
Abstract
Background
Walker-Warburg syndrome (WWS) is a rare form of alpha-dystroglycanopathy characterized by muscular dystrophy and severe malformations of the CNS and eyes. Bi-allelic pathogenic variants in POMK are the cause of a broad spectrum of alpha-dystroglycanopathies. POMK encodes protein-O-mannose kinase, which is required for proper glycosylation and function of the dystroglycan complex and is crucial for extracellular matrix composition.
Results
Here, we report on male monozygotic twins with severe CNS malformations (hydrocephalus, cortical malformation, hypoplastic cerebellum, and most prominently occipital meningocele), eye malformations and highly elevated creatine kinase, indicating the clinical diagnosis of a congenital muscular dystrophy (alpha-dystroglycanopathy). Both twins were found to harbor a homozygous nonsense mutation c.640C>T, p.214* in POMK, confirming the clinical diagnosis and supporting the concept that POMK mutations can be causative of WWS.
Conclusion
Our combined data suggest a more important role for POMK in the pathogenesis of meningoencephalocele. Only eight different pathogenic POMK variants have been published so far, detected in eight families; only five showed the severe WWS phenotype, suggesting that POMK-associated WWS is an extremely rare disease. We expand the phenotypic and mutational spectrum of POMK-associated WWS and provide evidence of the broad phenotypic variability of POMK-associated disease.
Similar content being viewed by others

Background
Encephalocele is a congenital malformation in which herniated meninges (with or without brain tissue) protrude outside the skull. The underlying cause has still not been completely elucidated [1]. Encephalocele is characteristic for some syndromal diseases, such as Knobloch syndrome (COL18A1), as well as certain ciliopathies such as Meckel-Gruber and Joubert syndromes, and several chromosomal aberrations [2]. Regarding diseases affecting proper glycosylation of alpha-dystroglycan, the manifestation of a meningoencephalocele has only rarely been associated with defects in known genes such as POMT1 and ISPD [3, 4]. Geis and colleagues discussed the presence of an encephalocele as possibly an indicator of the presence of pathogenic POMT1 mutations in terms of a phenotype-genotype correlation [3].
The protein-O-mannosyl kinase gene (POMK, OMIM *615247) encodes a protein involved in the glycosylation of the laminin-binding O-coupled carbohydrate chain of alpha-dystroglycan (α-DG). The α-DG in turn links the dystrophin complex via the sarcolemma to the extracellular matrix. POMK is expressed in various tissues including muscle, brain, retina, heart and kidney and is equally abundant in fetal and adult brain, heart and kidney tissues but reduced in skeletal muscle, with reduced expression at the end of the fetal period and an expression pattern predominantly in interstitial cells and blood vessels [5]. It is suspected that POMK plays an important role in the fetal development of myocytes, and indeed, embryonic pomk knockout zebrafish showed reduced embryonic motility and muscular dystrophy 3 days post fertilization [5]. In addition, Pomk-deficient zebrafish embryos showed small heads, delayed eye development, shortened and thickened tails and U-shaped somites as well as reduced embryo motility [5]. Notably, Pomk-deficient mouse models show severe, often lethal phenotypes with neuronal heterotopias in some brain areas, possibly as a consequence of defective neuronal migration [6]. The phenotypic presentation of both animal models accords with the hypothesis that POMK plays a significant role in muscle and nervous tissue, impacting on the differentiation of the respective cell types, and is in line with the concept of POMK as a gene occasionally associated with manifestation of brain malformations.
In humans, mutations within the POMK gene can lead to different alpha-dystroglycanopathy phenotypes ranging from the milder form (type MDDGC12 [7], OMIM #616094) to the most severe form, named Walker-Warburg syndrome (WWS, also named MDDGA12, OMIM #615249), which is a congenital muscular dystrophy associated with central nervous system and eye malformations. Variants in POMT1 are the main cause of WWS. Like POMK, the POMT1 gene also codes for an enzyme involved in the O-mannosylation pathway. The clinical picture of patients with POMT1 variants includes neural tube defects ranging from meningocele to meningoencephalocele [8]. Compared to POMT1, POMK variants leading to WWS are rarer and either result in the expression of a shortened, incorrectly folded protein or interfere with catalytic function [9]. POMK genotype-phenotype correlations are complex because even mutations leading to expression of a (massively) shortened protein can result in a mild phenotype. However, the functional and physiological mechanisms explaining the phenotypic variability still remain unclear [5].
To date, only 14 patients with POMK-associated alpha-dystroglycanopathy due to 8 different mutations have been described [5, 7, 10,11,12,13] and two additional pathogenic or likely pathogenic variants are listed in the POMK database [https://databases.lovd.nl/shared/view/POMK; accessed 7 April 2020] . An encephalocele was reported for only one patient, a 19-week fetus (termination of pregnancy; TOP) by Jae and colleagues [11]. Here, we describe monozygotic twins each with occipital meningoencephalocele and homozygous nonsense mutation in POMK.
Results
Clinical details
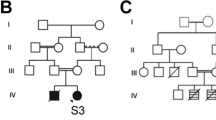
The mother and father are healthy consanguineous parents (first cousins) having in total nine children: six are healthy and one died postnatally due to complications of hydrocephalus (no further data or material available). Further family history was unremarkable apart from three paternal nephews with unspecific developmental delay of unknown origin (Fig. 1).

Pedigree. Healthy consanguineous parents. Six healthy siblings – one sibling died postnatally (congenital hydrocephalus). Three paternal first cousins (males) are affected by developmental delay (grey squares – no further information available)
Prenatal ultrasonography screening in the 23rd week of pregnancy revealed the presence of occipital encephaloceles, hydrocephalus and cerebellar hypoplasia in both twins (Fig. 2). Prenatal MRI scans confirmed these findings (Fig. 3). Monochorionic-diamniotic male twins were born in gestational week 35 + 2 by planned caesarean section and because of progressive contractions. Birth parameters were in the normal range. Gemini 1 (G1): weight 2330 g (− 0.6 SD, 20th percentile), length 47 cm (30th percentile, − 0.3 SD), and occipitofrontal circumference 31 cm (8th percentile, − 1.3 SD). - Gemini 2 (G2): weight 2230 g (15th percentile, − 0.9 SD), length 47 cm (30th percentile, − 0.3 SD), and occipitofrontal circumference 30.5 cm (4th percentile, − 1.6 SD). Postnatally, additional eye malformations were observed by ultrasound and posterior ophthalmoscopy in both siblings and lissencephaly (Fig. 4). G1 showed a microphthalmos of the right eye and a coloboma dorsolateral of the left bulbus. G2 showed a bilateral persistent hyperplastic primary vitreous body and a posterior staphyloma of the left eye. The eye malformations led to blindness in both twins.

Prenatal ultrasound of the brain (22th week of pregnancy): occipital encephalocele (white arrow) and hydrocephalus

Prenatal MRI scan at 23 + 4 weeks of gestation; a hypoplastic cerebellum (white arrow 1), occipital meningocele (white arrow 2); b persistent hypoplastic primary vitreous body (white arrow); c + d internal hydrocephalus

Postnatal ultrasound of the brain / eyes; A lissencephaly with polymicrogyria; b staphyloma; c persistent hypoplastic primary vitreous body
Brainstem evoked response audiometry (BERA) diagnosed severe sensorineural hearing loss in both patients.
Neuropediatric examination showed floppy infants with severe generalized muscle hypotonia, hyporeflexia, decreased spontaneous motor activity and muscular weakness observed when moving the muscles of the extremities against gravity. In addition to the ocular malformation, a prominent occipital protuberance was detected (histologically diagnosed as a meningoencephalocele) (Fig. 5).

a-b postnatal examination of both twins revealed prominent occipital meningocele (white arrows)
Laboratory analysis showed a significant increase of the creatine kinase level to 7159 U/l (G1) and 8769 U/l (G2), respectively, with an accompanying elevation of transaminase levels and LDH.
MRI of the brain (Fig. 6) and spine, each performed on the 2nd/3rd day of life, showed a median occipital meningocele with dorsally opened fourth ventricle and hypo- to aplastic cerebellar vermis, a long narrowed thoracic myelon with dorsal attachment of single caudal fibers, internal hydrocephalus as well as dysgyria with generalized polymicrogyria-like cobblestone malformations and bi-temporo-occipital subcortical band heterotopia.

Postnatal cranial MRI scan (performed at 2nd / 3rd day); clinical findings: occipital meningocele with dorsal enlarged 4th ventricle (white arrow picture a), vermis hypo−/aplasia (white arrow picture b), generalized polymicrogyria-like cobblestone malformation, temporo-occipital subcortical band heterotopia, eye malformations (G1: microphthalmia with coloboma and caudal cyst, G2: persistent hypoplastic primary vitreous body and posterior staphyloma). a/c = G1, b/d = G2
During the perinatal period, the meningoencephalocele was treated with neurosurgery and the hydrocephalus was treated with a ventriculo-peritoneal (VP) shunt. Both twins developed structural epilepsy with generalized seizures; G1 had repeated seizures with epileptic status refractory to antiepileptic therapy.
Both boys were provided with hearing aids. They received supportive therapy including physiotherapy and early remedial education and were cared for by our palliative care team. They had repeated hospitalizations due to therapy refractory epileptic seizures and severe respiratory infections. Psychomotor development was severely retarded and typical developmental milestones were not reached (even head control was not possible). Both boys could spontaneously move their heads, but the other extremities were tetraplegic. G2 was last measured at the age of 6 months [weight: 8430 g (0.44 SD), length 65.5 cm (− 0.95 SD)]. At the age of 17 months, G2 died of cardiorespiratory failure due to aspiration. Currently, at the age of 30 months, G1 continues to show no psychomotor development [weight: 16000 g (1.38 SD), length 94 cm (0.43 SD), occipitofrontal circumference: 44 cm (− 5.0 SD)].
Molecular genetic analysis
In consideration of the clinical findings, a molecular panel analysis for genes related to WWS was performed including FKRP, FKTN, ISPD, B3GNT1, COL4A1, LARGE, POMK and TMEM5.
Due to the combination of severe CNS- and eye-malformations in combination with high CK-levels in these floppy infants, an alpha-dystroglycanopathy was considered. This assumption was supported by the presence of elevated serum CK levels. Panel sequencing resulted in the identification of a homozygous nonsense mutation [c.640C>T; p.Gln214*; NM_032237.4, OMIM: 615247] within the POMK gene in both twins. Sanger sequencing confirmed the homozygous POMK variant in both siblings and showed the parents to be heterozygous mutation carriers. Unfortunately, no material of the deceased sibling presenting with a hydrocephalus was available for mutational screening.
Discussion
Walker-Warburg syndrome is a rare form of congenital muscular dystrophy (prevalence approximately 1:60,500) [14] with currently 18 known causative genes (POMT1, POMT2, POMGNT1, FKTN, FKRP, LARGE1, ISPD, POMGNT2, GMPPB, DAG1, TMEM5, B3GALNT2, POMK, B3GNT1, DOLK, DPM1, DPM2, DPM3) [15]. So far, only five families with WWS caused by variants in POMK have been described (in total eight individuals of whom four are terminated pregnancies; for details see families 3–7 in Table 1). All causative variants reported affected or diminished POMK activity, either by truncating the protein or impacting on the catalytic protein domain [5, 10,11,12,13,14,15].
The clinical manifestation of patients with pathogenic variants in POMK is rather broad (see introduction) and a varying spectrum of clinical manifestations and severity can even be observed within the subgroup of POMK-related WWS (classified as such by the authors/clinicians).
Renesse et al. described two siblings of a consanguineous family with a homozygous POMK nonsense mutation (c.325C>T, p.Q109X). Both siblings had secondary microcephaly, muscular hypotonia, feeding problems and developmental delay. In addition, they presented with hypomyelinization of the brain, mild hearing loss and intellectual disability [10]. The 15-year-old sibling has developed joint contractures, neuromuscular scoliosis and nocturnal hypoventilation. The 22-year-old sibling has used a wheelchair since the age of 17 and is dependent on comprehensive help in her everyday life. Ocular abnormalities were found in the form of large bulbi with reduced visual acuity, but did not meet the criteria of a megalocornea in this patient [10]. Given that the clinical presentation of these POMK siblings manifests in the spectrum between the milder MDDGC12 and the most severe MDDGA12 phenotype, these cases might be classified as MDDGB12.
Di Costanzo et al. reported on three patients from two different families with pathogenic POMK mutations. Patients from one family presented with the same homozygous POMK nonsense mutation (c.325C>T, p.Q109X) as the patients described by Von Renesse et al. [5, 10]. However, clinically, the patients described by Di Costanzo et al. had a milder limb-girdle muscular dystrophy phenotype in line with MDDGC12: both patients showed their first symptoms in infancy but are still able to walk at the age of 25 and 13 years, respectively. They show no ocular malformations and their IQ is slightly below average (IQ 80 and 83) [5]. In contrast, the second family characterized by Di Costanzo et al. had a compound heterozygous mutation (combination of frame shift and missense mutation, c.286delT, p.Phe96Phefs*19 and c.905T>A, p.Val302Asp) and presented clinically with WWS, similar to our two cases. The affected child was diagnosed in utero with macrocephaly and hydrocephalus. Postnatally, muscular hypotonia was present, as well as other CNS malformations including cobblestone lissencephaly, corpus callosum agenesis, vermis aplasia further complicated by eye malformations (glaucoma, retinal degeneration) and severe sensorineural hearing loss. The child showed a severe psychomotor developmental disorder, developed tonic seizures and died at the age of 4 years [5]. Jae et al. described additional compound heterozygous missense mutations in the POMK gene (p.Leu137Arg, p.Gln258Arg) in a patient with typical symptoms of WWS [11]. The second child of the family had severe brain malformations as well as an occipital encephalocele and died prenatally [11].
Very recently, Preiksaitiene et al. reported on two families with in total four WWS patients of which three were TOPs: brain malformations with hydrocephalus and ventriculomegaly due to a POMK nonsense mutation were detected in utero [12]. One mildly affected POMK patient (caused by a homozygous missense mutation) presenting with mirror movements of the hands was described by Ardicli et al.: the initial symptoms manifested during childhood with muscle weakness, easy fatigability, clumsiness and difficulties running and climbing. At the age of 12, proximal muscle weakness with calf hypertrophy was detected. On the MRI scan, several brain malformations were identified – surprisingly only associated with mild learning difficulties [13]. In addition, Strang-Karlsson et al. reported on a family with two siblings with a homozygous POMK missense mutation resulting in mild congenital muscular dystrophy: during childhood hip and neck cramps (triggered by yawning) were described together with proximal muscle weakness with calf hypertrophy. Investigation of a muscle biopsy obtained from one patient revealed normal histological findings whereas the biopsy from the sibling showed moderate chronic myopathic changes with small groups of regenerating fibers and sparse inflammatory cell infiltrates [7].
Based on our findings as well as previous findings of meningoencephaloceles in patients with POMT1 and ISPD mutations, we recommend an initial laboratory analysis of CK in newborns which present clinically with the combined symptoms of muscular weakness and meningoencephalocele [3, 4].
POMK is an atypical kinase that phosphorylates the 6-position of O-mannose after mannose has been modified by both GTDC2 and B3GALNT2 (two proteins encoded by genes leading to overlapping neurological phenotypes). The glycan structure resulting from POMK-modulated phosphorylation appears to be relevant for binding to the extracellular matrix (ECM) [10, 16]. Although the basic biochemical function of POMK is well understood, further research on larger POMK patient populations is needed to improve understanding of the phenotypic variability, which might be caused by the activation of compensatory mechanisms (warranting proper protein glycosylation and ECM assembly) and/ or the presence of further molecular genetic alterations of relevance as modifiers.
Conclusion
Given that encephaloceles are occasionally associated with other genetic defects causative of alpha-dystroglycanopathies, including genes encoding for proteins involved in O-mannosylation of α-DG such as POMT1 [3], the presence of a meningoencephalocele in our POMK patients supports the concept that perturbed post-translational modification of α-DG has a detrimental impact on α-DG-function and affects correct maturation of the neural tube during fetal development. Our combined clinical and genetic findings thus expand the clinical spectrum of POMK patients and classify POMK as candidate gene for meningoencephalocele.
Methods
Study aim, design and setting of the study
The study aimed to combine clinical and diagnostical findings obtained in different patients with POMK mutations. The study took place at the university hospital Essen.
Characteristics of participitans
We analysed two monozygous twins with a mutation in POMK. We compared different patients published before and focused on mutations in c-DNA and protein level, age of onset, first symptoms, muscle weakness (localisation), CK level, biopsy findings, MRI findings, ocular findings, additional information and latest check-up.
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Abbreviations
- a-DG:
-
alpha-dystroglycan
- BERA:
-
Brainstem evoked response audiometry
- B3GALNT2:
-
Beta-1,3-N-Acetylgalactosaminyltransferase 2
- B3GNT1:
-
N-acetyllactosaminide beta-1,3-N-acetylglucosaminyltransferase
- CDG:
-
Congenital disorder of glycosylation
- CK:
-
Creatine kinase
- CMD:
-
Congenital muscle dystrophy
- CNS:
-
Central nervous system
- COL18A1:
-
Collagen type XVIII alpha 1 chain
- COL4A1:
-
Collagen type IV alpha 1 chain
- DAG1:
-
Dystroglycan 1
- DOLK:
-
Dolichol kinase
- DPM1:
-
Dolichyl-Phosphate Beta-D-Mannosyltransferase Subunit 1
- DPM2:
-
Dolichyl-Phosphate Beta-D-Mannosyltransferase Subunit 2
- DPM3:
-
Dolichyl-Phosphate Beta-D-Mannosyltransferase Subunit 3
- ECM:
-
Extraceullar matrix
- FKRP:
-
Fukutin-related protein
- FKTN:
-
Fukutin
- GMPPB:
-
GDP-Mannose Pyrophosphorylase B
- GTDC2:
-
Glycosyltransferase-like domain containing 2
- ISPD:
-
Isoprenoid synthase domain-containing protein
- LARGE:
-
Like-glycosyltransferase
- MDDGA12:
-
Muscular dystrophy-dystroglycanopathy Type A 12
- MDDGC12:
-
Muscular dystrophy-dystroglycanopathy Type C 12
- OMIM:
-
Online mendelian inheritance in man
- POMGNT1:
-
Protein O-linked mannose N-acetylglucosaminyltransferase 1
- POMGNT2:
-
Protein O-linked mannose N-acetylglucosaminyltransferase 2
- POMK:
-
Protein-O-Mannose Kinase
- POMT1:
-
Protein-O-mannosyltransferase 1
- POMT2:
-
Protein-O-mannosyltransferase 2
- SHH:
-
Sonig hedgehog gene
- TMEM5:
-
Transmembrane protein 5
- TOP:
-
Termination of pregnancy
- VC:
-
Vital capacity
- VP:
-
Ventriculo-peritoneal shunt
- WWS:
-
Walker-Warburg syndrome
References
Rolo A, Galea GL, Savery D, Greene NDE, Copp AJ. Novel mouse model of encephalocele: post-neurulation origin and relationship to open neural tube defects. Dis Models Mechan. 2019;12:dmm040683.
Seidahmed MZ, et al. Genetic, chromosomal, and syndromic causes of neural tube defects. Saudi Med J. 2014;35(Suppl 1):49–56.
Geis T, et al. Clinical long-time course, novel mutations and genotype-phenotype correlation in a cohort of 27 families with POMT1-related disorders. Orphanet J Rare Dis. 2019;14:179, https://pubmed.ncbi.nlm.nih.gov/31311558.
Bönnemann CG, et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul Disord. 2014;24:289–311, http://www.sciencedirect.com/science/article/pii/S0960896614000029.
Di Costanzo S, et al. POMK mutations disrupt muscle development leading to a spectrum of neuromuscular presentations. Hum Mol Genet. 2014;23:5781–92.
Moore SA, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature. 2002;418:422–5. https://doi.org/10.1038/nature00838.
Strang-Karlsson S, et al. A novel compound heterozygous mutation in the POMK gene causing limb-girdle muscular dystrophy-dystroglycanopathy in a sib pair. Neuromuscul Disord. 2018;28:614–8.
Devisme L, et al. Cobblestone lissencephaly: neuropathological subtypes and correlations with genes of dystroglycanopathies. Brain. 2012;135:469–82.
Nagae M, et al. 3D structural analysis of protein O-mannosyl kinase, POMK, a causative gene product of dystroglycanopathy. Genes Cells. 2017;22:348–59.
von Renesse A, et al. POMK mutation in a family with congenital muscular dystrophy with merosin deficiency, hypomyelination, mild hearing deficit and intellectual disability. J Med Genet. 2014;51:275.
Jae LT, et al. Deciphering the Glycosylome of Dystroglycanopathies using haploid screens for Lassa virus entry. Science. 2013;340:479.
Preiksaitiene E, et al. Pathogenic homozygous variant in POMK gene is the cause of prenatally detected severe ventriculomegaly in two Lithuanian families. Am J Med Genet. 2020;182:536–42.
Ardicli D, et al. Congenital mirror movements in a patient with alpha-dystroglycanopathy due to a novel POMK mutation. Neuromuscul Disord. 2017;27:239–42, http://www.sciencedirect.com/science/article/pii/S0960896616310203.
Genetics Home References. Walker-Warburg-syndrome. Available at https://ghr.nlm.nih.gov/condition/walker-warburg-syndrome#genes (2019).
Johnson K, et al. Detection of variants in dystroglycanopathy-associated genes through the application of targeted whole-exome sequencing analysis to a large cohort of patients with unexplained limb-girdle muscle weakness. Skelet Muscle. 2018;8:23, https://pubmed.ncbi.nlm.nih.gov/30060766.
Yoshida-Moriguchi T, et al. SGK196 is a glycosylation-specific O-mannose kinase required for Dystroglycan function. Science. 2013;341:896.
Acknowledgements
We thank Dr. Rachel Thompson for proofreading the manuscript.
Funding
This study was not supported by funding.
Author information
Authors and Affiliations
Contributions
USS, KR, HK, LP and ADM performed the clinical examination of the patients. UH conducted the genetic testing. AKu and ME performed the human genetic consultation and counselling. AKö, AI, AS examined the patients with ultrasound. BS generated the MRI-report. USS, AR, LP, AKu and FK drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The parents of the twins consented to publication of clinical, diagnostical and molecular findings.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Paul, L., Rupprich, K., Della Marina, A. et al. Further evidence for POMK as candidate gene for WWS with meningoencephalocele. Orphanet J Rare Dis 15, 242 (2020). https://doi.org/10.1186/s13023-020-01454-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-020-01454-0