
Abstract
Background
Dengue virus (DENV) infection caused by international visitors has become a public health concern in China. Although sporadic imported cases of DENV have been documented in Yunnan, China since 2000, a complete genome sequence of dengue virus serotype 3 (DENV-3) imported from Laos is still not available. Here, we report the first complete genome sequence and genomic characterization of a DENV-3 strain (YNPE3) isolated from a patient returned from Laos.
Methods
Viral isolation from the patient’s serum was performed using mosquitoes C6/36 cells. Reverse transcriptase polymerase chain reaction (RT-PCR) was used for identification and serotyping of the virus. The complete sequence was determined by Sanger dideoxy sequencing. Homology analysis was implemented by NCBI-BLAST. Multiple sequence alignment was performed using MegAlign module of the Lasergene 7 software package DNASTAR. MFOLD software was used to predict the RNA secondary structure of 5′ untranslated region (UTR) and 3′ UTR. Phylogenetic analysis, which was based on envelope gene and complete coding sequence, was performed by Maximum-Likelihood method.
Results
RT-PCR analysis confirmed that the virus belonged to dengue virus serotype 3, which was named YNPE3 strain. The full-length genome of the YNPE3 strain was 10,627 nucleotides (nts) with an open reading frame (ORF) encoding 3390 amino acids. Strain YNPE3 shared 98.6–98.8% nucleotide identity with the closely related strains isolated in India (JQ922556, KU216209, KU216208). We observed the deletion of about 40 nts in the 5′ UTR and 3′ UTR of strain YNPE3, and 11 nts (ACGCAGGAAGT) insertion that was present in the 3′ UTR of YNPE3. Compared with prototype strain H87, abundant amino acid substitutions in the YNPE3 strain were observed. Phylogenetic analysis revealed that the YNPE3 strain belonged to genotype III of DENV-3, and that it might be closely related with genotype III strains isolated in Laos and India.
Conclusions
This is the first report of the complete genome sequence and molecular characterization of a DENV-3 isolate imported from Laos. The presented results can further promote disease surveillance, and epidemiological and evolutionary studies of the DENV-3 in Yunnan province of China.
Similar content being viewed by others


Background
Dengue infection is a rapidly re-emerging mosquito-borne infectious disease caused by dengue virus (DENV), that affects approximately 390 million people annually [1]. More than 100 countries, spanning from the Americas, the Western Pacific, South-East Asia, and Africa to Europe, have reported severe epidemics of dengue. In fact, more than 3.9 billion people worldwide are estimated to be at transmission risk [2]. Dengue infections cause varying degrees of dengue disease, with major clinical manifestations ranging from asymptomatic Dengue Fever (DF) to serious Dengue hemorrhagic fever (DHF) and Dengue shock syndrome (DSS) [3]. Unfortunately, approximately 50–100 million DF cases and hundreds of thousands of DHF cases occur annually [4, 5]. Since there are still no licensed vaccines or specific antiviral drugs, and since each patient with dengue hemorrhagic fever needs to face exorbitant medical costs, dengue infections have become a serious global health problem, and a heavy socioeconomic burden affecting healthcare systems worldwide.
As the etiologic agent of dengue fever, DENV belongs to the genus Flavivirus and family Flaviviridae [6, 7] primarily transmitted by Aedes aegyptis and Aedes albopictus [8, 9], that are known to circulate in the tropical and subtropical regions of the world [8]. DENV is an enveloped, positive-sense, single-stranded RNA virus. The genome of DENV is approximately 11 kb in length, with one open reading frame (ORF) flanked by 5′ and 3′ non-coding regions. A polyprotein is encoded by the ORF, which is cleaved into three structural proteins: C: Capsid glycoprotein; M: membrane glycoprotein; E: envelope glycoprotein; and seven non-structural proteins (NS): NS1, NS2A, NS2B, NS3, NS4A, NS4B and NS5 [10]. Furthermore, DENV is divided into four serotypes(DENV- 1 to DENV-4)based on antigen cross-reactivity. Since there is no cross protection among serotypes, secondary infection of heterologous DENV serotypes often results in severe disease [11]. In addition, each serotype is further divided into distinct genotypes based on viral genome sequencing and evolutionary analysis [12].
Over the past decade, dengue infection has expanded over the vast geographical range [13,14,15,16], This may be caused by more frequent international travel, climate change, virus evolution, unplanned urbanization, globalization, population mobility and failure of Aedes mosquitoes control [13, 14, 17, 18]. In Mainland China, the first recorded DENV infection occurred in Guangdong in 1978 [19]. Over the past 30 years, DENV has spread throughout the country. Although the four serotypes have been circulating in Guangdong, Guangxi, Hainan, Fujian, Zhejiang and Yunnan provinces of China [20, 21], the DENV-1 has been a predominate serotype since 1990s [21]. Geographically, provinces such as Guangdong, Guangxi, Hainan, Fujian, and Zhejiang are situated in the southeast coast of China, whereas Yunnan borders with Southeast Asian countries, such as Myanmar, Laos, Thailand, Vietnam which are all considered endemic risk areas [21,22,23,24,25]. The spread of DENV by tourists returning from Southeast countries is common in China, especially in Yunnan Province. Since 2000, most of the DF documented in Yunnan have been sporadic imported cases. In 2013, a large-scale DENV-3 outbreak comprising 1287 indigenous cases and 44 imported cases occurred in Xishuangbanna, Jinghong, Yunnan [26]. Epidemiological analysis showed that this outbreak was closely related with the imported dengue cases from Laos and Myanmar [26, 27], which suggested that imported dengue cases were mainly responsible for outbreaks and the circulation of DENV-3 in Yunnan, China. Due to the special geographical location, humid climate, booming international tourism, population mobility and the spread of the Aedes mosquitoes, Yunnan is at risk of becoming an endemic risk area in the future.
Although dengue cases imported from Laos have been reported in previous studies, they mainly focused on the envelope (E) gene of the imported DENV-3. The complete genome sequence and molecular characterization of the DENV-3 strain imported from Laos are still not available. In the current research, we reported the complete genome sequence and genomic characterization of a DENV-3 strain imported from Laos. The results from this study provide valuable clues for disease surveillance, transmission control and vaccine design.
Methods
Source of virus
A 10 year old student, who resides in Pu’er, Yunnan Province, China, went on a 7 day trip to Laos from August 10th to August 16th, 2013. She experienced slight fever during the 4th day of the trip, and influenza like symptoms on the third day after returning home. In August 20th, 2013, clinical diagnosis showed a positive result for NS1 antigen of patient’s serum based on the viral NS1 antigen colloidal gold test using the Dengue Ag Rapid Test (CTK Biotech, Inc., San Diego, CA), in the Jinghong People’s Hospital in China. The medical history of the patient was unremarkable with dengue fever and other flaviviruses disease. Written informed consent was obtained from this patient and her legal guardian. A serum sample was collected from the patient within 3–7 days after the onset of the illness and stored in −80 °C until use.
Virus isolation and serotyping
The patient’s serum was inoculated into the C6/36 Aedes albopictus cell line in RPMI 1640 medium (Biological Industries, USA) supplemented with 2% fetal bovine serum (Biological Industries, USA) at 28 °C in 5% CO2. When 70% cytopathic effect (CPE) was observed, the virus was passaged twice more to prepare viral stocks. Supernatant from infected cells was harvested and stored at −80 °C until used. Viral RNA was extracted from 500 μL of infected culture supernatant by Trizol reagent (Tiangen, Beijing, China) according to the manufacturer’s instructions. For virus identification, reverse transcriptase polymerase chain reaction (RT-PCR) amplification was carried out as previously described [28]. Briefly, viral RNA was reverse transcribed in a BioRad C1000 Cycler system using GoScript™ Reverse Transcription System (Promega, WI, USA) according to the manufacturer’s instructions. The cDNA was used for PCR analysis. Five pairs of primers, which were designated as universal primers D1 and D2 of DENV, and four typing primers D1 and TS1 of DENV-1, D1 and TS2 of DENV-2, D1 and TS3 of DENV-3, D1 and TS4 of DENV-4 (primers were described in Additional file 1: Table S1), were employed to amplify the partial capsid/membrane glycoprotein precursor (PrM) gene of DENV. PCR amplification was conducted with the following program: 2 min at 94 °C, followed by 35 cycles at 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, and a final extension step at 72 °C for 10 min. The specific PCR amplicons were further confirmed by agarose gel electrophoresis. PCR products were visualized by a biospectrum® 815 imaging system (UVP, USA).
Genome amplification and sequencing
A total of nine overlapping sub-genomic fragments that spanned the complete genomic region were amplified using nine pairs of primers (Additional file 2: Table S2) [29] by RT-PCR. First, viral RNA was reverse transcribed using the GoScript Reverse Transcription System (Promega, WI, USA) following the manufacturer’s instructions. The RT reaction was conducted under the following conditions: 25 °C for 5 min, followed by 1 h at 42 °C, then 72 °C for 15 min. Next, the PCR was carried out in a reaction mixture of 50 μL containing 5 μL of 10 × La Taq Buffer (Mg2+ Plus), 8 μL of dNTP mixture (2.5 mM), 2 μM of forward and backward primers, and one unit of high fidelity La Taq DNA polymerase (TaKaRa, Dalian, China). PCR amplification was performed under the following parameters [29]: 94 °C for 5 min, followed by 35 cycles of denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 2 min and final extension at 72 °C for 5 min. The PCR results were analyzed by 1% agarose gel electrophoresis and visualized by a biospectrum® 815 imaging system (UVP, USA). Finally, the PCR products were used as templates for bi-directional DNA sequencing by the Sanger dideoxy sequencing method (Invitrogen Ltd., Shanghai).
Genomic characterization and sequencing analysis
The sequences of nine sub-genomic fragments were assembled into a complete genome by removing overlapping sequences with the DNAMAN software program (v. 8.0.8.789) (Lynnon Biosoft, USA). The assembled viral genome was further aligned by NCBI-BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to determine the similarity with other virus strains. Multiple sequences alignment analyses of nucleotides and amino acids for six closely related DENV-3 isolates from China, Laos and other countries were performed based on MegAlign module of Lasergene 7 software package DNASTAR (DNASTAR Inc., USA). Further, the secondary structure of 3′ UTR and 5′ UTR of the YNPE3 strain and prototype strain H87 (M93130) of DENV-3 were determined by the MFOLD software package (http://unafold.rna.albany.edu/?q=mfold) [30] using default folding parameters.
Phylogenetic analysis
Two phylogenetic trees were drawn based on the complete coding sequence (CDS) and E gene of DENV-3. For the E gene, a total of 76 representative strains isolated from different geographical regions in different years were retrieved from GenBank (Additional file 3: Table S3). For the CDS, a total of 75 representative DENV-3 isolates were prepared (Additional file 4: Table S4), the two sets of strains were different with a 10% overlap. The evolutionary history was inferred using the Maximum-Likelihood method [31] of MEGA software (version 7) [32], with a bootstrap test (1000 replicates) [33]. The evolutionary distance was calculated using the Tamura-Nei model [34]. The mutation rate at each site was modeled with a gamma distribution (shape parameter =5). DENV-1 strain Hawaii, DENV-2 strain New Guinea C and DENV-4 strain H241 served as outgroups.
Results
Identification and serotyping of virus
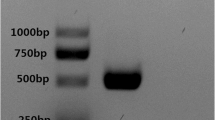
In order to isolate the virus, the patient’s serum was inoculated in cells of the C6/36 cell line until the occurrence of typical CPE, followed by passage twice to increase virus stocks. Compared with normal cells, a typical CPE with the fusion, shedding, aggregation, breaking and suspension were observed in infected cells after 9 days of infection. Furthermore, two fragments with 511 and 290 bp were amplified specifically by RT-PCR with universal primes for DENV and typing primes for DENV-3 respectively, and no specific amplification appeared in the D1TS1, D1TS2 and D1TS4, indicating that the isolate belonged to the serotype 3 of DENV (Fig. 1).

Electrophoresis of typical primer identification of DENV-3. The symbol M indicates DL 2000 DNA marker; The D1D2 indicates the universal amplicon that amplified the partial capsid/premembrane gene (511 bp) of DENV by upstream primer D1 and downstream primer D2. The D1TS1 (482 bp), D1TS2 (119 bp), D1TS3 (290 bp) and D1TS4 (392 bp) are specific amplicons of DENV-1, DENV-2, DENV-3 and DENV-4, respectively
Complete genome sequence and genomic characterization of YNPE3
To amplify the complete genome sequence of the YNPE3 strain, nine sets of primers were designed to amplify nine overlapping fragments across the whole viral genome. As shown in Fig. 2, the nine overlapping fragments were amplified specifically by RT-PCR. Moreover, the sequences of all the overlapping amplicons assembly revealed a full-length genome with 10,627 nts for the YNPE3 strain. The complete genome sequence was submitted to the GenBank database under the accession number MF370226.1. The YNPE3 strain contained a single ORF that encoded 3390 amino acids (aa) flanked by 5′ UTR (55 nts) and 3′ UTR (399 nts). The nucleotide acid composition of the YNPE3 strain was 32.11% A, 26.03% G, 21.28% T and 20.58% C. The percentage of purine (58.14%) was higher than that of pyrimidine (41.86%).

Electrophoresis of the whole genome sequence amplification from YNPE3 strain on a 1% agarose gel. The symbol M indicates DL 2000 DNA marker; and then from left to right the primer sets are: F1:3F1/3R1, F2: 3F2/3R2, F3: 3F3/3R3, F4: 3F4/3R4, F5: 3F5/3R5, F6: 3F6/3R6, F7: 3F7/3R7, F8: 3F8/3R8, F9: 3F9/3R9
Further similarity analysis by BLAST showed that the YNPE3 strain had the highest identity with the SG(EHI)D3/15095Y15 strain (KY921907) isolated from Singapore in 2015, it possessed 99.4% nucleotide identity and 99.6% amino acid identity. Followed by three strains from Indian (JQ922556, KU216209, KU216208), they showed to share 98.6–98.8% nucleotide identity with YNPE3 strain. Compared with the prototype strain H87, there was 94.0% nucleotide identity and 97.8% amino acid identity. The strains from Laos (KY849769, KY849770, KY849771, KY849772, KY849773, KY849774 and KY849775) shared 93.1% nucleotide identity and 97.8–97.9% amino acid identity with YNPE3 strain. Yet, the strains from China YN01 (KF824902) and YN02 (KF824903) isolated from Yunnan in 2013, shared 92.9% nucleotide identity and 97.7–97.8% amino acid identity.
Moreover, two fragments of about 40 nts, including 1-AGTTGTTAGTCTACGTGGACCGACAAGAACAGTTTCGAC-39 for 5′ UTR and 389-ACGCCAGAAAATGGAATGGTG CTGTTGAATCAACAGGTTCT-429 for 3′ UTR were deleted from the YNPE3 strain, and 11 nts 9-ACGCAGGAAGT-19 insertion presented in the hyper-variable region of the 3′ UTR of the YNPE3 strain when compared with strain H87. In view of the important role of the 3′ and 5′ UTR of Flaviviruses in viral replication and pathogenesis [35, 36], we analyzed the RNA secondary structures of the 3′ and 5′ UTR of the YNPE3 strain by MFOLD software that was used to predict RNA secondary structures. The results showed that two stem-loops were observed in the 5′ UTR of the YNPE3 and strain H87 (Fig. 3a). However, due to the deletion of 39 nts in the 5′ UTR of the YNPE3 strain, the large stem loop showed a truncated secondary structure. In addition, the secondary structure in variable region (VR) of the 3′ UTR of the YNPE3 strain was significantly different from that of the prototype H87 strain, the strain YNPE3 presented a unique secondary structure (Fig. 3b). As show in Fig. 3c, the overall secondary structure of 3′ UTR of the YNPE3 strain was consistent with H87; only at the terminal part it presented a distinctive structure due to the nucleotides deletions and insertions.

Prediction of secondary structure of 5′ UTR and 3′ UTR of strain YNPE3 and compared with the standard strain H87. a Prediction secondary structure of the whole 5′ UTR from the YNPE3 strain (nucleotides 1 to 55) and the prototype strain H87 (nucleotides 1 to 94). b Prediction secondary structure of the 3′ UTR variable region from the YNPE3 strain (nucleotides 1 to115) and strain H87 (nucleotides 1 to 104). c prediction secondary structure of the whole 3′ UTR from the prototype strain H87 and the YNPE3 strain
Amino acid substitution analysis of YNPE3
To determine the variations of the amino acids of the YNPE3 strain, the complete amino acid sequences of eight closely related reference strains were retrieved from GenBank database (http://www.ncbi.nlm.nih.gov). They were subjected to multiple sequence alignment analysis. As shown in Table 1, compared with strain H87, a total of 75 amino acid substitutions were found in the whole polyprotein of the YNPE3 strain with a substitution rate of 2.2%. These substitutions occurred mainly in the E, NS1, NS3 and NS5 proteins of YNPE3. However, the PrM/M protein was relatively conserved, and there were no amino acid substitutions found. Also, less amino acids substitutions were found in the capsid, NS2B and NS4A proteins of YNPE3. Compared with strain H87, many of the amino acid substitutions in the YNPE3 strain were conservative, and they mainly consisted of exchange of amino acids with similar physicochemical properties; Whereas, few amino acids mutations were involved in changes in polarity, such as E169: A(Ala) → T(Thr), E301: L(Leu) → T(Thr), NS193: I(Ile) → T(Thr) and NS5188: T(Thr) → A(Ala). Moreover, the strain YNPE3 and Balotra87-s shared the same unique amino acid replacements in some sites.
Phylogenetic analysis
To determine the evolutionary history of the YNPE3 strain, two phylogenetic trees were drawn based on the E gene and the CDS of DENV-3. The phylogenetic tree based on the E gene revealed that DENV-3 strains had five distinct genotypic groups (Fig. 4). The YNPE3 strain was clustered into genotype III and was closely related to the contemporaneous DENV-3 strains from Laos, Vietnam, Thailand and India. Interestingly, seven Yunnan isolates (KR347359, KX262915, KJ438298, KR347397, KR347420, KX262916, KX262914), and nine strains from Laos (KF816148, KF816160, KF816162, LC147060, KF816158, KF816159, KF816161, LC147061, LC147059) that were identified in 2013, belonged to genotype II. Moreover, the phylogenetic tree based on CDS revealed that the YNPE3 strain was grouped into genotype III along with other DENV-3 isolates from different regions (Fig. 5). Phylogenetically, the strains Balotra87-s (KU216209), IND/58760 (JQ992556), DENV-3/IND/59826 (JQ992556) from India and two strains from China (JF504679, GU363549) were closely related to the YNPE3 strain. Interestingly, the YN01 and YN02 strains isolated in Yunnan in 2013 were classified into genotype II and were located in the same clades with seven strains isolated in Laos in 2010. Overall, the two trees suggested that the current strain belonged to the genotype III of DENV-3.

Phylogenetic tree based on envelope gene of DENV-3 strains obtained by Maximum-Likelihood method in MEGA7.0. Black dot represents DENV-3 sequence isolated in current study, the Laos and other Chinese DENV-3 isolates are marked with black squares and triangles, respectively. A total of 76 reference strains of the five genotypes available in NCBI GenBank were used for comparison. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The scale bar expresses the genetic distance. DENV strains are named as follows: GenBank accession number/strain/country/year

Phylogenetic tree based on the complete coding sequences of DENV-3 strains obtained by Maximum-Likelihood method in MEGA7.0. Black dot represents DENV-3 sequence isolated in current study. The Laos and other Chinese DENV-3 isolates are marked with black squares and triangles, respectively. A total of 75 reference strains of the four genotypes available in NCBI GenBank are retrieved. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. The scale bar expresses the genetic distance. DENV strains are named as follows: GenBank accession number/strain/country/year
Discussion
Over recent years, dengue has become a serious public health concern in China. The imported cases of dengue from other countries, and especially Southeast Asia countries, have caused a regional epidemic in Yunnan, China [26, 27, 37]. Therefore, disease surveillance and prevention employing genome sequencing and molecular and evolutionary studies of DENV isolates are of great importance. As stated in the introduction, dengue outbreak in Yunnan was related to the outbreak in Laos. In current research, the phylogenetic analysis based on E gene suggested that the YNPE3 strain was closely related to the contemporaneous strains isolated in Laos. Previous study has revealed a strong relationship between the Laos 2013 isolates genotype III and strains from Bhutan (2007) and India (2005) [22]. Especially, the phylogenetic tree based on E gene indicated that strains from Thailand, Laos, Vietnam and Pakistan had a strong relationship with the India isolates of genotype III (from 2005 to 2013). The phylogenetic tree based on CDS also supported that YNPE3 strain had a close relationship with India isolates in 2005 (strains IND/58760 and DENV-3/IND/59826) and 2013 (strain Balotra87-s). These strains shared 98.2–98.9% nucleotide identity with YNPE3 strain. Nevertheless, the precise evolutionary relationship between strain YNPE3 and strains from Laos and India could not be confirmed due to lack of complete coding sequences information on genotype III from Laos during 2005–2013. Yet, previous studies have reported the emergence of genotype III from 2009 to 2011 in India and China [38, 39]. This genotype has re-emerged in Southeast Asian countries and it has been in circulation for a very long time. Moreover, the two phylogenetic trees indicated genotype II isolated in Yunnan was closely related with Laos strains isolated in 2010 and 2013, which suggested that this genotype most likely had common ancestor with the genotype II strains circulating in Laos.
Previous studies have indicated an important role of the secondary structure of the 5′ and 3′ UTR of the Flaviviruses in viral replication [40, 41], The deletion and insertion of bases in the 5′ and 3′ UTR probably leads to changes in RNA secondary structures. For most DENV isolates, the 3′ UTR of the genome contains three functional domains [40, 42]. The nucleotide insertions and mutations usually occur in domain I, which is close to the NS5 protein and is regarded as the variable region of the 3′ UTR of DENV-3 [35, 42], The potential effect of the insertion and mutation has shown to be related with viral replication efficiency [36, 43, 44]. Our results revealed that the insertion of 11 nts occurred in domain I of the 3′ UTR of the YNPE3 strain, while the secondary structure of VR was different from that of strain H87, which was consistent with the results of a previous study [45]. However, no significant change occurred in the whole secondary structure of 3′ UTR. The 5′ UTR has two domains, including the short stem loop domain II and large stem loop domain I, which are separated by a functional spacer oligo U sequence. Domain I is the promoter of the viral RNA-dependent RNA polymerase (RdRp). It comprises several conserved structures: three helix regions (Stem 1, Stem 2 and Stem 3), a top loop and a side stem loop [35, 46]. Most of the variations in sequence and structure occur in the side loop and Stem 3 helix region. The replication of the virus is affected if the side loop is deleted [35, 46]. Interestingly, the side loop and Stem 3 helix region of the YNPE3 strain were deleted. We further compared the differences in secondary structure of 5′ UTR and 3′ UTR between Yunnan strains YN01, YN02 and our strain YNPE3. The YN01 and YN02 revealed a same structure with H87 in 5′ UTR but a distinctive structure in 3′ UTR (data not show). Due to lack of a complete genome sequence of DENV-3 from Laos, no comparison could be made between YNPE3 and strains from Laos. Hence, further investigation is needed to elucidate the biological implication of these deletions and insertions.
As major viral antigen, the E protein can be recognized by host cells, thus inducing neutralizing antibodies and blocking membrane fusion and virion assembly [47,48,49]. Moreover, the virulence of dengue virus mainly depends on the E region. The mutations of amino acids in conserved regions of the E protein affect the viral attachment in host cells [47]. The E383: K(Lys) → N(Asn) and E391R(Arg) → K(Lys) might be related to the severity of the disease of DENV-3 [50]. The mutations that occur in the E124: S(Ser) → P(Pro), E169: A(Ala) → T(Thr) and E383: K(Lys) → N(Asn) are usually characteristics of genotype III [51]. The E169: A(Ala) → T(Thr) and E301: L(Leu) → T(Thr) are two important non-conservative replacements that involve the nonpolar hydrophobic amino acid to polar amino acid. The E-169, as a positively selected site [52], has been shown to be located in a murine B and T cell epitope [53]. These mutations might be related to the transmission capacity of these isolates [54]. Similar substitutions referring to shifts of polarity of amino acids were observed in other proteins, particularly in non-structural proteins NS1, NS2A, NS3 and NS5. Importantly, further studies are needed to confirm the actual functions of these substitutions. Like genotype III, genotype II strains from Laos and China also have exclusive amino acid substitution characteristics. We concluded that the same genotype, or the highly related strains shared some common characteristics in amino acid substitutions.
Conclusions
In conclusion, a full-length genome of an imported DENV-3 strain YNPE3 from Laos was reported for the first time. Deletions of nucleotides in the 3′ and 5′ UTR were observed. Studying the amino acid substitutions can lead to a better understanding of viral pathogenesis, which in turn might promote the development of new DENV vaccine. The genotype III and genotype II of DENV-3 from Yunnan were strongly related to those DENV-3 strains from Southeast Asia countries. Thus, the dengue surveillance and warning systems in Yunnan province of China need to be strengthened by timely monitoring of the two DENV-3 genotypes, that are prevalent in Southeast Asia.
Abbreviations
- Aa:
-
Amino acids
- BLAST:
-
Basic local alignment search tool
- C:
-
Capsid protein
- CDS:
-
Complete coding sequence
- CPE:
-
Cytopathic effect
- DENV:
-
Dengue virus
- DENV-3:
-
Dengue virus serotype 3
- DF:
-
Dengue fever
- DHF:
-
Dengue hemorrhagic fever
- DSS:
-
Dengue shock syndrome
- E:
-
Envelope glycoprotein
- GI:
-
Genotype I
- GII:
-
Genotype II
- GIII:
-
Genotype III
- GV:
-
Genotype V
- M:
-
Membrane glycoprotein
- NS:
-
Nonstructural proteins
- nts:
-
nucleotides
- ORF:
-
Open reading frame
- PrM:
-
Membrane glycoprotein precursor
- RdRp:
-
RNA-dependent RNA polymerase
- RPMI:
-
Roswell Park Memorial Institute
- RT-PCR:
-
Reverse transcriptase polymerase chain reaction
- UTR:
-
Untranslated region
- VR:
-
Variable region
References
Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, Geroge DB, Jaenisch T, Wint GRW, Simmons CP, Scott TW, Farrar JJ, Hay SI. The global distribution and burden of dengue. Nature. 2013;496(7446):504–7.
World Health Organization. Dengue and severe dengue. 2015. http://www.who.int/news-room/fact-sheets/detail/dengue-and-severe-dengue. Accessed 2 Feb 2018.
Bäck AT, Lundkvist A. Dengue viruses – an overview. Infect Ecol Epidemiol. 2013;3(3):19839.
Gubler DJ. Epidemic dengue/dengue hemorrhagic fever as a public health, social and economic problem in the 21st century. Trends Microbiol. 2002;10(2):100–3.
Rigau-Perez JG, Clark GG, Gubler DJ, Reiter P, Sanders EJ, Vorndam AV. Dengue and dengue Haemorrhagic fever. Lancet. 1998;352(9132):971–7.
Holmes EC, Twiddy SS. The origin, emergence and evolutionary genetics of dengue virus. Infect Genet Evol. 2003;3(1):19–28.
Kuno G, Chang GJJ, Tsuchiya KR, Karabatsos N, Cropp CB. Phylogeny of the genus Flavivirus. J Virol. 1998;72(1):73–83.
Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. 1998;11(3):480–96.
Ranjit S, Kissoon N. Dengue hemorrhagic fever and shock syndromes. Pediatr Crit Care Med. 2011;12(1):90–100.
Chambers TJ, Chang SH, Galler R, Rice CM. Flavivirus genome organization, expression, and replication. Annu Rev Microbiol. 1990;44(1):649–88.
Halstead SB. Dengue. Lancet. 2007;370(9599):1644–52.
Ricohesse R, Chambers TJ, Monath TP. Microevolution and virulence of dengue viruses. Adv Virus Res. 2003;59(1):315–41.
Gubler DJ. Dengue, urbanization and globalization: the unholy trinity of the 21(st) century. Trop Med Health. 2011;39(4 Suppl):3–11.
Kraemer MU, Sinka ME, Duda KA, Mylne AQ, Shearer FM, Barker CM, Moore CG, Carvalho RG, Coelho GE, Van Bortel W, Hendrickx G, Schaffner F, Elyazar IR, Teng HJ, Brady OJ, Messina JP, Pigott DM, Scott TW, Smith DL, Wint GR, Golding N, Hay SI. The global distribution of the arbovirus vectors Aedes aegypti and Ae. albopictus. Elife. 2015;4:e08347.
Lai S, Huang Z, Hang Z, Anders KL, Perkins TA, Yin W, Yu L, Di M, Chen Q, Zhang Z. The changing epidemiology of dengue in China, 1990–2014: a descriptive analysis of 25 years of nationwide surveillance data. BMC Med. 2015;13(1):100.
Wang C, Yang W, Fan J, Wang F, Jiang B, Liu Q. Spatial and temporal patterns of dengue in Guangdong Province of China. Asia Pac J Public Health. 2015;27(2):844–53.
Sim S, Hibberd ML. Genomic approaches for understanding dengue: insights from the virus, vector, and host. Genome Biol. 2016;17(1):1–15.
Wildersmith A, Schwartz E. Dengue in travelers. New Engl J Med. 2005;353(9):924–32.
Qiu FX, Gubler DJ, Liu JC, Chen QQ. Dengue in China: a clinical review. Bull World Health Organ. 1993;71(3–4):349–59.
Wu W, Bai Z, Zhou H, Tu Z, Fang M, Tang B, Liu J, Liu L, Liu J, Chen W. Molecular epidemiology of dengue viruses in southern China from 1978 to 2006. Virol J. 2011;8(1):1–9.
Wu JY, Lun ZR, James AA, Chen XG. Dengue fever in mainland China. Am J Trop Med Hyg. 2010;83(3):664–71.
Lao M, Caro V, Thiberge JM, Bounmany P, Vongpayloth K, Buchy P, Duong V, Vanhlasy C, Hospied JM, Thongsna M. Co-circulation of dengue virus type 3 genotypes in Vientiane capital, Lao PDR. PLoS One. 2014;9(12):e115569.
Fan W, Yu S, Cosgriff TM. The re-emergence of dengue in China. Rev Infect Dis. 1989;11(Suppl 4):S847–53.
Rabaa MA, Hang VTT, Wills B, Farrar J, Simmons CP, Holmes EC. Phylogeography of recently emerged DENV-2 in southern Viet Nam. PLoS Neglect Trop Dis. 2010;4(7):e766.
Thu HM, Lowry K, Myint TT, Shwe TN, Han AM, Khin KK, Thant KZ, Thein S, Aaskov J. Myanmar dengue outbreak associated with displacement of serotypes 2, 3, and 4 by dengue 1. Emerg Infect Dis. 2004;10(4):593–7.
Hu TS, Zhang HL, Feng Y, Fan JH, Tang T, Liu YH, Zhang L, Yin XX, Chen G, Li HC, Zu J, Li HB, Li YY, Yu J, Zhang FQ, Fan QS. Epidemiological and molecular characteristics of emergent dengue virus in Yunnan Province near the China-Myanmar-Laos border, 2013–2015. BMC Infect Dis. 2017;17(1):331.
Guo X, Yang H, Wu C, Jiang J, Fan J, Li H, Zhu J, Yang Z, Li Y, Zhou H, Zhang J. Molecular characterization and viral origin of the first dengue outbreak in Xishuangbanna, Yunnan Province, China, 2013. Am J Trop Med Hyg. 2015;93(2):390–3.
Ministry of Health, People’s Republic of China. WS 216-2008 diagnostic criteria and principle of management of dengue fever. Beijing: China Standard Publishing House; 2008.
Chen N, Yong-Xin YU, Hong-Shan XU, Liu XY, Jia LL, Dong GM, Yu-Hua LI. Complete genomic sequence of dengue type 3 virus strains isolated from Guangxi, China. J Pathogen Biol. 2014;9(5):394–9.
Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31(13):3406–15.
Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17(6):368–76.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870–4.
Felsenstein J. Confidence limits on phylogenies: an approach using the bootstrap. Evolution. 1985;39(4):783–91.
Tamura K, Nei M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol Biol Evol. 1993;10(3):512–26.
Gebhard LG, Filomatori CV, Gamarnik AV. Functional RNA elements in the dengue virus genome. Viruses. 2011;3(9):1739–56.
Clarke BD, Roby JA, Slonchak A, Khromykh AA. Functional non-coding RNAs derived from the flavivirus 3′ untranslated region. Virus Res. 2015;206:53–61.
Wang B, Yang H, Feng Y, Zhou H, Dai J, Hu Y, Zhang L, Wang Y, Baloch Z, Xia X. The distinct distribution and phylogenetic characteristics of dengue virus serotypes/genotypes during the 2013 outbreak in Yunnan, China: phylogenetic characteristics of 2013 dengue outbreak in Yunnan, China. Infect Genet Evol. 2015;37(2016):1–7.
Tao J, Yu XD, Hong WX, Zhou WZ, Man Y, Deng YQ, Zhu SY, Qin ED, Jian W, Qin CF, Zhang FC. Co-circulation of two genotypes of dengue virus serotype 3 in Guangzhou, China, 2009. Virol J. 2012;9(1):1–8.
Gupta E, Mohan S, Bajpai M, Choudhary A, Singh G. Circulation of dengue virus-1 (DENV-1) serotype in Delhi, during 2010–11 after dengue virus-3 (DENV-3) predominance: a single Centre hospital-based study. J Vector Borne Dis. 2012;49(2):82–5.
Alvarez DE, Ezcurra ALDL, Fucito S, Gamarnik AV. Role of RNA structures present at the 3′UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology. 2005;339(2):200–12.
Markoff L. 5′- and 3′-noncoding regions in Flavivirus RNA. Adv Virus Res. 2003;59:177–228.
Iglesias NG, Gamarnik AV. Dynamic RNA structures in the dengue virus genome. RNA Biol. 2011;8(2):249–57.
Clyde K, Kyle JL, Harris E. Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol. 2006;80(23):11418–31.
Men RH, Bray M, Clark D, Chanock RM, Lai CJ. Dengue type 4 virus mutants containing deletions in the 3′ noncoding region of the RNA genome: analysis of growth restriction in cell culture and altered viremia pattern and immunogenicity in rhesus. J Virol. 1996;70(6):3930–7.
Castro MGD, Nogueira FBD, Nogueira RMR, Lourençodeoliveira R, Santos FBD. Genetic variation in the 3′ untranslated region of dengue virus serotype 3 strains isolated from mosquitoes and humans in Brazil. Virol J. 2013;10(1):1–11.
Lodeiro MF, Filomatori CV, Gamarnik AV. Structural and functional studies of the promoter element for dengue virus RNA replication. J Virol. 2009;83(2):993–1008.
Diamond MS, Pierson TC. Molecular insight into dengue virus pathogenesis and its implications for disease control. Cell. 2015;162(3):488–92.
Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003;10(11):907–12.
Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108(5):717–25.
Aquino VH, Anatriello E, Gonçalves PF, Silva EVDA, Vasconcelos PF, Vieira DS, Batista WC, Bobadilla ML, Vazquez C, Moran M. Molecular epidemiology of dengue type 3 virus in Brazil and Paraguay, 2002–2004. Am J Trop Med Hyg. 2006;75(4):710–5.
Patil JA, Cherian S, Walimbe AM, Bhagat A, Vallentyne J, Kakade M, Shah PS, Cecilia D. Influence of evolutionary events on the Indian subcontinent on the phylogeography of dengue type 3 and 4 viruses. Infect Genet Evol. 2012;12(8):1759–69.
Twiddy SS, Woelk CH, Holmes EC. Phylogenetic evidence for adaptive evolution of dengue viruses in nature. J Gen Virol. 2002;83(Pt 7):1679–89.
Leclerc C, Dériaud E, Megret F, Briand JP, Van Regenmortel MH, Deubel V. Identification of helper T cell epitopes of dengue virus E-protein. Mol Immunol. 1993;30(7):613–25.
Sharma S, Dash PK, Agarwal S, Shukla J, Parida MM, Rao PV. Comparative complete genome analysis of dengue virus type 3 circulating in India between 2003 and 2008. J Gen Virol. 2011;92(Pt 7):1595–600.
Acknowledgements
We would like to thank Dr. Nigel W. Fraser of Department of Microbiology, Perelman School of Medicine, University of Pennsylvania, Philadelphia, USA, for proof-reading and reviewing our manuscript.
Funding
This work was supported by the CAMS Innovation Fund for Medical Sciences (2017-I2M-3-022), the Peking Union Medical College Youth Fund (3332016113), the National Key Research and Development Program of the Ministry of Science and Technology of China (2016YFC1202300), the State Project for Essential Drug Researchand Development (SQ2018ZX091510), the National Natural Science Foundation of China (31500724), and the Yunnan Applied Basic Research Projects (2017FB115).
Availability of data and materials
All data generated or analyzed during this study are induced in this published article.
Author information
Authors and Affiliations
Contributions
YH, HZ and JS designed the study, XG and HZ provided blood samples and collected clinical data. LM, ZZ, XG and JS performed the experiments and drafted the paper. JS, NH, JS and MW help to interpret the results, JS, HZ and YH contributed to the writing. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
All methods in this study were approved the Institutional Ethics Committee (Institute of Medical Biology, Chinese Academy of Medical Sciences, and Peking Union Medical College) and were in accordance with the Declaration of Helsinki for Human Research of 1974 (last modified in 2000).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Typical primers of dengue virus. (DOC 32 kb)
Additional file 2:
Table S2. Primers used for the complete genome amplification. (DOC 38 kb)
Additional file 3:
Table S3. Sequence information used in E gene phylogenetic tree construction. (DOC 88 kb)
Additional file 4:
Table S4. Sequence information used in the complete coding sequence phylogenetic tree construction. (DOC 88 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Mo, L., Shi, J., Guo, X. et al. Molecular characterization and phylogenetic analysis of a dengue virus serotype 3 isolated from a Chinese traveler returned from Laos. Virol J 15, 113 (2018). https://doi.org/10.1186/s12985-018-1016-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12985-018-1016-5