
Abstract
Type 2 diabetes mellitus (T2DM) steadily increases in prevalence since the 1950’s, the period of widespread distribution of refrigerated pasteurized cow’s milk. Whereas breastfeeding protects against the development of T2DM in later life, accumulating epidemiological evidence underlines the role of cow’s milk consumption in T2DM. Recent studies in rodent models demonstrate that during the breastfeeding period pancreatic β-cells are metabolically immature and preferentially proliferate by activation of mechanistic target of rapamycin complex 1 (mTORC1) and suppression of AMP-activated protein kinase (AMPK). Weaning determines a metabolic switch of β-cells from a proliferating, immature phenotype with low insulin secretion to a differentiated mature phenotype with glucose-stimulated insulin secretion, less proliferation, reduced mTORC1- but increased AMPK activity. Translational evidence presented in this perspective implies for the first time that termination of milk miRNA transfer is the driver of this metabolic switch. miRNA-148a is a key inhibitor of AMPK and phosphatase and tensin homolog, crucial suppressors of mTORC1. β-Cells of diabetic patients return to the postnatal phenotype with high mTORC1 and low AMPK activity, explained by continuous transfer of bovine milk miRNAs to the human milk consumer. Bovine milk miRNA-148a apparently promotes β-cell de-differentiation to the immature mTORC1-high/AMPK-low phenotype with functional impairments in insulin secretion, increased mTORC1-driven endoplasmic reticulum stress, reduced autophagy and early β-cell apoptosis. In contrast to pasteurized cow’s milk, milk’s miRNAs are inactivated by bacterial fermentation, boiling and ultra-heat treatment and are missing in current infant formula. Persistent milk miRNA signaling adds a new perspective to the pathogenesis of T2DM and explains the protective role of breastfeeding but the diabetogenic effect of continued milk miRNA signaling by persistent consumption of pasteurized cow’s milk.
Similar content being viewed by others

Introduction
Milk, sugar and saturated fat are substantial components of Western diet. The per capita milk consumption in the United States and Germany in 2017 was 65.2 L and 52.2 L, respectively [1, 2]. An increasing number of epidemiological studies identified the consumption of commercial cow’s milk with an increased risk of insulin resistance and type 2 diabetes mellitus (T2DM) [3,4,5,6,7,8,9] (Table 1). In contrast, fermented milk products have been associated with a decreased risk of T2DM [10]. A nested case-cohort within 8 European countries of the European Prospective Investigation into Cancer and Nutrition (EPIC) Study (n = 340,234) analyzed the amount and type of dairy product intake and incident T2DM and identified an increased risk for T2DM by milk consumption in 5 of 8 countries [7]. The prospective Dutch Lifeline Cohort Study (n = 112,086) investigated the association of non-fermented milk products, milk and fermented milk products on participants with prediabetes (defined as fasting plasma glucose between 5.6 and 6.9 mmol/L or HbA1c of 5.7–6.4%) and newly diagnosed T2DM (defined as fasting plasma glucose > 7.0 mmol/L or HbA1c > 6.5%) [8]. A positive association between full-fat milk consumption (150 g/day) as well as non-fermented dairy products with prediabetes and T2DM has been shown [8]. Apparently, there is a diabetogenic ingredient in non-fermented milk compared to fermented milk products. A substantial difference between non-fermented pasteurized milk and fermented milk products is the fact that pasteurized milk contains bioactive miRNA-enriched exosomes and extracellular vesicles that survive degradation in the gastrointestinal tract [11,12,13,14,15]. Milk exosomes are taken up by endocytosis in intestinal and endothelial cells [16, 17], are bioavailable for human milk consumers [18], and reach the systemic circulation of the human milk consumer in a dose-dependent manner [19]. After oral administration, bovine milk exosomes are transported into various tissues and organs as recently demonstrated in several mouse models [20, 21]. Therefore, milk exosomes are regarded as promising new drug carriers to reach distant tissues for pharmacological intervention [22, 23]. Milk miRNAs including bovine miRNA-148a and miRNA-29b, which are identical to human miRNAs (mirbase.org), resist pasteurization, homogenization and refrigerated storage [24,25,26]. A recent study focusing on human skim milk identified 10 miRNAs that accounted for > 70% of the reads mapped to miRNAs. Among them miRNA-148a represented around 30% of the reads [27]. Notably, miRNA-148a is the most abundant miRNA in human and bovine milk fat and milk exosomes [25,26,27,28,29].
Accumulating evidence supports the role of milk-derived exosomal miRNAs in systemic metabolic regulation [30,31,32,33,34,35,36,37,38]. In contrast to pasteurized commercial milk, exosome integrity and miRNA content of fermented milk products such as yoghurt are degraded by bacterial fermentation [39]. The protein content of milk exosomes and their miRNA expression monitored by miRNA-29b and miRNA-21 were significantly reduced after fermentation [39]. Thus, commercial pasteurized milk in contrast to fermented milk products is a donor of bioactive exosomal miRNAs.
Weaning triggers β-cell maturation
Recent evidence from murine models indicates that weaning, i.e., the termination of milk intake, triggers a critical maturation step of pancreatic β-cells [40, 41]. Weaning coincides with enhanced glucose-stimulated insulin secretion (GSIS) from islets [40]. Jaafar et al. recently demonstrated that a switch from the nutrient sensor mechanistic target of rapamycin complex 1 (mTORC1) to the energy sensor 5′-adenosine monophosphate-activated protein kinase (AMPK) is of critical importance for functional maturation of β-cells during weaning [41]. AMPK was activated by the dietary transition taking place during weaning, and this in turn inhibits mTORC1 activity to promote the adult β-cell phenotype [41]. Notably, milk has been characterized as a postnatal activator of mTORC1 for postnatal growth, translation and anabolism [42]. mTORC1 activation is required for the development and growth of β-cells during embryonic and early postnatal life [43,44,45,46,47]. AMPK is a potent inhibitor of mTORC1 activation [48, 49] and is the key target of the common antidiabetic drug metformin, which activates AMPK and inhibits mTORC1 [50, 51]. Jaafar et al. hypothesized that postnatal β-cell maturation may represent an adaptation to the cessation of milk consumption, and that mTORC1 repression through AMPK activation may act as a physiological mediator of this process [41]. Intriguingly, allowing mice to continue assimilating milk fat throughout their entry into adulthood, a period during which this is usually declining, was sufficient to allow β-cells to maintain neonatal levels of mTORC1 activity, which was otherwise completely repressed in milk-free control mice [41]. In addition, recent evidence supports the view that miRNAs play a key role in regulating β-cell differentiation and β-cell identity. A shift in miRNA expression has been associated with postnatal β-cell differentiation [52,53,54,55,56,57].
These observations lead to the three questions: 1) Is the cessation of milk-derived miRNAs the responsible mechanism for the metabolic switch of immature to differentiated β-cells? 2) Does continued bovine miRNA signaling by persistent consumption of cow’s milk induce the immature β-cell phenotype of T2DM? 3) Does miRNA-deficient infant formula feeding disturb appropriate β-cell proliferation?
Weaning-dependent loss of milk miRNAs: the maturation signal for β-cells
Exosomal miRNA traffic plays an important role in β-cell regulation, especially cell-to-cell communication between β-cells as well as β-cell cross-talk with circulatory exosomes derived from distant tissues involved in the regulation of glucose homeostasis [58,59,60,61,62]. Notably, exosomes released from lipid-induced insulin-resistant muscles have been shown to modulate miRNA-dependent gene expression and proliferation of recipient β-cell cells in mice [61]. Furthermore, pancreatic cancer cells release exosomes into the systemic circulation that readily reach β-cells and impair insulin secretion (paraneoplastic diabetes) [63]. Remarkably, the islet capillary network exhibits five times higher density than the capillary network of the exocrine counterpart and shows high permeability [64]. The islet microvascular endothelial cells have about 10 times more fenestrations than those of the exocrine tissue [64]. These fenestrae have an unusually extensive pore size of 100 nm in diameter [64] allowing a rapid passage of macromolecules and exosomes most likely including milk-derived exosomes (30–100 nm) [65]. Bovine milk exosomes have been detected in multiple tissues and organs after oral administration to mice [20, 21]. It is thus conceivable that milk-derived exosomes maintain a signaling cross-talk to the β-cells of the milk recipient, who physiologically is the newborn infant requiring maternal milk exosomes for adequate mTORC1-dependent β-cell proliferation and β-cell mass extension associated with suppression of AMPK.
Milk-derived miRNAs: suppressors of AMPK and activators of mTORC1
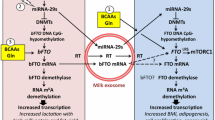
AMPK phosphorylates the mTORC1 binding partner raptor [48] and tuberous sclerosis protein TSC2, the upstream suppressor of mTORC1 and thereby suppresses mTORC1 [49]. AMPK mediates cellular energy responses to control mTORC1-dependent cell growth and survival [48, 49]. It has recently been demonstrated in breast cancer cells that upregulation of miRNA-148a inhibits the expression of AMPK [66]. In a highly conserved manner with strong binding affinity, miRNA-148a targets the catalytic subunit α 1 of AMPK (PRKAA1) as well as the AMPK regulatory subunit γ 2 (PRKAG2) (targetscan.org) (Table 2). Reif et al. recently demonstrated that milk exosome-derived miRNA-148a also suppresses phosphatase and tensin homolog (PTEN), a pivotal inhibitor of the phosphatidylinositol-3 kinase (PI3K)-AKT-mTORC1 signaling pathway [67]. miRNA-29b and miRNA-29c, other two miRNA components of bovine milk [11], target the AMPK subunit β 2 (PRKAB2) (targetscan.org) (Fig. 1a). Consumption of pasteurized commercial milk increased the levels of miRNA-29b in plasma and blood monocytes [19], whereas bacterial fermentation of milk decreased miRNA-29b concentration [39]. miRNA-29b also targets DBT (dihydrolipoamide branched-chain transacylase) [68], the E2 core component of branched-chain α-keto acid dehydrogenase, the rate-limiting enzyme of branched-chain amino acid (BCAA) catabolism. BCAAs are key activators of mTORC1 [69,70,71,72] and are increased in plasma of patients with T2DM [73,74,75,76,77,78,79,80].

a Milk exosome-mediated miRNA signaling of pancreatic β-cells during physiologic breastfeeding and persistence of bovine milk-derived miRNA signaling by continued consumption of pasteurized cow’s milk. miRNA-148a suppresses AMPK and PTEN. miRNA-29b inhibits the catabolism of branched-chain amino acids (BCAAs). Reduced AMPK- and PTEN activity combined with increased BCAA levels activate mTORC1, which promotes β-cell proliferation and mass expansion. miRNA-148a together with miRNA-130a suppress the transcription factor complex EER γ /PGC1 α, which controls multiple mitochondrial genes involved in ATP production required for glucose-stimulated insulin secretion (GSIS). miRNA-29b suppresses ONECUT2, an inhibitor of granuphilin resulting in enhanced suppression of GSIS. miRNA-29b suppresses DNMT3A increasing the expression of “forbidden” genes of mature β-cells such as HK1 and LDHA. b Weaning terminates milk miRNA signaling. The disappearance of miRNA-148a enhances the activity of AMPK and PTEN resulting in increased suppression of mTORC1, whereas AMPK-dependent gene-regulation is upregulated. EER γ /PGC1 α activates mitochondrial genes involved in ATP production which in combination with ONECUT2-mediated granuphilin suppression enhance GSIS. However, this mature β-cell phenotype de-differentiates again by persistent intake of bovine milk exosomes
It is thus conceivable that weaning-associated termination of milk miRNA-mediated suppression of AMPK, PTEN and DBT decreases β-cell mTORC1 activity promoting the metabolic switch to AMPK-regulated β-cell maturation with appropriate GSIS (Fig. 1b).
Milk miRNA-148a: regulator of AMPK-controlled disallowed genes
In the β-cell, a small group of genes, which are abundantly expressed in most if not all other mammalian tissues, are highly selectively repressed such as lactate dehydrogenase A (LDHA) and monocarboxylate transporter-1 (SLC16A1). Their inactivation ensures that pyruvate and lactate, derived from muscle during exercise, do not stimulate inappropriate insulin release [81, 82]. Loss of AMPK from β-cells up-regulates the β-cell-disallowed gene family resulting in β-cell de-differentiation characterized by increased expression of normally repressed (“disallowed”) genes, such as LDHA, SLC16A1, MGST1, PDGFRA leading to aberrant fuel sensing [83]. Elevated AMPK activity, which is stimulated by metformin, is required to suppress these disallowed β-cell genes [83,84,85]. Milk-miRNA-148a-mediated suppression of AMPK may thus compromise β-cell identity and function to maintain the postnatal immature, proliferating phenotype (Fig. 1a ).
miRNA-148a: critical suppressor of estrogen-related receptor- γ
AMPK activation triggers a switch to oxidative metabolism in mature islets [41]. Yoshihara et al. recently identified estrogen-related receptor- γ (ERR γ) encoded on ESRRG as a master regulator of β-cell maturation that is expressed in the adult, but not neonatal β-cell [86]. ERR γ is a crucial mediator of multiple endocrine and metabolic signals and plays important roles in β-cell maturation [86, 87]. Postnatal induction of ERR γ drives a transcriptional network activating mitochondrial oxidative phosphorylation, tricarboxylic acid (TCA) cycle, fatty acid oxidation, the electron transport chain (ETC), and ATP production needed to drive GSIS. Mice deficient in β-cell-specific ERR γ expression are glucose intolerant and fail to secrete insulin in response to a glucose challenge [86]. During the postnatal period before weaning, ATP-dependent insulin secretion is low [86]. However, during β-cell maturation, ERR γ upregulates several mitochondrial genes (MDH1, COX6A2, ATP2A2, NDUFS2, and ATP6V0A2) [87]. Notably, ERR γ binds to its co-activator PGC-1 α (peroxisome proliferator-activated receptor- γ co-activator 1 α; PPARGC1A) producing a stable transcription factor ERR γ /PGC-1 α complex [88, 89]. Both mRNAs of ESRRG and PPARGC1A exhibit highly conserved binding sites (8-mer) for miRNA-148a, miRNA148b and miRNA-152, whereas PPARGC1A exhibits further conserved binding sites for miRNA-130a, miRNA-29a, miRNA-29b, and miRNA-29c (targetscan.org) (Fig. 1a). Remarkably, it has been shown that elevated expression of miRNA-130a, miRNA-130b and miRNA-152 suppresses GSIS via modulation of intracellular ATP levels [54]. miRNA-130a is another abundant miRNA of cow’s milk [11]. miRNA-152 is a member of the miRNA-148a/miRNA-148b/miRNA-152 family, which all share identical seed sequences [90]. Thus, termination of miRNA-148a/miRNA-130a/miRNA-29b signaling of milk most likely explains the critical switch to the adult metabolically mature β-cell phenotype with adequate GSIS (Fig. 1b). It is conceivable that this pathway may operate in concert with other miRNAs and RNA signaling networks, but due to its abundance in milk, miRNA-148a may play the leading role.
Cessation of milk miRNA signaling stabilizes β-cell identity
DNA methylation directs functional maturation of pancreatic β-cells. It has been demonstrated that β-cell identity is maintained by DNA methylation-mediated repression of the lineage determination gene aristaless-related homeobox, X-linked (ARX) [91]. β-Cells deficient in DNA methyltransferase 1 (DNMT1), the maintenance DNA methyltransferase that propagates DNA methylation patterns during cell division, were converted to α-cells [91]. Propagation of DNA methylation during cell division is essential for repression of α-cell lineage determination genes to maintain β-cell identity. Milk miRNA-148a, which targets DNMT1 [35, 92], may thus prevent DNMT1-mediated β-cell differentiation.
DNMT3A also promotes β-cell differentiation. In a murine model, β-cell-specific deletion of DNMT3A prevented the metabolic switch, resulting in loss of GSIS [93]. DNMT3A binds to the promoters of the genes encoding hexokinase 1 (HK1) and lactate dehydrogenase A (LDHA), both of which regulate the metabolic switch. Knockdown of these two key DNMT3A targets restored GSIS response in islets from animals with β-cell-specific DNMT3A deletion. Furthermore, DNA methylation-mediated repression of glucose-secretion decoupling genes to modulate GSIS was conserved in human β-cells. Remarkably, DNMT3A is a target of miRNA-29b [94,95,96]. Together, these observations reveal a critical epigenetic role for weaning, i.e., the loss of milk miRNAs, for DNA methylation-mediated β-cell maturation (Fig. 1b).
Milk miRNA-148a: suppressor of β-cell MAFB
The transcription factor MAFB plays a key role in late events essential for β-cell maturation and activates genes involved in mature endocrine functions including those significant for glucose sensing (SCL2A2), vesicle maturation (SCL30A6), Ca2+ signaling (CAMK2B) and insulin secretion (NNAT) [97, 98]. A switch from MAFB to MAFA expression accompanies differentiation to pancreatic β-cells [99]. MAFB increases the expression of MAFA, which is important to maintain pancreatic β-cell function in adults [97, 100]. MAFB is a direct target of miRNA-148a [101]. miRNA-148a has been detected as a major miRNA component of bovine milk and milk fat [25, 26]. Notably, MAFA was lower in islets of mice that continuously received milk fat [40]. Thus, persistent milk consumption in adults may maintain an immature β-cell phenotype with compromised MAFB to MAFA conversion (Table 2).
Milk miRNA-deficient infant formula and impaired β-cell maturation
There is general agreement that breastfeeding protects against T2DM [102,103,104,105,106]. Human breastmilk and milk lipids contain and transfer bioactive miRNAs including miRNA-148a [26,27,28,29, 106, 107]. In contrast, infant formula exhibits severe deficiencies of miRNAs including miRNA-148a (Fig. 2b, d) [108,109,110]. It is thus conceivable that miRNA-deficient formula feeding in comparison to breastfeeding may impair postnatal β-cell proliferation with appropriate acquisition of β-cell mass increasing the risk of T2DM later in life. The systemic availability of bioactive miRNAs in breastmilk may explain the superiority of breastfeeding compared to miRNA-deficient formula feeding in the prevention of T2DM. However, the beneficial effect on milk miRNA signaling during the breastfeeding period may turn into an adverse effect, when this miRNA signaling process is not terminated.

a Physiological termination of breastmilk miRNA signaling after weaning. There is an appropriate metabolic switch from mTORC1-driven β-cell proliferation and mass expansion to AMPK-driven mature β-cell function with GSIS. b Infant formula feeding with deficient milk miRNA signaling may impair adequate β-cell mass expansion and differentiation to regularly matured β-cells. Metabolic challenges of an impaired pool of β-cells in later life may enhance the risk of type 2 diabetes. c Persistence of milk miRNA signaling after regular breastfeeding by intake of bioactive exosomes of bovine milk may de-differentiate β-cell back to a mTORC1 ↑ /AMPK ↓ progenitor phenotype enhancing the risk of type 2 diabetes mellitus. d The worst scenario: miRNA-deficient formula-fed infants start into life with a postnatally compromised pool of β-cells, have no appropriate miRNA-dependent metabolic switch to mature β-cells and may thus experience early de-differentiation of their β-cells to the mTORC1 ↑ /AMPK ↓ progenitor phenotype, that increases mTORC1-driven endoplasmic reticulum (ER) stress with reduced autophagy and early β-cell apoptosis
Milk miRNAs: potential drivers of β-cell de-differentiation in type 2 diabetes
The top 20 most abundant miRNAs are shared between mammalian species including humans, especially miRNA-148a and members of the let-7 family, which points to their key regulatory functions [111]. It has recently been demonstrated that whole blood levels of miRNA-148a, miRNA-122, miRNA-144, miRNA-589, and let-7a were associated with glycemic status [112]. In addition, miR-148a and miRNA-144 were associated with elevated glucose levels, and miRNA-148a and other miRNAs were associated with HbA1c levels [112]. These data underline the diabetogenic role of miRNA-148a, which represents the most abundant miRNA species of human and bovine milk exosomes and milk fat [25,26,27,28,29]. Accumulating evidence suggests that a functional deficiency, involving de-differentiation of the mature β-cell towards a more progenitor-like state, may be an important driver for impaired insulin secretion in T2DM [113]. In fact, Jaafar et al. provided recent evidence that T2DM, a condition marked by both mitochondrial degeneration and dysregulated GSIS, was associated with a remarkable reversion of the normal AMPK-dependent adult β-cell signature to a more neonatal one characterized by mTORC1 activation (Fig. 2a, b) [41]. Persistently over-activated mTORC1 has been associated with increased endoplasmic reticulum (ER) stress and impaired autophagy resulting in early β-cell apoptosis [70, 71, 114,115,116,117,118,119,120].
Persistent consumption of pasteurized milk is apparently the ideal endocrine signaling mechanism that maintains mTORC1 over-activation combined with milk-miRNA-148a/miRNA-29b-mediated suppression of AMPK, ERR γ, DNMT1, DNMT3A and MAFB promoting both mTORC1-dependent β-cell stress as well as β-cell de-differentiation with functional impairments of GSIS. These observations shed a new light on milk exosomal miRNAs operating as critical environmental promoters of T2DM, i.e., milk-derived diabetogenic miRNA toxicity.
Milk transforming growth factor- β in β-cell de-differentiation
Functional β-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3 (UCN3) [121]. UCN3 is expressed in adult β-cells in both mouse and humans and appears late in β-cell differentiation [122]. Loss of UCN3 expression is an early event in β cell de-differentiation in T2DM [123]. A small molecule inhibitor of TGF-β receptor 1 (TGFBR1) has been shown to protect β-cells from the loss of key β-cell transcription factors and restores a mature β-cell identity including UCN3 expression even after exposure to prolonged and severe T2DM. Inhibition of TGF- β receptor 1 was found to protect against β-cell de-differentiation and to restore the identity of mature β-cells. Notably, exosomes of commercial milk contain and transfer bioactive TGF- β to the milk recipient [124]. Upregulation of SMAD7, a downstream mediator of TGF- β signaling, promotes β-cell proliferation [125]. Milk exosome-mediated TGF- β signaling in synergy with milk miRNAs may thus promote proliferation and de-differentiation of β-cells (Fig. 1a).
Milk miRNA-29: insulin resistance and disturbed energy homeostasis
The preferred uptake of milk exosomes by the liver may enhance hepatic miRNA-29b levels [20, 23]. Aberrantly enhanced expression of miRNA-29b has been reported across five common rodent models of insulin resistance (IR) and diabetes (OB = leptin-deficient ob/ob mice; STZ = streptozotocin-treated mice; ZF = fa/fa Zucker Fatty rats; UCD = rat model of late-onset type diabetes (UC Davis); LIRKO = liver-specific insulin receptor knock-out mice; NOD mice) [126,127,128,129,130]. In skeletal muscle of diabetic Goto-Kakizaki rats, miRNA-29a, miRNAR-29b, and miRNA-29c were significantly upregulated [131]. miRNA-29a and miRNA-29c are increased in skeletal muscle from patients with T2DM and are decreased following endurance training in healthy young men and in rats [132]. miRNA-29b via its seed sequence ACCACGA targets key players of IR such as insulin receptor substrate-1 (IRS1) (Table 3). Hung et al. recently demonstrated that acute suppression of IR-associated hepatic miRNA-29 in vivo using locked nucleic acid (LNA) technology improved glycemic control in adult mice [133]. miRNA-29 suppression resulted in increased expression miRNA-29b targets DNMT3A and ENHO (Table 3). As outlined, DNMT3A is important for β-cell differentiation [93], whereas ENHO encodes the energy homeostasis hormone adropin. Remarkably, decreased serum adropin levels have been detected in T2DM patients that negatively correlate with body mass index (BMI) [134]. Adropin deficiency worsens high-fat diet-induced metabolic defects [135]. Milk-miRNA-29b-mediated suppression of adropin may thus increase BMI, a desired mechanism for the growing infant but not intended for body mass homeostasis in adulthood. Milk-mediated transfer of miRNA-29b may thus aggravate both IR and BMI. In fact, IR has been demonstrated in 8-year-old boys consuming 53 g protein daily provided as 1.5 L skim milk compared to boys receiving 53 g protein provided as low-fat meat [3]. In accordance, the NHANES 1999–2004 study identified a correlation between milk consumption and BMI in children [136].
miRNA-29b and β-cell exocytosis
Overexpression of miRNA-29a/b/c in the MIN6 β-cell line and dissociated islet cells led to impairment in GSIS [137]. Defective insulin release was associated with diminished expression of the transcription factor ONECUT2, and a consequent rise of granuphilin, an inhibitor of β-cell exocytosis. ONECUT2 binds to the granuphilin promoter and represses its transcriptional activity (Fig. 1a) [137]. Silencing of ONECUT2 mimicked the effects of miRNA-9 on stimulus-induced exocytosis and on granuphilin expression [137]. Notably, ONECUT2 is a target gene of miRNA-29a/b/c [138, targetscan.org] (Table 3). Milk-miRNA-29 may thus attenuate insulin secretion during the physiologically restricted period of breastfeeding.
Milk miRNA-148a synergizes with high-fat diet-induced adipogenesis
T2DM and obesity (diabesity) with associated IR are intimately related. miRNA-148a is increased in adipose tissues from obese individuals and mice fed a high-fat diet (HFD) [138]. miRNA-148a suppresses its target gene WNT1, an endogenous inhibitor of adipogenesis [139, 140]. Ectopic expression of miRNA-148a, a potential constellation evoked by milk miRNA intake, accelerates differentiation and partially rescued WNT1-mediated inhibition of adipogenesis, whereas knockdown of miRNA-148a inhibited adipogenesis [138, 139]. miRNA-148a also silences WNT10b, a further endogenous inhibitor of adipogenesis [140]. Furthermore, increased expression of miRNA-148a via suppression of DNMT1 enhanced adipocyte differentiation, whereas in the absence of DNMT1 adipocyte-specific gene expression and lipid accumulation occurred precociously [141]. DNA methylation biphasically regulates 3 T3-L1 preadipocyte differentiation [142]. Inhibition of DNA methylation at late stage of preadipocyte differentiation promotes lipogenesis and the adipocyte phenotype in 3 T3-L1 cells, which may be mediated by induction of sterol regulatory element-binding transcription factor 1c (SREBF1c), whose promoter activity is upregulated by DNA demethylation during adipogenesis [143]. Persistent transfer of milk exosomal miRNA-148a may thus enhance SREBF1c-mediated lipid accumulation in adipocytes. In accordance, the MIR148A gene has been identified as an obesity risk gene in humans exhibiting single nucleotide polymorphisms which enhance miRNA-148a expression [144, 145].
Furthermore, milk exosome-derived miRNA-148a may induce a state of hyperphagia, which is meaningful for the growing infant during the anabolic time of breastfeeding. The accumulation of milk exosomes in the brain may allow miRNA-mediated fine-tuning of hypothalamic centers regulating satiety control [20, 33]. Cholecystokinin (CCK), released by duodenal I-cell during intestinal nutrient abundance, is an important hormone that induces satiety signals in the hypothalamus via binding to CCK receptor 2 (CCKBR). Remarkably, CCKBR is a direct target of miR-148a [146]. CCKBR deletion was associated with increased body weight and hypothalamic neuropeptide Y (NPY) content, which explains the increased food intake in CCK2R knockout mice [147, 148]. Thus, persistent intake of milk exosomes may directly promote adipogenesis and indirectly maintain a “hungry brain”, synergistic mechanisms promoting diabesity and increased BMI.
Environmental risk factors promoting milk-miRNA-driven type 2 diabetes
When Neolithic humans took milk from other mammalian species as a nutrient source about 10,000 years ago, the majority of collected milk was processed by natural microbial fermentation, which degrades milk exosomes and their miRNAs [39]. However, with the introduction of pasteurization (78 ° C), bioactive exosomal miRNAs of milk survived and reached the human food chain by refrigerated storage since the 1950s´. Remarkably, since that time, T2DM prevalence increased progressively from 1% to more than 8.5% today with a prognosis of 10% in the near future [149, 150]. Pasteurization and cooling technologies are not the only changes that increase diabetogenic miRNA exposure. Importantly, MIR148A has been identified as a domestication gene of dairy cattle that together with MIR29B1 increases milk yield [151,152,153]. Genetic selection for high performance dairy cows may thus has increased the miRNA-related diabetogenic toxicity of commercial milk [32, 33].
Conclusions
Presented translational evidence sheds a new light on the physiological role of milk signaling in postnatal and adult regulation of β-cell homeostasis. Milk-derived exosomes and their cargo, especially miRNA-148a, miRNA-29b, miRNA-29c, miRNA-130a and TGF- β, altogether suppress β-cell differentiation and insulin secretion promoting mTORC1-dependent β-cell proliferation during the postnatal growth period of β-cells. Weaning can be regarded as a physiological “turn-off” signal restricting maternal exosomal miRNA signaling. Accumulated translational evidence supports the hypothesis that the termination of exosomal milk miRNA signaling during weaning presents the appropriate signals for AMPK/DNMT1/DNMT3A-driven β-cell maturation towards the adult GSIS phenotype. However, unnatural persistence of milk signaling by continued consumption of pasteurized cow’s milk maintains the immature postnatal progenitor-like state of β-cells promoting β-cell de-differentiation with over-activation of mTORC1 and suppression of AMPK. Persistently over-activated mTORC1 and insufficient secretory function of milk-miRNA-dedifferentiated β-cells finally enhances ER stress, impairs autophagy and promotes early β-cell apoptosis explaining the epidemiological link between milk consumption and T2DM (Fig. 2c, d). It should be kept in mind that milk-driven miRNA-148a/miRNA-29b signaling is not the only pathway controlling the balance of mTORC1 and AMPK. Other RNAs may act in concert for adjustments of mTORC1 and AMPK activity. Especially long noncoding RNAs (lncRNAs) are in the focus of RNA research in T2DM [154]. Notably, lncRNAs have recently been detected in bovine milk exosomes and have been shown to be stable during in vitro digestion [155].
In contrast to persistent milk miRNA signaling in adulthood, the absence of milk miRNAs in artificial infant formula may impair regular early postnatal β-cell development increasing the risk of T2DM, a plausible new explanation for the diabetes-preventive effect of breastfeeding (Fig. 2c, d) [32, 33].
Before new treatment options for T2DM with exosomes and extracellular vesicles are employed [62, 156], milk’s physiological functions in maternal-neonatal β-cell communication should be studied in more detail. The environmental exposure of the human milk consumer to bioactive bovine miRNAs that apparently compromise β-cell homeostasis has to be terminated. Milk miRNAs are potential diabetogenic biohazards that should not reach the human food chain [32, 33, 37].
Accumulated evidence allows to conclude that milk-derived exosomes of pasteurized milk represent critical pathogens of Western civilization promoting T2DM and explain the epidemic diabetes as a communicable milk-exosome-mediated disease.
Boiling, ultra-heat-treatment, ultra-sonication and fermentation of milk are effective and required methods to reduce the diabetogenic effects of this common food component of developed societies. Among the known roles of gluco- and lipotoxicity of Western diet, presented translational evidence identified milk miRNA toxicity as a new preventable factor in the pathogenesis of T2DM.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Abbreviations
- AMPK:
-
5′-adenosine monophosphate-activated protein kinase
- ARX:
-
aristaless-related homeobox, X-linked
- BCAA:
-
branched-chain amino acid
- BCKA:
-
branched-chain α-keto acid
- BCKD:
-
branched-chain α-keto acid dehydrogenase
- DBT:
-
dihydrolipoamide branched-chain acyltransferase
- DNMT1:
-
DNA methyltransferase 1
- EPIC:
-
European Prospective Investigation into Cancer and Nutrition
- ER:
-
endoplasmic reticulum
- ERRγ:
-
estrogen-related receptor-γ
- ETC:
-
electron transport chain
- EV:
-
extracellular vesicle
- GSIS:
-
glucose-stimulated insulin secretion
- HK1:
-
hexokinase 1
- HPFS:
-
Health Professionals Follow-up Study
- IR:
-
insulin resistance
- IRS-1:
-
insulin receptor substrate 1
- LNA:
-
locked nucleic acid
- lncRNA:
-
long noncoding RNA
- MAFA:
-
V-MAF avian musculoaponeurotic fibrosarcoma oncogene homolog A
- MAFB:
-
V-MAF musculoaponeurotic fibrosarcoma oncogene family, protein B
- miRNA:
-
micro-ribonucleic acid
- MONICA:
-
monitoring trends and determinants in cardiovascular diseases
- mRNA:
-
messenger ribonucleic acid
- mTORC1:
-
mechanistic target of rapamycin complex 1
- NHS:
-
Nurses’ Health Study
- PI3K:
-
phosphatdylinositol-3 kinase
- PPARGC1A:
-
peroxisome proliferator-activated receptor- γ, coactivator 1 α (PGC1 α)
- PRKAA1:
-
AMP-activated protein kinase, catalytic subunit α 1
- PTEN:
-
phosphatase and tensin homolog
- T2DM:
-
type 2 diabetes mellitus
- TCA:
-
tricarboxylic acid
- TGFBR1:
-
TGF-β receptor 1
- UCN3:
-
urocortin 3
- WNT1:
-
wingless-type MMTV integration site family, member 1
- WNT10B:
-
wingless-type MMTV integration site family, member 10B;
References
https://www.statista.com/statistics/448642/per-capita-consumption-of-milk-in-the-us/. Accessed 28 Sept 2019.
https://milchindustrie.de/wpcontent/uploads/2018/11/ProkopfDeutschland_Mop ro_2010-2018x_Homepage.pdf. Accessed 28 Sept 2019.
Hoppe C, Mølgaard C, Vaag A, Barkholt V, Michaelsen KF. High intakes of milk, but not meat, increase s-insulin and insulin resistance in 8-year-old boys. Eur J Clin Nutr. 2005;59:393–8.
Lawlor DA, Ebrahim S, Timpson N, Davey SG. Avoiding milk is associated with a reduced risk of insulin resistance and the metabolic syndrome: findings from the British Women’s Heart and Health Study. Diabet Med. 2005;22:808–11.
Bergholdt HK, Nordestgaard BG, Ellervik C. Milk intake is not associated with low risk of diabetes or overweight-obesity: a Mendelian randomization study in 97,811 Danish individuals. Am J Clin Nutr. 2015;102:487–96.
Song Y, Chavarro JE, Cao Y, Qiu W, Mucci L, Sesso HD, et al. Whole milk intake is associated with prostate cancer-specific mortality among U.S. male physicians. J Nutr. 2013;143:189–96.
Sluijs I, Forouhi NG, Beulens JW, van der Schouw YT, Agnoli C, Arriola L, et al. The amount and type of dairy product intake and incident type 2 diabetes: results from the EPIC-InterAct study. Am J Clin Nutr. 2012;96:382–90.
Brouwer-Brolsma EM, Sluik D, Singh-Povel CM, Feskens EJM. Dairy product consumption is associated with pre-diabetes and newly diagnosed type 2 diabetes in the lifelines cohort study. Br J Nutr. 2018;119:442–55.
Hruby A, Ma J, Rogers G, Meigs JB, Jacques PF. Associations of dairy intake with incident prediabetes or diabetes in middle-aged adults vary by both dairy type and glycemic status. J Nutr. 2017;147:1764–75.
Guo J, Givens DI, Astrup A, Bakker SJL, Goossens GH, Kratz M, et al. The impact of dairy products in the development of type 2 diabetes: where does the evidence stand in 2019? Adv Nutr. 2019;00:1–10.
Izumi H, Kosaka N, Shimizu T, Sekine K, Ochiya T, Takase M. Bovine milk contains microRNA and messenger RNA that are stable under degradative conditions. J Dairy Sci. 2012;95:4831–41.
Hata T, Murakami K, Nakatani H, Yamamoto Y, Matsuda T, Aoki N. Isolation of bovine milk-derived microvesicles carrying mRNAs and microRNAs. Biochem Biophys Res Commun. 2010;396:528–33.
Izumi H, Tsuda M, Sato Y, Kosaka N, Ochiya T, Iwamoto H, et al. Bovine milk exosomes contain microRNA and mRNA and are taken up by human macrophages. J Dairy Sci. 2015;98:2920–33.
Benmoussa A, Lee CH, Laffont B, Savard P, Laugier J, Boilard E, et al. Commercial dairy cow milk microRNAs resist digestion under simulated gastrointestinal tract conditions. J Nutr. 2016;146:2206–15.
Benmoussa A, Ly S, Shan ST, Laugier J, Boilard E, Gilbert C, et al. A subset of extracellular vesicles carries the bulk of microRNAs in commercial dairy cow’s milk. J Extracell Vesicles. 2017;6:1401897.
Wolf T, Baier SR, Zempleni J. The intestinal transport of bovine milk exosomes is mediated by endocytosis in human colon carcinoma Caco-2 cells and rat small intestinal IEC-6 cells. J Nutr. 2015;145:2201–6.
Kusuma RJ, Manca S, Friemel T, Sukreet S, Nguyen C, Zempleni J. Human vascular endothelial cells transport foreign exosomes from cow’s milk by endocytosis. Am J Physiol Cell Physiol. 2016;310:C800–7.
Wang L, Sadri M, Giraud D, Zempleni J. RNase H2-dependent polymerase chain reaction and elimination of confounders in sample collection, storage, and analysis strengthen evidence that microRNAs in bovine milk are bioavailable in humans. J Nutr. 2018;148:153–9.
Baier SR, Nguyen C, Xie F, Wood JR, Zempleni J. MicroRNAs are absorbed in biologically meaningful amounts from nutritionally relevant doses of cow milk and affect gene expression in peripheral blood mononuclear cells, HEK-293 kidney cell cultures, and mouse livers. J Nutr. 2014;144:1495–500.
Manca S, Upadhyaya B, Mutai E, Desaulniers AT, Cederberg RA, White BR, et al. Milk exosomes are bioavailable and distinct microRNA cargos have unique tissue distribution patterns. Sci Rep. 2018;8:11321.
Arntz OJ, Pieters BC, Oliveira MC, Broeren MG, Bennink MB, de Vries M, et al. Oral administration of bovine milk derived extracellular vesicles attenuates arthritis in two mouse models. Mol Nutr Food Res. 2015;59:1701–12.
Munagala R, Aqil F, Jeyabalan J, Gupta RC. Bovine milk-derived exosomes for drug delivery. Cancer Lett. 2016;371:48–61.
Betker JL, Angle BM, Graner MW, Anchordoquy TJ. The potential of exosomes from cow milk for oral delivery. J Pharm Sci. 2019;108:1496–505.
Howard KM, Jati Kusuma R, Baier SR, Friemel T, Markham L, Vanamala J, et al. Loss of miRNAs during processing and storage of cow's (Bos taurus) milk. J Agric Food Chem. 2015;63:588–92.
Kirchner B, Pfaffl MW, Dumpler J, von Mutius E, Ege MJ. microRNA in native and processed cow's milk and its implication for the farm milk effect on asthma. J Allergy Clin Immunol. 2016;137:1893–5.
Golan-Gerstl R, Elbaum Shiff Y, Moshayoff V, Schecter D, Leshkowitz D, Reif S. Characterization and biological function of milk-derived miRNAs. Mol Nutr Food Res. 2017;61(10).
Rubio M, Bustamante M, Hernandez-Ferrer C, Fernandez-Orth D, Pantano L, Sarria Y, et al. Circulating miRNAs, isomiRs and small RNA clusters in human plasma and breast milk. PLoS One. 2018;13:e0193527.
Simpson MR, Brede G, Johansen J, Johnsen R, Storrø O, Sætrom P, et al. Human breast milk miRNA, maternal probiotic supplementation and atopic dermatitis in offspring. PLoS One. 2015;10:e0143496.
Munch EM, Harris RA, Mohammad M, Benham AL, Pejerrey SM, Showalter L, et al. Transcriptome profiling of microRNA by next-gen deep sequencing reveals known and novel miRNA species in the lipid fraction of human breast milk. PLoS One. 2013;8:e50564.
Melnik BC. Milk: an epigenetic amplifier of FTO-mediated transcription? Implications for Western diseases. J Transl Med. 2015;13:385.
Melnik BC, Kakulas F, Geddes DT, Hartmann PE, John SM, Carrera-Bastos P, et al. Milk miRNAs: simple nutrients or systemic functional regulators? Nutr Metab (Lond). 2016;13:42.
Melnik BC, Schmitz G. MicroRNAs: Milk’s epigenetic regulators. Best Pract Res Clin Endocrinol Metab. 2017;31:427–42.
Melnik BC, Schmitz G. Milk’s role as an epigenetic regulator in health and disease. Diseases. 2017;5:E12.
Zempleni J, Aguilar-Lozano A, Sadri M, Sukreet S, Manca S, Wu D, et al. Biological activities of extracellular vesicles and their cargos from bovine and human milk in humans and implications for infants. J Nutr. 2017;147:3–10.
Melnik BC. Milk disrupts p53 and DNMT1, the guardians of the genome: implications for acne vulgaris and prostate cancer. Nutr Metab (Lond). 2017;14:55.
Benmoussa A, Provost P. Milk microRNAs in health and disease. Compr Rev Food Sci Food Safety. 2019;18:703–22.
Melnik BC, Schmitz G. Exosomes of pasteurized milk: potential pathogens of Western diseases. J Transl Med. 2019;17:3.
Zempleni J, Sukreet S, Zhou F, Wu D, Mutai E. Milk-derived exosomes and metabolic regulation. Annu Rev Anim Biosci. 2019;7:245–62.
Yu S, Zhao Z, Sun L, Li P. Fermentation results in quantitative changes in milk-derived exosomes and different effects on cell growth and survival. J Agric Food Chem. 2017;65:1220–8.
Stolovich-Rain M, Enk J, Vikesa J, Nielsen FC, Saada A, Glaser B, et al. Weaning triggers a maturation step of pancreatic β cells. Dev Cell. 2015;32:535–45.
Jaafar R, Tran S, Shah AN, Sun G, Valdearcos M, Marchetti P, et al. mTORC1 to AMPK switching underlies β-cell metabolic plasticity during maturation and diabetes. J Clin Invest. 2019;130:4124–37.
Melnik BC. Milk - a nutrient system of mammalian evolution promoting mTORC1-dependent translation. Int J Mol Sci. 2015;16:17048–87.
Mori H, Inoki K, Opland D, Munzberg H, Villanueva EC, Faouzi M, et al. Critical roles for the TSC-mTOR pathway in beta-cell function. Am J Physiol Endocrinol Metab. 2009;297:E1013–22.
Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, et al. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol. 2008;28:2971–9.
Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, et al. Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat Commun. 2017;8:16014.
Chau GC, Im DU, Kang TM, Bae JM, Kim W, Pyo S, et al. mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress. J Cell Biol. 2017;216:2091–105.
Ni Q, Gu Y, Xie Y, Yin Q, Zhang H, Nie A, et al. Raptor regulates functional maturation of murine beta cells. Nat Commun. 2017;8:15755.
Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell. 2008;30:214–26.
Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–90.
Melnik BC, Schmitz G. Metformin: an inhibitor of mTORC1 signaling. J Endocrinol Diabetes Obes. 2014;2:1029.
Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia. 2017;60:1577–85.
Alejandro EU, Gregg B, Wallen T, Kumusoglu D, Meister D, Chen A, et al. Maternal diet-induced microRNAs and mTOR underlie β cell dysfunction in offspring. J Clin Invest. 2014;124:4395–410.
Dumortier O, Fabris G, Van Obberghen E. Shaping and preserving β-cell identity with microRNAs. Diabetes Obes Metab. 2016;18(Suppl 1):51–7.
Ofori JK, Salunkhe VA, Bagge A, Vishnu N, Nagao M, Mulder H, et al. Elevated miR-130a/miR130b/miR-152 expression reduces intracellular ATP levels in the pancreatic beta cell. Sci Rep. 2017;7:44986.
Esguerra JLS, Nagao M, Ofori JK, Wendt A, Eliasson L. MicroRNAs in islet hormone secretion. Diabetes Obes Metab. 2018;20(Suppl 2):11–9.
Jacovetti C, Matkovich SJ, Rodriguez-Trejo A, Guay C, Regazzi R. Postnatal β-cell maturation is associated with islet-specific microRNA changes induced by nutrient shifts at weaning. Nat Commun. 2015;6:8084.
Jacovetti C, Rodriguez-Trejo A, Guay C, Sobel J, Gattesco S, Petrenko V, et al. MicroRNAs modulate core-clock gene expression in pancreatic islets during early postnatal life in rats. Diabetologia. 2017;60:2011–20.
Guay C, Menoud V, Rome S, Regazzi R. Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun Signal. 2015;13:17.
Osmai M, Osmai Y, Bang-Berthelsen CH, Pallesen EM, Vestergaard AL, Novotny GW, et al. MicroRNAs as regulators of beta-cell function and dysfunction. Diabetes Metab Res Rev. 2016;32:334–49.
Guay C, Regazzi R. Exosomes as new players in metabolic organ cross-talk. Diabetes Obes Metab. 2017;19(Suppl 1):137–46.
Jalabert A, Vial G, Guay C, Wiklander OP, Nordin JZ, Aswad H, et al. Exosome-like vesicles released from lipid-induced insulin-resistant muscles modulate gene expression and proliferation of beta recipient cells in mice. Diabetologia. 2016;59:1049–58.
Ge Q, Xie XX, Xiao X, Li X. Exosome-like vesicles as new mediators and therapeutic targets for treating insulin resistance and β-cell mass failure in type 2 diabetes mellitus. J Diabetes Res. 2019;2019:3256060.
Javeed N, Sagar G, Dutta SK, Smyrk TC, Lau JS, Bhattacharya S, et al. Pancreatic cancer-derived exosomes cause paraneoplastic β-cell dysfunction. Clin Cancer Res. 2015;21:1722–33.
Zanone MM, Favaro E, Camussi G. From endothelial to beta cells: insights into pancreatic islet microendothelium. Curr Diabetes Rev. 2008;4:1–9.
Reinhardt TA, Lippolis JD, Nonnecke BJ, Sacco RE. Bovine milk exosome proteome. J Proteome. 2012;75:1486–92.
Gao W, Ge S, Sun J. Ailanthone exerts anticancer effect by up-regulating miR-148a expression in MDA-MB-231 breast cancer cells and inhibiting proliferation, migration and invasion. Biomed Pharmacother. 2019;109:1062–9.
Reif S, Elbaum Shiff Y, Golan-Gerstl R. Milk-derived exosomes (MDEs) have a different biological effect on normal fetal colon epithelial cells compared to colon tumor cells in a miRNA-dependent manner. J Transl Med. 2019;17:325.
Mersey BD, Jin P, Danner DJ. Human microRNA (miR29b) expression controls the amount of branched chain alpha-ketoacid dehydrogenase complex in a cell. Hum Mol Genet. 2005;14:3371–7.
Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–602.
Jewell JL, Russell RC, Guan KL. Amino acid signalling upstream of mTOR. Nat Rev Mol Cell Biol. 2013;14:133–9.
Bar-Peled L, Sabatini DM. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014;24:400–6.
Zheng X, Liang Y, He Q, Yao R, Bao W, Bao L, et al. Current models of mammalian target of rapamycin complex 1 (mTORC1) activation by growth factors and amino acids. Int J Mol Sci. 2014;15:20753–69.
Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35.
Melnik BC. Leucine signaling in the pathogenesis of type 2 diabetes and obesity. World J Diabetes. 2012;3:38–53.
Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol. 2014;10:723–36.
Melnik BC. The pathogenic role of persistent milk signaling in mTORC1- and milk-microRNA-driven type 2 diabetes mellitus. Curr Diabetes Rev. 2015;11:46–62.
Yoon MS. The emerging role of branched-chain amino acids in insulin resistance and metabolism. Nutrients. 2016;8(7):E405.
Bloomgarden Z. Diabetes and branched-chain amino acids: what is the link? J Diabetes. 2018;10:350–2.
Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol. 2018;596:623–45.
Solon-Biet SM, Cogger VC, Pulpitel T, Wahl D, Clark X, Bagley EE, et al. Branched-chain amino acids impact health and lifespan indirectly via amino acid balance and appetite control. Nat Metab. 2019;1:532–45.
Pullen TJ, Sylow L, Sun G, Halestrap AP, Richter EA, Rutter GA. Overexpression of monocarboxylate transporter-1 (SLC16A1) in mouse pancreatic β-cells leads to relative hyperinsulinism during exercise. Diabetes. 2012;61:1719–25.
Pullen TJ, Rutter GA. When less is more: the forbidden fruits of gene repression in the adult β-cell. Diabetes Obes Metab. 2013;15:503–12.
Kone M, Pullen TJ, Sun G, Ibberson M, Martinez-Sanchez A, Sayers S, et al. LKB1 and AMPK differentially regulate pancreatic β-cell identity. FASEB J. 2014;28:4972–85.
Rutter GA, Leclerc I. The AMP-regulated kinase family: enigmatic targets for diabetes therapy. Mol Cell Endocrinol. 2009;297:41–9.
Rourke JL, Hu Q, Screaton RA. AMPK and friends: central regulators of β cell biology. Trends Endocrinol Metab. 2018;29:111–22.
Yoshihara E, Wei Z, Lin CS, Fang S, Ahmadian M, Kida Y, et al. ERRγ is required for the metabolic maturation of therapeutically functional glucose-responsive β cells. Cell Metab. 2016;23:622–34.
Misra J, Kim DK, Choi HS. ERRγ: a junior orphan with a senior role in metabolism. Trends Endocrinol Metab. 2017;28:261–72.
Devarakonda S, Gupta K, Chalmers MJ, Hunt JF, Griffin PR, Van Duyne GD, et al. Disorder-to-order transition underlies the structural basis for the assembly of a transcriptionally active PGC-1α/ERRγ complex. Proc Natl Acad Sci U S A. 2011;108:18678–83.
Takacs M, Petoukhov MV, Atkinson RA, Roblin P, Ogi FX, Demeler B, et al. The asymmetric binding of PGC-1α to the ERRα and ERRγ nuclear receptor homodimers involves a similar recognition mechanism. PLoS One. 2013;8:e67810.
Chen Y, Song YX, Wang ZN. The microRNA-148/152 family: multi-faceted players. Mol Cancer. 2013;12:43.
Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic β cell identity is maintained by DNA methylation-mediated repression of Arx. Dev Cell. 2011;20:419–29.
Pan W, Zhu S, Yuan M, Cui H, Wang L, Luo X, et al. MicroRNA-21 and microRNA-148a contribute to DNA hypomethylation in lupus CD4+ T cells by directly and indirectly targeting DNA methyltransferase 1. J Immunol. 2010;184:6773–81.
Dhawan S, Tschen SI, Zeng C, Guo T, Hebrok M, Matveyenko A, et al. DNA methylation directs functional maturation of pancreatic β cells. J Clin Invest. 2015;125:2851–60.
Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood. 2009;113:6411–8.
Starlard-Davenport A, Kutanzi K, Tryndyak V, Word B, Lyn-Cook B. Restoration of the methylation status of hypermethylated gene promoters by microRNA-29b in human breast cancer: a novel epigenetic therapeutic approach. J Carcinog. 2013;12:15.
Zhang Z, Cao Y, Zhai Y, Ma X, An X, Zhang S, et al. MicroRNA-29b regulates DNA methylation by targeting Dnmt3a/3b and Tet1/2/3 in porcine early embryo development. Develop Growth Differ. 2018;60:197–204.
Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, et al. MafA and MafB regulate genes critical to beta-cells in a unique temporal manner. Diabetes. 2010;59:2530–9.
Artner I, Blanchi B, Raum JC, Guo M, Kaneko T, Cordes S, et al. MafB is required for islet beta cell maturation. Proc Natl Acad Sci U S A. 2007;104:3853–8.
Nishimura W, Kondo T, Salameh T, El Khattabi I, Dodge R, Bonner-Weir S, et al. A switch from MafB to MafA expression accompanies differentiation to pancreatic beta-cells. Dev Biol. 2006;293:526–39.
Hang Y, Stein R. MafA and MafB activity in pancreatic β cells. Trends Endocrinol Metab. 2011;22:364–73.
Cheng P, Chen C, He HB, Hu R, Zhou HD, Xie H, et al. miR-148a regulates osteoclastogenesis by targeting V-maf musculoaponeurotic fibrosarcoma oncogene homolog B. J Bone Miner Res. 2013;28:1180–90.
Gouveri E, Papanas N, Hatzitolios AI, Maltezos E. Breastfeeding and diabetes. Curr Diabetes Rev. 2011;7:135–42.
Kalra B, Gupta Y, Kalra S. Breast feeding: preventive therapy for type 2 diabetes. J Pak Med Assoc. 2015;65:1134–6.
Pereira PF, Alfenas Rde C, Araújo RM. Does breastfeeding influence the risk of developing diabetes mellitus in children? A review of current evidence. J Pediatr (Rio J). 2014;90:7–15.
Horta BL, de Lima NP. Breastfeeding and type 2 diabetes: systematic review and meta-analysis. Curr Diab Rep. 2019;19:1.
Alsaweed M, Lai CT, Hartmann PE, Geddes DT, Kakulas F. Human milk miRNAs primarily originate from the mammary gland resulting in unique miRNA profiles of fractionated milk. Sci Rep. 2016;6:20680.
Alsaweed M, Hartmann PE, Geddes DT, Kakulas F. MicroRNAs in breastmilk and the lactating breast: potential immunoprotectors and developmental regulators for the infant and the mother. Int J Environ Res Public Health. 2015;12:13981–4020.
Chen X, Gao C, Li H, Huang L, Sun Q, Dong Y, et al. Identification and characterization of microRNAs in raw milk during different periods of lactation, commercial fluid, and powdered milk products. Cell Res. 2010;20:1128–37.
Lönnerdal B. Human milk microRNAs/exosomes: composition and biological effects. Nestle Nutr Inst Workshop Ser. 2019;90:83–92.
Leiferman A, Shu J, Upadhyaya B, Cui J, Zempleni J. Storage of extracellular vesicles in human milk, and microRNA profiles in human milk exosomes and infant formulas. J Pediatr Gastroenterol Nutr. 2019;69:235–8.
van Herwijnen MJC, Driedonks TAP, Snoek BL, Kroon AMT, Kleinjan M, Jorritsma R, et al. Abundantly present miRNAs in milk-derived extracellular vesicles are conserved between mammals. Front Nutr. 2018;5:81.
Mononen N, Lyytikäinen LP, Seppälä I, Mishra PP, Juonala M, Waldenberger M, et al. Whole blood microRNA levels associate with glycemic status and correlate with target mRNAs in pathways important to type 2 diabetes. Sci Rep. 2019;9:8887.
Rutter GA, Pullen TJ, Hodson DJ, Martinez-Sanchez A. Pancreatic β-cell identity, glucose sensing and the control of insulin secretion. Biochem J. 2015;466:203–18.
Hur KY, Jung HS, Lee MS. Role of autophagy in β-cell function and mass. Diabetes Obes Metab. 2010;12(Suppl 2):20–6.
Bartolomé A, Kimura-Koyanagi M, Asahara S, Guillén C, Inoue H, Teruyama K, et al. Pancreatic β-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes. 2014;63:2996–3008.
Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, et al. Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia. 2017;60:668–78.
Bartolomé A, García-Aguilar A, Asahara SI, Kido Y, Guillén C, Pajvani UB, et al. MTORC1 regulates both general autophagy and mitophagy induction after oxidative phosphorylation uncoupling. Mol Cell Biol. 2017;37(23):e00441–17.
Ali M, Bukhari SA, Ali M, Lee HW. Upstream signalling of mTORC1 and its hyperactivation in type 2 diabetes (T2D). BMB Rep. 2017;50:601–9.
Ardestani A, Lupse B, Kido Y, Leibowitz G, Maedler K. mTORC1 signaling: a double-edged sword in diabetic β cells. Cell Metab. 2018;27:314–31.
Guillén C, Benito M. mTORC1 overactivation as a key aging factor in the progression to type 2 diabetes mellitus. Front Endocrinol (Lausanne). 2018;9:621.
Blum B, Hrvatin S, Schuetz C, Bonal C, Rezania A, Melton DA. Functional beta-cell maturation is marked by an increased glucose threshold and by expression of urocortin 3. Nat Biotechnol. 2012;30:261–4.
van der Meulen T, Xie R, Kelly OG, Vale WW, Sander M, Huising MO. Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic alpha and beta cells. PLoS One. 2012;7:e52181.
Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, et al. Reversal of β cell de-differentiation by a small molecule inhibitor of the TGFβ pathway. Elife. 2014;3:e02809.
Pieters BC, Arntz OJ, Bennink MB, Broeren MG, van Caam AP, Koenders MI, et al. Commercial cow milk contains physically stable extracellular vesicles expressing immunoregulatory TGF-β. PLoS One. 2015;10:e0121123.
Xiao X, Wiersch J, El-Gohary Y, Guo P, Prasadan K, Paredes J, et al. TGFβ receptor signaling is essential for inflammation-induced but not β-cell workload-induced β-cell proliferation. Diabetes. 2013;62:1217–26.
Silva VA, Polesskaya A, Sousa TA, Corrêa VM, André ND, Reis RI, et al. Expression and cellular localization of microRNA-29b and RAX, an activator of the RNA-dependent protein kinase (PKR), in the retina of streptozotocin-induced diabetic rats. Mol Vis. 2011;17:2228–40.
Roggli E, Gattesco S, Caille D, Briet C, Boitard C, Meda P, et al. Changes in microRNA expression contribute to pancreatic β-cell dysfunction in prediabetic NOD mice. Diabetes. 2012;61:1742–51.
Baran-Gale J, Fannin EE, Kurtz CL, Sethupathy P. Beta cell 5′-shifted isomiRs are candidate regulatory hubs in type 2 diabetes. PLoS One. 2013;8:e73240.
Kurtz CL, Peck BC, Fannin EE, Beysen C, Miao J, Landstreet SR, et al. MicroRNA-29 fine-tunes the expression of key FOXA2-activated lipid metabolism genes and is dysregulated in animal models of insulin resistance and diabetes. Diabetes. 2014;63:3141–8.
Dooley J, Garcia-Perez JE, Sreenivasan J, Schlenner SM, Vangoitsenhoven R, Papadopoulou AS, et al. The microRNA-29 family dictates the balance between homeostatic and pathological glucose handling in diabetes and obesity. Diabetes. 2016;65:53–61.
He A, Zhu L, Gupta N, Chang Y, Fang F. Overexpression of micro ribonucleic acid 29, highly up-regulated in diabetic rats, leads to insulin resistance in 3T3-L1 adipocytes. Mol Endocrinol. 2007;21:2785–94.
Massart J, Sjögren RJO, Lundell LS, Mudry JM, Franck N, O'Gorman DJ, et al. Altered miR-29 expression in type 2 diabetes influences glucose and lipid metabolism in skeletal muscle. Diabetes. 2017;66:1807–18.
Hung YH, Kanke M, Kurtz CL, Cubitt R, Bunaciu RP, Miao J, et al. Acute suppression of insulin resistance-associated hepatic miR-29 in vivo improves glycemic control in adult mice. Physiol Genomics. 2019;51:379–89.
Zang H, Jiang F, Cheng X, Xu H, Hu X. Serum adropin levels are decreased in Chinese type 2 diabetic patients and negatively correlated with body mass index. Endocr J. 2018;65:685–91.
Chen S, Zeng K, Liu QC, Guo Z, Zhang S, Chen XR, et al. Adropin deficiency worsens HFD-induced metabolic defects. Cell Death Dis. 2017;8:e3008.
Wiley AS. Dairy and milk consumption and child growth: is BMI involved? An analysis of NHANES 1999-2004. Am J Hum Biol. 2010;22:517–25.
Plaisance V, Abderrahmani A, Perret-Menoud V, Jacquemin P, Lemaigre F, Regazzi R. MicroRNA-9 controls the expression of granuphilin/Slp4 and the secretory response of insulin-producing cells. J Biol Chem. 2006;281:26932–42.
Shi C, Zhang M, Tong M, Yang L, Pang L, Chen L, et al. miR-148a is associated with obesity and modulates adipocyte differentiation of mesenchymal stem cells through Wnt signaling. Sci Rep. 2015;5:9930.
Shi C, Pang L, Ji C, Wang J, Lin N, Chen J, et al. Obesity-associated miR148a is regulated by cytokines and adipokines via a transcriptional mechanism. Mol Med Rep. 2016;14:5707–12.
Cho YM, Kim TM, Hun Kim D, Hee Kim D, Jeong SW, Kwon OJ. miR-148a is a downstream effector of X-box-binding protein 1 that silences Wnt10b during adipogenesis of 3T3-L1 cells. Exp Mol Med. 2016;48:e226.
Londoño Gentile T, Lu C, Lodato PM, Tse S, Olejniczak SH, Witze ES, et al. DNMT1 is regulated by ATP-citrate lyase and maintains methylation patterns during adipocyte differentiation. Mol Cell Biol. 2013;33:3864–78.
Yang X, Wu R, Shan W, Yu L, Xue B, Shi H. DNA methylation biphasically regulates 3T3-L1 preadipocyte differentiation. Mol Endocrinol. 2016;30:677–87.
Schwenk RW, Vogel H, Schürmann A. Genetic and epigenetic control of metabolic health. Mol Metab. 2013;2:337–47.
Monda KL, Chen GK, Taylor KC, Palmer C, Edwards TL, Lange LA, et al. A meta-analysis identifies new loci associated with body mass index in individuals of African ancestry. Nat Genet. 2013;45:690–6.
Voisin S, Almén MS, Zheleznyakova GY, Lundberg L, Zarei S, Castillo S, et al. Many obesity-associated SNPs strongly associate with DNA methylation changes at proximal promoters and enhancers. Genome Med. 2015;7:103.
Yu B, Lv X, Su L, Li J, Yu Y, Gu Q, et al. MiR-148a functions as a tumor suppressor by targeting CCK-BR via inactivating STAT3 and Akt in human gastric cancer. PLoS One. 2016;11:e0158961.
Clerc P, Coll Constans MG, Lulka H, Broussaud S, Guigné C, Leung-Theung-Long S, et al. Involvement of cholecystokinin 2 receptor in food intake regulation: hyperphagia and increased fat deposition in cholecystokinin 2 receptor-deficient mice. Endocrinology. 2007;148:1039–49.
Chen H, Kent S, Morris MJ. Is the CCK2 receptor essential for normal regulation of body weight and adiposity? Eur J Neurosci. 2006;24:1427–33.
CDC’s Division of Diabetes Translation. National Diabetes Surveillance System available at http://www.cdc.gov/diabetes/statistics. Accessed 3 Oct 2019.
Xu G, Liu B, Sun Y, Du Y, Snetselaar LG, Hu FB, et al. Prevalence of diagnosed type 1 and type 2 diabetes among US adults in 2016 and 2017: population based study. BMJ. 2018;362:k1497.
Do DN, Li R, Dudemaine PL, Ibeagha-Awemu EM. MicroRNA roles in signalling during lactation: an insight from differential expression, time course and pathway analyses of deep sequence data. Sci Rep. 2017;7:44605.
Do DN, Dudemaine PL, Li R, Ibeagha-Awemu EM. Co-expression network and pathway analyses reveal important modules of miRNAs regulating milk yield and component traits. Int J Mol Sci. 2017;18(7):E1560.
Bian Y, Lei Y, Wang C, Wang J, Wang L, Liu L, et al. Epigenetic regulation of miR-29s affects the lactation activity of dairy cow mammary epithelial cells. J Cell Physiol. 2015;230:2152–63.
Lin Z, Li X, Zhan X, Sun L, Gao J, Cao Y, et al. Construction of competitive endogenous RNA network reveals regulatory role of long non-coding RNAs in type 2 diabetes mellitus. J Cell Mol Med. 2017;21:3204–13.
Zeng B, Chen T, Xie MY, Luo JY, He JJ, Xi QY, et al. Exploration of long noncoding RNA in bovine milk exosomes and their stability during digestion in vitro. J Dairy Sci. 2019;102:6726–37.
Beuzelin D, Kaeffer B. Exosomes and miRNA-loaded biomimetic nanovehicles, a focus on their potentials preventing type-2 diabetes linked to metabolic syndrome. Front Immunol. 2018;9:2711.
Acknowledgements
The author thanks his academic teacher in molecular pathology Gerd Schmitz, former director of the Institute for Clinical Chemistry and Laboratory Medicine, University of Regensburg, for many inspiring discussions and all scientific support and dedicates this article on the occasion of his 70th birthday.
Author details
Department of Dermatology, Environmental Medicine and Health Theory,
University of Osnabrück, Osnabrück, Germany.
Funding
There is no source of funding.
Author information
Authors and Affiliations
Contributions
BCM wrote the manuscript, performed translational research and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The author declares that he has no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Melnik, B.C. Milk exosomal miRNAs: potential drivers of AMPK-to-mTORC1 switching in β-cell de-differentiation of type 2 diabetes mellitus. Nutr Metab (Lond) 16, 85 (2019). https://doi.org/10.1186/s12986-019-0412-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12986-019-0412-1