemia fluctuation points towards the ABCC8/KCJN11 mutation: if not altered, firstly GCK and then HNF1α/4α should be tested. Mild hyperglycemia and the history of the previous HY point toward ABCC8/KCNJ11 or HNF1α/4α mutations, especially in late onset cases. HY occurring after physical exercise directs toward SCL16A1 mutation [20, 21]; post prandial HY, especially if associated with epilepsy or hyperammonemia, can suggest GLUD1 mutation [16, 17].

Clinical flow chart to diagnose CHI. GDM: Gestational Diabetes Mellitus; IGT: Impaired Glucose Tolerance; IFG: Impaired Fasting Glucose; Hypo: Hypoglycaemia; Hyper: Hyperglycaemia; Normo: Normoglycaemia
Discussion
Observations made on the 22 patients according to the literature are: a higher prevalence of familiar GMD, of LGA, of preterm birth and precocious HY during the first 72 h in ChD subjects; good Dx-responsiveness in MeD and unidentified defects, but only partial in ChD [22].
Literature concerning follow-up of CH cases generally focuses only on diabetes secondary to surgery: in our cases two subjects showed diabetes after subtotal pancreasectomy and the other two were the two brothers of the B. family who had the ABCC8 mutation. Interestingly 9/22 patients developed obesity during their follow up: 5 patients with ChD, 1 with GLUD1 and 3 with unidentified defects. This finding could be linked to ABCC8 role in susceptibility to obesity, as already reported in Type 2 Diabetes subjects [23], probably facilitated by hyperinsulinism; however, it is known that some ABCC8 Single Nucleotide Polymorphisms seem to be protective against metabolic syndrome [24]. Furthermore, the presence of neurological impairment, reported in 4 subjects affected by precocious and severe forms of ChD, 1 with GCK (case G.V. above described) and 1 with unidentified defect, is in agreement with the literature.
CH diagnostic algorithms start from the metabolic investigations [16, 17, 25, 26] and proceed with molecular diagnosis and genotype-phenotype correlation. Dx trial was considered the crucial step in CH treatment: resistant patients must be treated with octreotide and need to be characterized by 18F-DOPA PET-CT, to address the appropriate surgery (near-total pancreatectomy for diffuse forms or confined excision for focal forms) [2, 6]. On the other hand, the most recent approach starts from the research of peculiar metabolic findings to guide the genetic screening, which is the fundamental step, rather than the Dx-responsiveness, to select patients for 18F-DOPA PET-CT [9]. As a matter of fact, these authors have found that, although rare, some focal forms could be Dx-responsive and could be completely solved by surgery. Consequently, 18F-DOPA PET-CT is indicated in case of one recessive paternal ABCC8/KCNJ11 mutation in order to search for focal lesions, or when no mutation in the principal causative genes (ABCC8/KCNJ11, HNF1/4, GCK, HADH, SLC16A1, UCP2) is found, to search for atypical forms.
CH can occur throughout the entire childhood or adulthood. All available diagnostic algorithms don’t take into account late-presentation cases which could represent late forms but also misdiagnosed and mild forms. It is known that ABCC8 mutations can cause CH, evolving into hyperglycemia and gestational diabetes [10, 13, 27, 28]. Concerning our cases, the family of B.A. and B.B. is similar to a case previously described with a c.4435G > A mutation [12].
GCK activating mutations cause a heterogeneous phenotype: the severity of the symptoms and the age of presentation present substantial variations between affected individuals, even within the same family [22, 23]. For this reason, testing GCK gene is recommended in subjects with a non-specific pattern of HY. In our case, GCK c.1363G > T mutation has not been reported yet.
Our experience suggests that the algorithm to better diagnose CH should consider a wider approach, starting from the age of the first detection. When CH develops into diabetes, HY usually occurred during the early childhood and, in subsequent years, occurred in post-prandial time, after high glycemic index meals. In presence of family history of HY and/or Impaired Glucose Tolerance, Impaired Fasting Glucose or GDM, the hypothesis of ABCC8/KCNJ11, HNF1A or HNF4A gene mutations can be formulated. In particular, in ABCC8 gene mutations, hyperglycemia generally starts in adolescence, while in HNF1α/4α in prepubertal age [10,11,12,13,14].
As affirmed by others, 18F-DOPA PET-CT should be performed when a focal form is suspected in the presence of Dx-unresponsiveness (although some rare focal forms respond to diazoxide) and if no mutation is detected at all; in these patients images provided by 18F-DOPA PET-CT can address the choice of surgical treatment [8, 9, 27, 29]. Thus, in late presentation cases, we can assume that 18F-DOPA PET-CT should be performed only to exclude insulinoma, since it can occur at any age, but is very rare in pediatric age, and could represent a manifestation of Multiple endocrine neoplasia type 1 (MEN1) [30,31,32]. Insulinoma could either be detected by 18F-DOPA PET-CT or (68) Ga-DOTATATE [32]. Without a suspect of Insulinoma, it is difficult that 18F-DOPA PET-CT should be indicated for subjects with late presentation. ABCC8/KCNJ11 late diagnosed cases reported in literature are mostly due to dominant mutations which determine a diffused form; likewise MeD forms are all diffused [10,11,12,13,14].
In our cases, the rate of positivity of genetic tests is 73% (16/22), significantly higher than reported in literature (45–50%) [5]. It probably depends on a strict collaboration between experts of diabetes, metabolism and neonatal pathology in our University Hospital.
Conclusion
In conclusion, since CH may start not only in the neonatal period or infancy, but also in childhood or even later, it may be useful to present a diagnostic flow-chart that includes also these cases by considering the glycemic alterations of the whole family, both in the present and in the past.
Availability of data and materials
All data generated during this study are included in this published article and its supplementary information files.
Abbreviations
- CH:
-
Congenital Hyperinsulinism
- HY:
-
Hypoglycemia
- ChD:
-
Channel defect
- MeD:
-
Metabolic defect
- SUR1:
-
Sulfonylurea Receptor 1
- Dx:
-
Diazoxide
- GDM:
-
Gestational Diabetes Mellitus
- GMD:
-
Glucose metabolism derangements
- LGA:
-
Large for gestational age
References
Stanley CA, Baker L. Hyperinsulinism in infants and children: diagnosis and therapy. Adv Pediatr Infect Dis. 1976;23:315–55.
Vora S, Chandran S, Rajadurai VS, Hussain K. Hyperinsulinemic hypoglycemia in infancy: current concepts in diagnosis and management. Indian Pediatr. 2015;52(12):1051–9.
Faletra F, Athanasakis E, Morgan A, Biarnés X, Fornasier F, Parini R, et al. Congenital hyperinsulinism: clinical and molecular analysis of a large Italian cohort. Gene. 2013;521(1):160–5.
Sogno Valin P, Proverbio MC, Diceglie C, Gessi A, di Candia S, Mariani B, et al. Genetic analysis of Italian patients with congenital hyperinsulinism of infancy. Horm Res Paediatr. 2013;79:236–42.
Rahman SA, Nessa A, Hussain K. Molecular mechanisms of congenital hyperinsulinism. J Mol Endocrinol. 2015;54(2):R119–29.
Demirbilek H, Hussain K. Congenital Hyperinsulinism: diagnosis and treatment update. J Clin Res Pediatr Endocrinol. 2017;9:69–87.
De Leon DD, Stanley CA. Congenital Hypoglycemia Disorders: New Aspects of Etiology, Diagnosis, Treatment and Outcomes: Highlights of the Proceedings of the Congenital Hypoglycemia Disorders Symposium, Philadelphia April 2016. Pediatr Diabetes. 2017;18(1):3–9.
Roženková K, Güemes M, Shah P, Hussain K. The diagnosis and Management of Hyperinsulinaemic Hypoglycaemia. J Clin Res Pediatr Endocrinol. 2015;7(2):86–97.
Maiorana A, Barbetti F, Boiani A, Rufini V, Pizzoferro M, Francalanci P, et al. Focal congenital hyperinsulinism managed by medical treatment: a diagnostic algorithm based on molecular genetic screening. Clin Endocrinol. 2014;81(5):679–88.
Abdulhadi-Atwan M, Bushman J, Tornovsky-Babaey S, Perry A, Abu-Libdeh A, Glaser B, et al. Novel de novo mutation in sulfonylurea receptor 1 presenting as hyperinsulinism in infancy followed by overt diabetes in early adolescence. Diabetes. 2008;57(7):1935–40.
Vieira TC, Bergamin CS, Gurgel LC, Moisés RS. Hyperinsulinemic hypoglycemia evolving to gestational diabetes and diabetes mellitus in a family carrying the inactivating ABCC8 E1506K mutation. Pediatr Diabetes. 2010;11(7):505–8.
Kapoor RR, Flanagan SE, James CT, McKiernan J, Thomas AM, Harmer SC, et al. Hyperinsulinaemic hypoglycaemia and diabetes mellitus due to dominant ABCC8/KCNJ11 mutations. Diabetologia. 2011;54(10):2575–83.
Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, et al. Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations. Diabetes. 2008;57(6):1659–63.
Stanescu DE, Hughes N, Kaplan B, Stanley CA, De León DD. Novel presentations of congenital hyperinsulinism due to mutations in the MODY genes: HNF1A and HNF4A. J Clin Endocrinol Metab. 2012;97(10):E2026–30.
Delvecchio M, Mozzillo E, Salzano G, Iafusco D, Frontino G, Patera PI, et al. Diabetes study Group of the Italian Society of pediatric endocrinology and diabetes (ISPED). Monogenic diabetes accounts for 6.3% of cases referred to 15 Italian pediatric diabetes centers during 2007 to 2012. J Clin Endocrinol Metab. 2017;102(6):1826–34.
Kapoor RR, Flanagan SE, Fulton P, Chakrapani A, Chadefaux B, Ben-Omran T, et al. Hyperinsulinism-hyperammonaemia syndrome: novel mutations in the GLUD1 gene and genotype-phenotype correlations. Eur J Endocrinol. 2009;161(5):731–5.
Palladino AA, Stanley CA. The hyperinsulinism/hyperammonemia syndrome. Rev Endocr Metab Disord. 2010;11(3):171–8.
Heslegrave AJ, Kapoor RR, Eaton S, Chadefaux B, Akcay T, Simsek E, et al. Leucine-sensitive hyperinsulinaemic hypoglycaemia in patients with loss of function mutations in 3-Hydroxyacyl-CoA dehydrogenase. Orphanet J Rare Dis. 2010;7:25.
Ferrara CT, Boodhansingh KE, Paradies E, Giuseppe F, Steinkrauss LJ, Topor LS, et al. Novel hypoglycemia phenotype in congenital Hyperinsulinism due to dominant mutations of uncoupling protein 2. J Clin Endocrinol Metab. 2017;102(3):942–9.
Meissner T, Otonkoski T, Feneberg R, Beinbrech B, Apostolidou S, Sipilä I, et al. Exercise induced hypoglycaemic hyperinsulinism. Arch Dis Child. 2001;84(3):254–7.
Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, et al. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate induced insulin release. Diabetes. 2003;52(1):199–204.
Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant KATP channel mutations. J Clin Invest. 2008;118(8):2877–86.
Hani EH, Clément K, Velho G, Vionnet N, Hager J, Philippi A, et al. Genetic studies of the sulfonylurea receptor gene locus in NIDDM and in morbid obesity among French Caucasians. Diabetes. 1997;46(4):688–94.
Chen YL, Pei D, Hung YJ, Lee CH, Hsiao FC, Wu CZ, Lin JD, Hsu CH, Chang JB, Hsieh CH. Associations between genetic variants and the severity of metabolic syndrome in subjects with type 2 diabetes. Genet Mol Res. 2015;14(1):2518–26.
Arnoux JB, Verkarre V, Saint-Martin C, Montravers F, Brassier A, Valayannopoulos V, Brunelle F, Fournet JC, Robert JJ, Aigrain Y, Bellanné-Chantelot C, de Lonlay P. Congenital hyperinsulinism: current trends in diagnosis and therapy. Orphanet J Rare Dis. 2011;6:63. https://doi.org/10.1186/1750-1172-6-63 Review. PMID: 21967988 Free PMC Article.
Senniappan S, Shanti B, James C, Hussain K. Hyperinsulinaemic hypoglycaemia: genetic mechanisms, diagnosis and management. J Inherit Metab Dis. 2012;35(4):589–601. https://doi.org/10.1007/s10545-011-9441-2 Epub 2012 Jan 10. Review. PMID: 22231386.
Gutgold A, Gross DJ, Glaser B, Szalat A. Diagnosis of ABCC8 congenital Hyperinsulinism of infancy in a 20-year-old man evaluated for factitious hypoglycemia. J Clin Endocrinol Metab. 2017;102(2):345–9. https://doi.org/10.1210/jc.2016-3254.
Morishita K, Kyo C, Yonemoto T, Kosugi R, Ogawa T, Inoue T. Asymptomatic congenital Hyperinsulinism due to a Glucokinase-activating mutation, Treated as Adrenal Insufficiency for Twelve Years. Case Rep Endocrinol. 2017;2017:4709262. https://doi.org/10.1155/2017/4709262 Epub 2017 Jan 9.
Yorifuji T. Congenital hyperinsulinism: current status and future perspectives. Ann Pediatr Endocrinol Metab. 2014;19(2):57–68.
Gϋemes M, Rahman SA, Kapoor RR, Flanagan S, Houghton JAL, Misra S, Oliver N, Dattani MT, Shah P. Hyperinsulinemic hypoglycemia in children and adolescents: recent advances in understanding of pathophysiology and management. Rev Endocr Metab Disord. 2020. https://doi.org/10.1007/s11154-020-09548-7.
Jani N, Moser AJ, Khalid A. Pancreatic endocrine tumors. Gastroenterol Clin N Am. 2007;36(2):431–9 x-xi. Review. PMID: 17533088.
Tuzcu SA, Pekkolay Z, Kılınç F, Tuzcu AK. 68Ga-DOTATATE PET/CT can be an alternative imaging method in Insulinoma patients. J Nucl Med Technol. 2017;45(3):198–200. https://doi.org/10.2967/jnmt.117.192708 Epub 2017 Aug 10.
Acknowledgements
Not applicable.
Funding
There is no funding source.
Author information
Authors and Affiliations
Contributions
AC and ADM researched data and wrote the manuscript. EM and VF contributed to the discussion and revised the manuscript. FMR and PB researched data. AF conceived and revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable: this article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
Subjects and their caregivers gave their written informed consent to publish their case.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Casertano, A., De Matteis, A., Mozzillo, E. et al. Diagnosis of congenital Hyperinsulinism can occur not only in infancy but also in later age: a new flow chart from a single center experience. Ital J Pediatr 46, 131 (2020). https://doi.org/10.1186/s13052-020-00894-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13052-020-00894-5