
Abstract
Gastrointestinal cancer, the most common solid tumor, has a poor prognosis. With the development of high-throughput sequencing and detection technology, recent studies have suggested that many chemical modifications of human RNA are involved in the development of human diseases, including cancer. m6A, the most abundant modification, was revealed to participate in a series of aspects of cancer progression. Recent evidence has shown that methyltransferase-like 3 (METTL3), the first identified and a critical methyltransferase, catalyzes m6A methylation on mRNA or non-coding RNA in mammals, affecting RNA metabolism. Abnormal m6A levels caused by METTL3 have been reported to be involved in different aspects of cancer development, including proliferation, apoptosis, and metastasis. In this review, we will shed light on recent findings regarding the biological function of METTL3 in gastrointestinal cancer and discuss future research directions and potential clinical applications of METTL3 for gastrointestinal cancer.
Similar content being viewed by others

Background
It is well known that cancer is a multistage genetic and epigenetic disease with a complex etiology involving mutation, upregulation, downregulation, and deletion of oncogenes and tumor suppressor genes [1,2,3,4]. Gene amplification/deletion/mutation or chromosomal translocation is abnormal genetic changes that lead to tumorigenesis and tumor development [5, 6]. Recently, there has been increasing evidence that epigenetic regulation plays a major role in cancer [7, 8]. Epigenetic modifications are heritable and reversible and can regulate gene expression and cancer progression without DNA sequence changes [1, 5]. Previous studies mostly focused on the role of DNA methylation, histone modification (methylation and acetylation), and non-coding RNAs in the biological function of cancer [9,10,11] (Fig. 1).

Abnormal genetics and epigenetics contribute to cancer development. Genetic changes mainly include gene amplification, deletion, mutation, and chromosomal translocation, while epigenetic changes include DNA methylation, histone modification (methylation, acetylation, etc.), non-coding RNA, and RNA methylation/acetylation. Abnormal genetics and epigenetics lead to oncogene activation and tumor suppressor gene inactivation, which result in uncontrolled cell growth and decreased apoptosis. With the development of cancer, tumor cells evade the immune system and promote angiogenesis; tumor cells can also invade the stroma via migration and invasion and enter the lymph vessels or blood vessels to cause distant metastasis
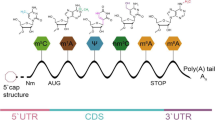
Similar to DNA/histone modifications, more than 100 chemical modifications have been found in human RNA, which has become a hot research topic in the biological sciences and extends to a novel field of RNA modification-mediated epigenetic regulation [12,13,14,15] (Fig. 1). RNA methylation is the main chemical modification of human RNA; the types of RNA methylation include 5-methylcytosine (m5C), 1-methylguanosine (m1G), m2G, m6G, m7G, N6-methyladenosine (m6A), and m1A [13]. m6A modification is the most abundant RNA modification and ubiquitously occurs in eukaryotic RNA [16, 17]. m6A modifications account for approximately 0.1–0.4% of adenosine molecules in the isolated RNA from mammals [18]. m6A modification can regulate RNA stability, splicing, transport, localization, or translation and has been reported to play a critical role in different diseases, including cancer [19]. Most m6A sites are found within the consensus sequence RRm6ACH (R = G or A, H = A, C, or U) [20, 21]. m6A modification is reversible and dynamic in mammalian cells and can be installed by m6A methyltransferases (writers) and removed by m6A demethylases (erasers). In addition, specific RNA-binding proteins, also called readers, can recognize and bind to the m6A motif and influence RNA metabolism processes, including RNA stabilization, decay, splicing, translation, and nuclear export [21, 22] (Fig. 2). To date, an increasing number of novel multiple m6A regulatory enzymes (writers, erasers, and readers) have been identified to be involved in the regulation of m6A [20].

Summary of the m6A modification mechanism mediated by writers, erasers, and readers. The methyltransferase complex composed of the METTL3-METTL14-WTAP core component and other regulatory cofactors (KIAA1429, RBM15, ZC3H13, and METTL16) catalyses methylation at the N6 adenosine. Moreover, m6A can be reversibly removed by m6A eraser proteins (FTO and ALKBH5). m6A can also be recognized by m6A-binding proteins to affect mRNA fate. YTHDC1 can affect the exportation of m6A-modified mRNA transcripts from the nucleus to the cytoplasm, while METTL3, EIF3, IGF2BP1/2/3, YTHDF1/3, and YTHDC2 can promote the translation of RNA. YTHDC1, HNRNPA2B1, and HNRNPC can promote RNA splicing. IGF2BP1/2/3 can enhance RNA stability, while YTHDF2/3 and YTHDC2 accelerate the decay of RNA
m6A methyltransferases are multicomponent methyltransferase complexes that consist of at least 7 “writer” proteins, including methyltransferase-like 3/14/16 (METTL3/14/16), WT1-associated protein (WTAP), vir-like m6A methyltransferase-associated (VIRMA, also called KIAA1429), zinc finger CCCH-type containing 13 (ZC3H13), and RNA-binding motif protein 15 (RBM15) [21, 23]. Among the complexes, METTL3 is the sole catalytic subunit that binds to the methyl donor S-adenosylmethionine (SAM) and catalyzes methyl group transfer [23]. In addition to METTL3, METTL14 serves as a pseudomethyltransferase to support METTL3 and recognizes target RNAs, while WTAP ensures the localization of the METTL3-METTL14 heterodimer to the nuclear speckle and promotes catalytic activity [24, 25]. METTL16 catalyzes m6A modification in U6-snRNA and participates in pre-RNA splicing, while RBM15 binds the m6A complex and recruits it to special RNA sites [26, 27]. ZC3H13 can enhance m6A by bridging WTAP to the mRNA-binding factor Nito [28, 29]. KIAA1429 directs m6A in the 3′-UTR and near the stop codon by recruiting the methyltransferase complex to modulate region-selective methylation [30]. Moreover, RNA m6A modification could also be removed by the 2 demethylases, including alpha-ketoglutarate-dependent dioxygenase (FTO) and alkB homolog 5 RNA demethylase (ALKBH5), thus conferring reversible and dynamic regulation of m6A methylation [23] (Fig. 2).
Among the m6A methyltransferases, METTL3 was first identified and acts as the major catalytic enzyme (writer) to catalyze m6A modification on mRNA and non-coding RNA [31, 32]. In recent years, the biological functions of METTL3 have been widely studied to be involved in various types of cancer development, including gastric cancer (GC), colorectal cancer (CRC), liver cancer (LC), and pancreatic cancer (PC). In the present review, we will focus on the functional role of METTL3 in gastrointestinal cancer identified by the recent findings of our and other laboratories and discuss directions for future research and potential clinical application of METTL3 for gastrointestinal cancer.
The role of METTL3 in gastrointestinal cancer
Recent studies have shown that METTL3 is closely associated with the processes involved in the progression of gastrointestinal cancer, including tumor proliferation, apoptosis, metastasis, angiogenesis, chemo/radiotherapy resistance, glycolipid metabolism, and cancer stem cell (CSC) maintenance (Fig. 3). We herein present a summary of the recent findings of METTL3 in gastrointestinal cancer (Table 1).

The biological function of METTL3 in gastrointestinal cancer. METTL3 regulates the differential expression of oncogenes and tumor suppressor genes at posttranscriptional levels by mediating RNA stability and translation, which contributes to processes involved in the development of gastrointestinal cancer, including cell proliferation, apoptosis, invasion, migration, metastasis, angiogenesis, radiochemotherapy resistance, glycolysis/lipid metabolism, and CSC maintenance in gastrointestinal cancer
The role of METTL3 in the proliferation and apoptosis of gastrointestinal cancer
The basic characteristics of cancer include the ability to proliferate indefinitely and evade apoptosis, which are the hallmarks of cancer [55]. Many studies have demonstrated that METTL3 promotes cell proliferation and inhibits apoptosis in gastrointestinal cancer by regulating several different targets or pathways, including mRNAs and non-coding RNAs [56]. Our study showed that METTL3 protein levels were significantly upregulated in GC, contributing to poor prognosis [33]. In addition, overexpression of METTL3 accelerated GC cell proliferation both in vitro and in vivo. Furthermore, we confirmed that elevated METTL3 promoted cell proliferation using a GC organoid model. Mechanistically, METTL3 promotes m6A methylation on HDGF mRNA, and the reader insulin-like growth factor 2 mRNA-binding protein 3 (IGF2BP3) directly binds to the m6A site and enhances hepatoma-derived growth factor (HDGF) mRNA stability. Further, secreted HDGF promotes tumor angiogenesis, while nuclear HDGF activates glycolysis-related proteins, including enolase 2 (ENO2) and solute carrier family 2 member 4 (GLUT4), followed by an increase in glycolysis to cause tumor growth in GC [33]. Other studies also showed that METTL3 promotes GC cell proliferation and inhibits apoptosis through alterations of other targets and pathways, including an increase in preprotein translocation factor (SEC62) mRNA stability [36] and the activation of the AKT/MYC-related pathway [39, 40]. In addition to regulating mRNA, METTL3 also influences non-coding RNA metabolism in GC. For example, METTL3 interacts with the non-coding RNA LINC00470 to suppress phosphatase and tensin homolog (PTEN) mRNA stability, resulting in GC cell proliferation [34]. Recent findings showed that METTL3 expression was higher in CRC tissues than in normal tissues and that this feature indicated poor prognosis; upregulation of METTL3 promoted CRC tumor growth by stabilizing SRY-box 2 (SOX2) [43] and cyclin E1 (CCNE1) mRNA in an m6A-dependent manner [45]. However, another study showed that METTL3 was a tumor suppressor that inhibited CRC cell proliferation [42]. In human hepatocellular carcinoma (HCC), METTL3 was found to be significantly upregulated and contributed to the poor prognosis of HCC patients [50]. Functionally, knockdown or knockout of METTL3 inhibited HCC growth, while the opposite result was observed when METTL3 was overexpressed. Mechanistically, METTL3 inhibited suppressor of cytokine signaling 2 (SOCS2) expression via m6A-YTHDF2-dependent mRNA degradation. In addition to regulating mRNA, METTL3 also promoted HCC cell proliferation by enhancing miR-6079 expression or LINC00958 transcript stability [48]. METTL3 was also an oncogene that promoted PC cell proliferation by accelerating miR-25 processing and maturation [53]. In summary, substantial evidence has revealed that METTL3 is an independent prognostic factor for gastrointestinal cancer and that METTL3 is essential for the proliferation of gastrointestinal cancer as it regulates the stability, degradation, and maturation of mRNA or non-coding RNA.
The role of METTL3 in the migration, invasion, and metastasis of gastrointestinal cancer
One of the main hallmarks of cancer is the activation of invasion and metastasis [55]. Metastasis is responsible for more than 90% of cancer-related deaths related to solid tumors [57]. In our study [33], overexpression of METTL3 promoted GC cell migration and invasion in vitro and liver metastasis in vivo through enhancing the stability of HDGF mRNA. Others also confirmed that METTL3 promoted the migration and invasion of GC by targeting the MYC-related pathway [40] and interacting with LINC00470 to suppress PTEN mRNA in GC [34]. Tumor epithelial-mesenchymal transition (EMT) refers to the process by which epithelial cells lose polarity, close connections, and cell-cell adhesion properties and acquire infiltration and migration abilities, which leads cells to adopt interstitial cell morphology and characteristics; this is an early and crucial step in metastasis progression [58]. Recent findings showed that METTL3 was required for the EMT process in vitro and for metastasis in vivo as METTL3 enhances zinc finger MYM-type containing 1 (ZMYM1) mRNA stability [35, 37]. In HCC, METTL3 promoted EMT and metastasis through triggering polysome-mediated translation of Snail family transcriptional repressor (Snail) mRNA [49]. In addition, knockdown of METTL3 inactivated the AKT pathway to reduce GC cell migration and invasion [39], and upregulation of METTL3 facilitated metastasis of CRC via the miR-1246/SPRED2/MAPK pathway [41] and the stabilization of SOX2 expression [43]. However, Deng et al. showed that METTL3 suppressed CRC cell migration via p38/ERK pathways [42]. In HCC, METTL3 also accelerated HCC cell metastasis by promoting the stability of the oncogenic non-coding LINC00958 RNA transcript [48] and reducing the mRNA expression of tumor suppressor SOCS2 [50]. A recent study also revealed that METTL3 promoted cell invasion and migration in PC [52], but the mechanism still needs to be further studied. Collectively, these findings reveal that METTL3 could serve as an oncogene in the EMT and metastasis of gastrointestinal cancer.
The role of METTL3 in angiogenesis of gastrointestinal cancer
Angiogenesis is one of the most basic factors in tumor growth and metastasis and can provide nutrition for tumor tissue metabolism. In our study [33], we found that microvessel density was significantly higher in tumor tissues with high METTL3 expression than in those with low expression. In vitro, we found that the upregulation of METTL3 promoted human umbilical vein endothelial cell (HUVEC) growth and tube formation via the secretion of HDGF. Our results indicated that METTL3 may promote GC growth and metastasis by promoting angiogenesis. However, whether METTL3 is involved in the angiogenesis of other gastrointestinal cancers and thus affects the malignant process of tumors still needs further study.
The role of METTL3 in chemo- and radiotherapy resistance of gastrointestinal cancer
Chemotherapy and radiotherapy are widely used in the treatment of solid tumors [59]. However, resistance to chemotherapy and radiotherapy due to a series of genetic and epigenetic alterations limits its efficacy [59]. A recent study revealed that lncRNA ARHGAP5-AS1 was significantly increased in chemoresistant GC cells, which contributed to chemoresistance. Furthermore, ARHGAP5-AS1 stabilized ARHGAP5 mRNA by recruiting METTL3 to stimulate m6A modification of ARHGAP5 mRNA and contribute to drug resistance, indicating that METTL3 was involved in chemotherapy resistance in GC [38]. In addition, the upregulation of METTL3 promoted preferential pre-mRNA splicing to produce the p53 R273H mutant protein and resulted in acquired multidrug resistance in CRC [44]. In PC, overexpression of METTL3 also contributed chemo- and radioresistance via activation of the MAPK, ubiquitin, and RNA splicing pathways [54]. These observations suggest that METTL3 is involved in chemoradiotherapy resistance in gastrointestinal cancer, indicating that METTL3 may be a potential target for reversing chemoradiotherapy resistance.
The role of METTL3 in glycolipid metabolism in gastrointestinal cancer
Abnormal energy metabolism (glucose metabolism, lipid metabolism, and amino acid metabolism) is one of the main characteristics of cancer [55, 60]. Cancer cells reprogram metabolism to support malignant tumor initiation and progression [61, 62]. It has been proven that the abnormal metabolism of tumor glucose and lipids is an important part of tumor metabolic reprogramming, which is closely related to tumor occurrence, development, metastasis, and recurrence [63]. In our study [33], the METTL3-HDGF axis activated glycolysis-related enzymes (ENO2 and GLUT4) at the transcriptional level to increase glycolysis, leading to GC cell proliferation and metastasis. In addition, a recent study also showed that METTL3-mediated induction of tumorigenesis in CRC was dependent upon on cell glycolysis. Mechanistically, METTL3 directly interacted with the 5′- or 3′-UTR regions of Hexokinase 2 (HK2) and the 3′-UTR region of solute carrier family 2 member 1 (GLUT1), further stabilizing these two genes and activating the glycolysis pathway [46]. Moreover, it has also been reported that a decrease in METTL3 downregulated intracellular glucose uptake and lactate production via inhibition of mTORC1 activity in HCC cells, indicating that METTL3 is involved in glycolysis activity in HCC [51]. Recent findings also suggested that increased expression of METTL3 could upregulate LINC00958 and increase lipogenesis to promote HCC progression [48]. These findings suggest that METTL3 may act as an oncogene to promote glycolysis and lipid synthesis by targeting related enzymes in gastrointestinal cancer.
The role of METTL3 in CSCs of gastrointestinal cancer
CSCs maintain the vitality of cancer cell growth via self-renewal and infinite proliferation [64,65,66]. The invasion and migration of tumor stem cells make metastasis possible. CSCs can be dormant for a long time and can confer drug resistance [65, 67]. Therefore, cancer often recurs in the period of time after most common tumor cells have been eliminated by conventional therapy. Recent findings have shown that m6A mRNA modification is critical for the self-renewal and tumorigenesis of glioblastoma stem cells [68] and breast cancer stem cells [69]. In CRC, METTL3 facilitates CRC self-renewal and increases stem cell frequency by preventing the mRNA degradation of SOX2, a cancer stem cell marker [43]. The above results support the oncogenic role of METTL3 in promoting CRC stemness. Whether METTL3 regulates cancer stemness in other gastrointestinal cancers deserves further study.
Upstream regulators of METTL3
Most studies focus on the function of METTL3 in cancer, and only a few studies have explored why METTL3 expression is abnormal in cancer. Current evidence suggests that histone modification and non-coding RNAs can influence the expression of METTL3. In GC, our study revealed that P300-mediated activation of H3K27 acetylation (H3K27ac) led to upregulated METTL3 expression in GC. In a cigarette smoke condensate-induced malignant transformation model of pancreatic duct epithelial cells, METTL3 was increased due to hypomethylation at the METTL3 promoter caused by the cigarette smoke condensate [53]. Through bioinformatics analysis, it was found that the transcription factor GFI-1 might activate METTL3 mRNA [37], but further functional verification is needed. It has been reported that miR-4429 reduces METTL3 expression in GC [36] and that miR-24-2 increases METTL3 expression in HCC (Fig. 4). Collectively, these results suggest that histone modification, promoter methylation, and non-coding RNAs can affect the expression of METTL3. Studying the upstream regulatory mechanism of METTL3 caused by epigenetic modification will allow us to better understand the biological function of METTL3 in cancers.

The upstream region of METTL3 in gastrointestinal cancer. Histone H3K27ac and hypomethylation at the promoter can increase METTL3 mRNA expression. The transcription factor GFI-1 might promote METTL3 mRNA expression. Non-coding RNA ARHGAP5-AS1 and miR-24-2 can promote METTL3 mRNA expression, while miR-4429 can inhibit METTL3 mRNA expression
Potential clinical application of METTL3
The above evidence shows that METTL3 plays a critical role in the progression of gastrointestinal cancer, indicating that METTL3 is a promising biomarker for clinical diagnosis and therapeutic response prediction and is a potential therapeutic target. According to our [33] and others’ studies [35, 40], METTL3 might be an independent prognostic factor for GC patients. We conducted a time-dependent receiver operating characteristic curve analysis, which indicated that the combination of the TNM stage and METTL3 risk score enhanced the ability of the model to predict prognosis. In addition, it has also been shown that overexpression of METTL3 contributes to a poor prognosis in CRC [43] and HCC [50] and correlates positively with tumor metastasis [41]. Therefore, METTL3 may be a biomarker of advanced GC, CRC, and HCC. Current studies are focused on the role of METTL3 in advanced gastrointestinal cancer and whether the expression of METTL3 is increased in the early stage of gastrointestinal cancer requires further study. If METTL3 is involved in the early stage of gastrointestinal cancer, it could improve early cancer diagnosis and prevention.
Given the oncogenic role of METTL3 in gastrointestinal cancer and its methyltransferase activity, it appears to be a promising therapeutic target for gastrointestinal cancer. Currently, no specific inhibitors of METTL3 have been found; thus, new potential candidates merit further exploration. Recently, Bedi et al. screened a library of 4000 analogs and derivatives of the adenosine moiety of SAM by high-throughput docking into METTL3, and two compounds showed good ligand efficiency [70]. They are further exploring potent and selective inhibitors of METTL3. Additionally, exploring inhibitors targeting molecules upstream or downstream of METTL3 is also a potential strategy for gastrointestinal cancer treatment. Chemotherapy and radiotherapy are still the main clinical treatments for gastrointestinal cancer; however, resistance to radio- and chemotherapy is inevitable and contributes to poor prognosis [59]. Therefore, the combination of chemo- and radiotherapy with inhibition of METTL3 expression or activity is a promising therapeutic strategy and is expected to be explored in the future.
Discussion
The present review suggests that expression of the methyltransferase METTL3 is significantly increased in various gastrointestinal cancer (GC, CRC, HCC, and PC) cells compared to normal cells and acts as an oncogene to promote the progression of gastrointestinal cancer; METTL3 can serve as a clinical diagnostic and therapeutic target. However, more large-scale and multicentre data are required to further explore the function of METTL3, which may lay a foundation for individualized precision therapy in gastrointestinal cancer.
Recent findings have shown that m6A modification and its regulators play important and diverse biological functions in the development of various cancers [23]. Among the m6A modulators, METTL3 is thoroughly and widely studied [23, 71]. As noted herein, METTL3 is involved in different aspects of gastrointestinal cancer progression, including cancer cell proliferation, apoptosis, invasion, migration, metastasis, angiogenesis, radiochemotherapy resistance, glycolysis/lipid metabolism, and CSC maintenance. The underlying mechanism of METTL3 is complex and involves multiple molecules and pathways in gastrointestinal cancer (Table 1). In GC, studies by our lab and others have confirmed the oncogenic role of METTL3 in promoting the malignant process of GC by regulating different targets or pathways. Our findings revealed that METTL3 expression is significantly increased in GC tissues and correlated with poor prognosis of GC patients and that the METTL3/HDGF/GLUT4/ENO2 axis promotes GC tumorigenesis and metastasis via an increase in glycolysis and angiogenesis [33]. METTL3 also promotes GC cell growth and metastasis by interacting with LINC00470 [34], enhancing ZMYM1 [35] and SEC62 mRNA stability [36]. It has also been reported that METTL3 can activate the AKT pathway [39], MYC-mediated pathway [40], and EMT process [37] to promote cell growth and metastasis and inhibit apoptosis in GC. LncRNA ARHGAP5-AS1 recruits METTL3 to stimulate m6A modification of ARHGAP5 to stabilize its mRNA, resulting in chemoresistance in GC [38]. In CRC, METTL3 is highly expressed in metastatic CRC and is associated with poor prognosis [43]. METTL3-mediated m6A modification is closely correlated with glycolysis pathway activation in CRC patient tissues [46]. Furthermore, METTL3 promotes CRC cell growth and metastasis by promoting CRC stemness by preventing SOX2 mRNA degradation [43]. METTL3 also directly stabilizes HK2 and GLUT1 expression through an IGF2BP2/3-dependent mechanism to promote CRC tumorigenesis via an increase in glycolysis [46]. It has also been shown that METTL3 promotes the malignant process of CRC by accelerating the maturation of pri-miR-1246 [41] or stabilizing CCNE1 mRNA. METTL3 also promotes CRC multidrug resistance via the acceleration of p53 R273H pre-mRNA splicing [44]. Interestingly, only one study indicated that METTL3 is a tumor suppressor that inhibits CRC cell proliferation, migration, and invasion, which may be attributed to differences in tumor tissue origin, intratumoral heterogeneity, and research methods [42]. In HCC, METTL3 is frequently upregulated and associated with poor prognosis of HCC patients [50]. METTL3 could promote glycolysis via activation of the mTORC1 pathway and accelerate lipogenesis by enhancing LINC00958 RNA transcript stability [48], which contributes to HCC progression. In addition, METTL3 promotes the malignant process of HCC through YTHDF2-dependent silencing of SOCS2 [50] and the promotion of miR6079 expression [47]. Furthermore, METTL3 triggers polysome-mediated translation of Snail mRNA to accelerate the EMT process of HCC [49]. In PC, METTL3 promotes PC tumorigenesis by accelerating the miR-25 process and maturation [53]. It has also been reported that METTL3 can activate MAPK, ubiquitin, and RNA splicing pathways to promote chemo- and radioresistance of PC [54]. In summary, METTL3 regulates the differential expression of oncogenes and tumor suppressor genes at posttranscriptional levels by influencing their RNA stability and translation, which contribute to the development of gastrointestinal cancer.
The present review showed that METTL3 influences the progression of gastrointestinal cancer through regulating the maturation, decay, stability, translation, and splicing of mRNA or non-coding RNA. However, little attention has been paid to the role of METTL3 in the crosstalk between cancer cells and tumor-associated fibroblasts and immune cells in the tumor microenvironment. A previous study showed that the deletion of METTL3 in mouse T cells disrupts T cell homeostasis and differentiation by targeting the IL-7/STAT5/SOCS pathways [72]. Another study also showed that METTL3-mediated mRNA m6A methylation promotes dendritic cell (DC) activation and function. Mechanistically, METTL3-mediated m6A of CD40, CD80, and TLR4 signaling adaptor Tirap transcripts enhances their translation in DCs to stimulate T cell activation and promote TLR4/NF-κB signaling-induced cytokine production [73]. Interestingly, in addition to m6A methyltransferase activity, METTL3 also promotes the translation of target transcripts in lung cancer cells independent of its catalytic activity [74]. The molecular mechanism of METTL3 in m6A regulation in gastrointestinal cancer biology still needs further exploration.
Most studies focus on the downstream effects of METTL3 in the development of gastrointestinal cancer, ignoring why METTL3 expression is dysregulated in gastrointestinal cancer. The current data suggest that H3K27ac of histones and hypomethylation at promoters can increase METTL3 expression in gastrointestinal cancer [33, 53]. Non-coding RNA ARHGAP5-AS1 [38], miR-24-2 [47], and miR-4429 [36] can also regulate METTL3 expression and influence the progression of gastrointestinal cancers. Whether there are other histone modifications or non-coding RNAs involved in the regulation of METTL3 needs further research and exploration. A recent study also reported that SUMOylation of METTL3 protein does not alter its stability or localization but significantly represses its m6A methyltransferase activity, leading to decreased m6A levels on mRNAs [75]. Whether there are other post-translational modifications, such as ubiquitination and glycosylation, involved in regulating the expression of METTL3 in gastrointestinal cancer needs further study.
Current studies refer to the biological function and mechanism of METTL3. Many studies have used METTL3 as a tumor biomarker, but the specificity and sensitivity of METTL3 in different types of gastrointestinal cancer need further study. In addition, few studies have focused on the screening of METTL3 inhibitors, which have great potential, but these studies are still in the early stages.
Conclusions
METTL3 plays a critical role in the development of gastrointestinal cancer, but there are still many problems that need further comprehensive study. Future research should be focused on (1) the role of METTL3 in the tumor microenvironment, (2) the molecular mechanism of modulating METTL3 expression and activity, and (3) the screening of specific inhibitors and their application in the clinic. Undoubtedly, METTL3 and its mediation of RNA m6A methylation in cancer are novel prognostic markers and predictive factors in gastrointestinal cancer.
Availability of data and materials
Not applicable.
Abbreviations
- ac4C:
-
N4-Acetylcytidine
- ALKBH5:
-
alkB Homolog 5, RNA demethylase
- ARHGAP5:
-
Rho GTPase-activating protein 5
- bcr/abl:
-
BCR activator of RhoGEF and GTPase/ABL proto-oncogene 1, non-receptor tyrosine kinase
- CCNE1:
-
Cyclin E1
- CRC:
-
Colorectal cancer
- CSC:
-
Cancer stem cell
- DC:
-
Dendritic cell
- EIF3:
-
Eukaryotic translation initiation factor 3 subunit A
- EMT:
-
Epithelial-mesenchymal transition
- ENO2:
-
Enolase 2
- FTO:
-
FTO alpha-ketoglutarate-dependent dioxygenase
- GC:
-
Gastric cancer
- GFI-1:
-
Growth factor independence 1
- GLUT1:
-
Solute carrier family 2 member 2
- GLUT4:
-
Solute carrier family 2 member 4
- H3K27ac:
-
Histone H3K27 acetylation
- HCC:
-
Hepatocellular carcinoma
- HDGF:
-
Hepatoma-derived growth factor
- HK2:
-
Hexokinase 2
- HNRNPA2B1:
-
Heterogeneous nuclear ribonucleoprotein A2/B1
- HNRNPC:
-
Heterogeneous nuclear ribonucleoprotein C
- HUVEC:
-
Human umbilical vein endothelial cells
- IGF2BP1/2/3:
-
Insulin-like growth factor 2 mRNA-binding protein 1/2/3
- IL-7:
-
Interleukin 7
- KIAA1429:
-
VIRMA, vir-like m6A methyltransferase associated
- LC:
-
Liver cancer
- m1A:
-
N1-Methyladenosine
- m1G:
-
1-Methylguanosine
- m2G:
-
1-Methylguanosine
- m5C:
-
5-Methylcytosine
- m6A:
-
N6-Methyladenosine
- m6G:
-
1-Methylguanosine
- m7G:
-
1-Methylguanosine
- MDM2:
-
MDM2 proto-oncogene
- METTL14:
-
Methyltransferase-like 14
- METTL16:
-
Methyltransferase-like 16
- METTL3:
-
Methyltransferase-like 3
- MYC:
-
MYC proto-oncogene, bHLH transcription factor
- NF-κB:
-
Nuclear factor kappa B subunit 1
- P53:
-
P53 tumor suppressor
- p53:
-
Tumor protein p53
- PC:
-
Pancreatic cancer
- PTEN:
-
Phosphatase and tensin homolog
- Rb:
-
RB transcriptional corepressor 1
- RBM15:
-
RNA-binding motif protein 15
- SAM:
-
S-Adenosylmethionine
- SEC62:
-
SEC62 homolog, preprotein translocation factor
- Snail:
-
Snail family transcriptional repressor
- SOCS:
-
Cytokine-inducible SH2-containing protein
- SOCS2:
-
Suppressor of cytokine signaling 2
- SOX2:
-
SRY-box 2
- SPRED2:
-
Sprouty-related EVH1 domain-containing 2
- STAT5:
-
Signal transducer and activator of transcription 5
- TLR4:
-
Toll-like receptor 4
- WTAP:
-
WT1-associated protein
- YTHDC1/2:
-
YTH domain-containing protein 1/2
- YTHDF1/2/3:
-
YTH N6-methyladenosine RNA-binding protein 1/2/3
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- ZMYM1:
-
Zinc finger MYM-type containing 1
References
Zhou S, Treloar AE, Lupien M. Emergence of the noncoding cancer genome: a target of genetic and epigenetic alterations. Cancer discovery. ss2016;6:1215–29.
Legoff L, D'Cruz SC, Tevosian S, Primig M, Smagulova F. Transgenerational inheritance of environmentally induced epigenetic alterations during mammalian development. Cells. 2019;8.
Mafficini A, Scarpa A. Genetics and epigenetics of gastroenteropancreatic neuroendocrine neoplasms. Endocrine reviews. 2019;40:506–36.
Zugazagoitia J, Guedes C, Ponce S, Ferrer I, Molina-Pinelo S, Paz-Ares L. Current challenges in cancer treatment. Clinical therapeutics. 2016;38:1551–66.
Chiba T, Marusawa H, Ushijima T. Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation. Gastroenterology. 2012;143:550–63.
Bashraheel SS, Domling A, Goda SK. Update on targeted cancer therapies, single or in combination, and their fine tuning for precision medicine. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2020;125:110009.
Li S, Kuo HD, Yin R, Wu R, Liu X, Wang L, et al. Epigenetics/epigenomics of triterpenoids in cancer prevention and in health. Biochemical pharmacology. 2020;113890.
Lin YT, Wu KJ. Epigenetic regulation of epithelial-mesenchymal transition: focusing on hypoxia and TGF-beta signaling. Journal of biomedical science. 2020;27:39.
Ning B, Li W, Zhao W, Wang R. Targeting epigenetic regulations in cancer. Acta biochimica et biophysica Sinica. 2016;48:97–109.
Kelly AD, Issa JJ. The promise of epigenetic therapy: reprogramming the cancer epigenome. Current opinion in genetics & development. 2017;42:68–77.
Kanwal R, Gupta K, Gupta S. Cancer epigenetics: an introduction. Methods in molecular biology. 2015;1238:3–25.
Traube FR, Carell T. The chemistries and consequences of DNA and RNA methylation and demethylation. RNA biology. 2017;14:1099–107.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nature reviews Molecular cell biology. 2017;18:31–42.
Gutbrod MJ, Martienssen RA. Conserved chromosomal functions of RNA interference. Nature reviews Genetics. 2020;21:311–31.
Arango D, Sturgill D, Alhusaini N, Dillman AA, Sweet TJ, Hanson G, et al. Acetylation of cytidine in mRNA promotes translation efficiency. Cell. 2018;175:1872–86 e24.
Yu S, Li X, Liu S, Yang R, Liu X, Wu S. N(6)-Methyladenosine: a novel RNA imprint in human cancer. Frontiers in oncology. 2019;9:1407.
Huang H, Weng H, Chen J. The biogenesis and precise control of RNA m(6)A methylation. Trends in genetics : TIG. 2020;36:44–52.
Desrosiers R, Friderici K, Rottman F. Identification of methylated nucleosides in messenger RNA from Novikoff hepatoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1974;71:3971–5.
Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nature reviews Molecular cell biology. 2014;15:313–26.
He L, Li H, Wu A, Peng Y, Shu G, Yin G. Functions of N6-methyladenosine and its role in cancer. Molecular cancer. 2019;18:176.
Chen XY, Zhang J, Zhu JS. The role of m(6)A RNA methylation in human cancer. Molecular cancer. 2019;18:103.
Meyer KD, Jaffrey SR. Rethinking m(6)A readers, writers, and erasers. Annual review of cell and developmental biology. 2017;33:319–42.
Huang H, Weng H, Chen J. m(6)A Modification in coding and non-coding RNAs: roles and therapeutic implications in cancer. Cancer cell. 2020;37:270–88.
Wang P, Doxtader KA, Nam Y. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Molecular cell. 2016;63:306–17.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell research. 2014;24:177–89.
Warda AS, Kretschmer J, Hackert P, Lenz C, Urlaub H, Hobartner C, et al. Human METTL16 is a N(6)-methyladenosine (m(6)A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO reports. 2017;18:2004–14.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m(6)A RNA methylation promotes XIST-mediated transcriptional repression. Nature 2016;537:369-373.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m(6)A machinery component Wtap/Fl(2)d. Genes & development. 2018;32:415–29.
Wen J, Lv R, Ma H, Shen H, He C, Wang J, et al. Zc3h13 regulates nuclear RNA m(6)A methylation and mouse embryonic stem cell self-renewal. Molecular cell. 2018;69:1028–38 e6.
Yue Y, Liu J, Cui X, Cao J, Luo G, Zhang Z, et al. VIRMA mediates preferential m(6)A mRNA methylation in 3′UTR and near stop codon and associates with alternative polyadenylation. Cell discovery. 2018;4:10.
Deng X, Su R, Weng H, Huang H, Li Z, Chen J. RNA N(6)-methyladenosine modification in cancers: current status and perspectives. Cell research. 2018;28:507–17.
Zhao W, Qi X, Liu L, Liu Z, Ma S, Wu J. Epigenetic regulation of m(6)A modifications in human cancer. Molecular therapy Nucleic acids. 2019;19:405–12.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2019. https://doi.org/10.1136/gutjnl-2019-319639 [Epub ahead of print].
Yan J, Huang X, Zhang X, Chen Z, Ye C, Xiang W, et al. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochemical and biophysical research communications. 2020;521:887–93.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Molecular cancer. 2019;18:142.
He H, Wu W, Sun Z, Chai L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m(6)A-caused stabilization of SEC62. Biochemical and biophysical research communications. 2019;517:581–7.
Liu T, Yang S, Sui J, Xu SY, Cheng YP, Shen B, et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. Journal of cellular physiology. 2020;235:548–62.
Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell death & disease. 2019;10:383.
Lin S, Liu J, Jiang W, Wang P, Sun C, Wang X, et al. METTL3 promotes the proliferation and mobility of gastric cancer cells. Open medicine. 2019;14:25–31.
Yang DD, Chen ZH, Yu K, Lu JH, Wu QN, Wang Y, et al. METTL3 promotes the progression of gastric cancer via targeting the MYC pathway. Frontiers in oncology. 2020;10:115.
Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, et al. Upregulated METTL3 promotes metastasis of colorectal cancer via miR-1246/SPRED2/MAPK signaling pathway. Journal of experimental & clinical cancer research : CR. 2019;38:393.
Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, et al. m(6)A methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. OncoTargets and therapy. 2019;12:4391–402.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m(6)A-IGF2BP2-dependent mechanism in colorectal carcinoma. Molecular cancer. 2019;18:112.
Uddin MB, Roy KR, Hosain SB, Khiste SK, Hill RA, Jois SD, et al. An N(6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochemical pharmacology. 2019;160:134–45.
Zhu W, Si Y, Xu J, Lin Y, Wang JZ, Cao M, et al. Methyltransferase like 3 promotes colorectal cancer proliferation by stabilizing CCNE1 mRNA in an m6A-dependent manner. Journal of cellular and molecular medicine. 2020;24:3521–33.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Molecular cancer 2020;19:72.
Yang Y, Song S, Meng Q, Wang L, Li X, Xie S, et al. miR24-2 accelerates progression of liver cancer cells by activating Pim1 through tri-methylation of histone H3 on the ninth lysine. Journal of cellular and molecular medicine. 2020;24:2772–90.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. Journal of hematology & oncology. 2020;13:5.
Lin X, Chai G, Wu Y, Li J, Chen F, Liu J, et al. RNA m(6)A methylation regulates the epithelial mesenchymal transition of cancer cells and translation of Snail. Nature communications. 2019;10:2065.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67:2254–70.
Lin Y, Wei X, Jian Z, Zhang X. METTL3 expression is associated with glycolysis metabolism and sensitivity to glycolytic stress in hepatocellular carcinoma. Cancer medicine. 2020;9:2859–67.
Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang J, et al. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathology, research and practice 2019;215:152666.
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, et al. Excessive miR-25-3p maturation via N(6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nature communications. 2019;10:1858.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. International journal of oncology. 2018;52:621–9.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Hu BB, Wang XY, Gu XY, Zou C, Gao ZJ, Zhang H, et al. N(6)-methyladenosine (m(6)A) RNA modification in gastrointestinal tract cancers: roles, mechanisms, and applications. Molecular cancer. 2019;18:178.
Nguyen DX, Massague J. Genetic determinants of cancer metastasis. Nature reviews Genetics. 2007;8:341–52.
Nieto MA, Huang RY, Jackson RA, Thiery JP. Emt: 2016. Cell. 2016;166:21–45.
Slade D. PARP and PARG inhibitors in cancer treatment. Genes & development. 2020;34:360–94.
Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Advanced materials. 2019;31:e1904197.
Sanderson SM, Gao X, Dai Z, Locasale JW. Methionine metabolism in health and cancer: a nexus of diet and precision medicine. Nature reviews Cancer. 2019;19:625–37.
Hoxhaj G, Manning BD. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nature reviews Cancer. 2020;20:74–88.
Heske CM. Beyond energy metabolism: exploiting the additional roles of NAMPT for cancer therapy. Frontiers in oncology. 2019;9:1514.
Peiris-Pages M, Martinez-Outschoorn UE, Pestell RG, Sotgia F, Lisanti MP. Cancer stem cell metabolism. Breast cancer research : BCR. 2016;18:55.
Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nature reviews Cancer. 2018;18:669–80.
Caglar HO, Biray AC. Alterations of cell cycle genes in cancer: unmasking the role of cancer stem cells. Molecular biology reports. 2020;47:3065–76.
Williams ED, Gao D, Redfern A, Thompson EW. Controversies around epithelial-mesenchymal plasticity in cancer metastasis. Nature reviews Cancer. 2019;19:716–32.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. m(6)A RNA methylation regulates the self-renewal and tumorigenesis of glioblastoma stem cells. Cell reports. 2017;18:2622–34.
Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m(6)A-demethylation of NANOG mRNA. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:E2047–56.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Caflisch A, Sledz P. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. 2020. https://doi.org/10.1002/cmdc.202000011 [Epub ahead of print].
Liu S, Li Q, Chen K, Zhang Q, Li G, Zhuo L, et al. The emerging molecular mechanism of m(6)A modulators in tumorigenesis and cancer progression. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie 2020;127:110098.
Li HB, Tong J, Zhu S, Batista PJ, Duffy EE, Zhao J, et al. m(6)A mRNA methylation controls T cell homeostasis by targeting the IL-7/STAT5/SOCS pathways. Nature. 2017;548:338–42.
Wang H, Hu X, Huang M, Liu J, Gu Y, Ma L, et al. Mettl3-mediated mRNA m(6)A methylation promotes dendritic cell activation. Nature communications. 2019;10:1898.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Molecular cell. 2016;62:335–45.
Du Y, Hou G, Zhang H, Dou J, He J, Guo Y, et al. SUMOylation of the m6A-RNA methyltransferase METTL3 modulates its function. Nucleic acids research. 2018;46:5195–208.
Acknowledgements
We thank the suggestions and guidance of Professor Beicheng Sun for this review.
Funding
This study was supported by the National Natural Science Foundation of China (81773383, 81370078, 81903085), the Science Foundation for Distinguished Young Scholars of Jiangsu Province (BK20170047), the Fundamental Research Funds for the Central Universities (021414380439), and the Project funded by the China Postdoctoral Science Foundation (2019 M651808).
Author information
Authors and Affiliations
Contributions
SW provided the direction of this review. QW, WG, HG, ZW, KX, and CC collected and analyzed the data. WG wrote the manuscript. SW revised this manuscript. The authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, Q., Geng, W., Guo, H. et al. Emerging role of RNA methyltransferase METTL3 in gastrointestinal cancer. J Hematol Oncol 13, 57 (2020). https://doi.org/10.1186/s13045-020-00895-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-020-00895-1