
Summary
Background
Dengue, a mosquito-borne viral infection caused by one of the four dengue virus (DENV) serotypes (DENV-1 to 4), replicate alternately on the mosquito vector and human host and are responsible for infections throughout tropical and subtropical regions of the world. In Brazil, the disease has become a major public health problem and the introduction of DENV-3 in 2000 in Rio de Janeiro (RJ) was associated with severe dengue epidemics. The potential emergence of strains associated with severe disease highlights the need for the surveillance of DENV in human host and vectors.
Methods
Aiming to contribute for DENV phylogenetic and vector-virus-human host studies, we sequenced the entire genome of one DENV-3 isolated from naturally infected Aedes aegypti from RJ in 2001 and characterized the 3’ UTR from strains isolated from mosquitoes and humans. Mosquitoes were pooled and submitted to virus isolation in Ae. albopictus C6/36 cells and the infecting serotype was identified by immunofluorescence using type-specific monoclonal antibody. Sequence analysis was performed using BioEdit software, the multiple alignments were performed using CLUSTAL W and the phylogenetic analysis by MEGA 5, using the Neighbor-joining method. Secondary structure prediction was performed by using the MFOLD program.
Results
Exclusive substitutions and a substitution leading to a stop codon on the NS5 gene were observed in the DENV-3 isolated from a naturally infected Ae. aegypti and fully sequenced. As an 8- nucleotides deletion was observed within the 11- nucleotides (nts) insertion on the variable region (VR) from the 3′UTR in this isolate, we further sequenced other DENV-3 from both mosquitoes and humans. The majority of DENV-3 from RJ analyzed were characterized by the 11-nts insertion in the VR of the 3′UTR, despite the observation of strains carrying the 8-nts deletion. The latter presented similar secondary structures, however not all strains presenting the 11-nts insertion were similar in the predicted secondary structure.
Conclusions
The phylogeny based on the analysis of the complete genome and 3′UTR characterized the DENV-3 isolated from both vector and human host as belonging to Genotype III (GIII), despite the differences observed on the 3’ UTR. Further studies are needed to address the role of those mutations in the transmission of the different viral populations and vector competence.
Similar content being viewed by others


Background
Dengue is a mosquito-borne viral infection caused by one of the four dengue virus serotypes (DENV-1 to 4), belonging to genus Flavivirus, family Flaviviridae. The viruses replicate alternately on the mosquito vector, mainly (Ae. aegypti) and human host and are responsible for infections throughout tropical and subtropical regions of the world[1, 2].
The rapid global spread of the four DENV serotypes in the last 50 years resulted in the dispersal of genotypes associated with increased disease severity[3]. In Brazil, dengue has been a major public health problem since DENV-1 introduction and spread in 1986[4], however the introduction of the genotype III of DENV-3, in December 2000, in Nova Iguaçu, State of Rio de Janeiro (RJ), caused one of the most severe epidemics reported in the country in 2002[5–7]. Despite the co-circulation of DENV-1, DENV-2 and DENV-3 in that area, DENV-3 was the only serotype detected in pools of Ae. aegypti during an entomological surveillance performed[8].
Sequencing of distinct DENV genomic regions has identified five genotypes for DENV-3: Genotypes I to III (GI to GIII) which are responsible for most DENV-3 human infections and have been associated with both dengue fever (DF) and dengue haemorrhagic fever (DHF) epidemics in Southeast Asia, Indian Subcontinent, South Pacific and East Africa and Americas, and Genotypes IV and V (GIV and GV) which were not associated with DHF epidemics and are only represented by few early sequences from Americas, South Pacific and Asia[9–13].
The DENV genome is composed by a positive single-stranded RNA of approximately 11 kb in length with an open reading frame encoding for the viral polyprotein, which is cleaved into three structural proteins (C, prM and E) and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4 and NS5) flanked by 5′ and 3′ untranslated regions (UTRs) of about 100 and 400 nucleotides, respectively[1]. The flaviviruses UTRs are predicted to form secondary stem-loop (SL) structures, which are highly conserved and play a role in viral replication[14–18].
According to predicted secondary structures, the DENV 3′UTR can be divided into three domains[18]. The domain I, which is located immediately after the NS5 stop codon, is considered the most variable region (VR) within the viral 3′UTR, as it shows large heterogeneity in both length and nucleotide sequences[19–21]. Mutations and deletions within these regions may alter infectivity and reduce efficiency of viral replication[22, 23] and differences between strains in these regions may correlate with DENV virulence and pathogenicity[24–27]. Furthermore, deletions and nucleotide variations were also described in the VR within the same serotype[28–30]. Domain II is of moderate conservation, comprising several hairpins motifs and where conserved sequence (CS2) and repeated CS2 (RCS2) are present. Domain III is the most conserved region of the 3′UTR with CS1 followed by a terminal stem-loop (3′SL)[18].
Here, aiming to contribute for the studies on human host-virus-vector interactions, we fully sequenced the genome of one DENV-3 isolated from naturally infected field-caught mosquitoes in RJ and characterized the viral 3′UTR in comparison to other sequenced DENV-3 isolated from naturally infected mosquitoes and human hosts.
Material and Methods
Ethical Statement
All human DENV-3 strains belong to a previously gathered collection from the Laboratory of Flavivirus, IOC/FIOCRUZ, RJ, Brazil obtained from acute phase human serum through the passive surveillance system from an ongoing Project approved by resolution number CSN196/96 from the Oswaldo Cruz Foundation Ethical Committee in Research (CEP 274/05), Ministry of Health, Brazil.
Ae. aegypti examined in this study were collected by the staff of the Dengue Control State Program for the determination of house infestation index, virological and entomological surveillance. No special permission or written consent is required for house entrance for mosquito collection and larval site treatment.
Viral strains
The DENV-3 strains isolated from Ae. aegypti adult mosquitoes (n= 4) and human hosts (n= 10) naturally infected in RJ were collected from epidemics occurred from 2001 to 2008. The first Brazilian DENV-3 strain (BR74886/02) isolated from a human fatal case fully sequenced[31] was used for comparison purposes and detailed information on the strains is provided on Table1.
DENV-3 human cases
From 2001 to 2008, the Laboratory of Flavivirus, as a Regional Reference Laboratory for the Brazilian Ministry of Health, received a total of 16,185 dengue suspected cases for routine diagnosis. Virus isolation was attempted in 9,405 cases and DENV-3 was the infecting serotype in 52.8% of the positive isolates. The samples analyzed in this study were chosen randomly and representative of each year, during and after the 2002 epidemic in RJ.
DENV-3 entomological surveillances
The three DENV-3 strains isolated (BR73354/01, BR73356/01 and BR73636/01) in 2001 from naturally infected Ae. aegypti adult mosquitoes used in this study were collected during an entomological survey performed in 35 districts of Nova Iguaçu, RJ, from July 2000 to June 2001. The other DENV-3 strain (BR81200/06) was isolated from naturally infected Ae. aegypti adult mosquitoes collected during an entomological survey conducted on 7 districts with different infestation index, randomly chosen in the municipality of Rio de Janeiro, RJ, from March 2005 to February 2006. Briefly, adult mosquitoes were collected twice a week, alternately in the morning and in the afternoon with manual and battery backpack aspirators and with nets, both indoor and in the yards and gardens, close to the dwellings. Mosquitoes were identified using a key as previously described[32], pooled according to gender, date, district of collection and stored in liquid nitrogen at the same day of collection. A total of 503 Ae. aegypti mosquitoes (352 females and 151 males) collected in 2000–2001 and 874 Ae. aegypti females collected in 2005–2006 were pooled (74 pools of 9–17 mosquitoes/pool in 2000–2001 and 27 pools of 2–10 mosquitoes/pool, jn 2005–2006) and all pools were submitted for virus isolation. Only positive pools were submitted to RT-PCR. for DENV serotype confirmation.
Preparation of vectors
Mosquitoes’ pools were macerated in 1 ml of Leibovitz L-15 medium (Sigma) plus antibiotics (penicillin-streptomycin, 10,000 units - Invitrogen) and centrifuged (6,000 rpm at 4°C for 30 min). Supernatant was transferred to an Eppendorf tube containing 100 mL of streptomycin / fungizone and penicillin, kept in an ice bath for 1 hour and centrifuged (3,000 rpm at 4°C for 15 min). Supernatant was transferred to an Eppendorf tube containing 0.3ml of fetal calf serum (Invitrogen) and frozen (−70°C).
Virus isolation
Virus isolation was performed by inoculation into monolayers of C6/36 Aedes albopictus cells[33] in Leibovitz L-15 medium (Sigma) supplemented with 2% fetal calf serum (Invitrogen) and 0.2 mM of nonessential amino acids (Invitrogen). Cells were incubated at 28°C for 5 to 7 days and observed for cytopathic effects. Isolates were identified by indirect fluorescent antibody test (IFAT) using serotype-specific monoclonal antibodies[34] and infected supernatant was clarified by centrifugation and virus stocks stored in 1-mL aliquots at −70°C.
RNA extraction
Viral RNA was extracted using QIAamp Viral RNA Mini kit (Qiagen) following the manufacturer’s instructions and stored at -70°C for DENV typing and sequencing. For the viral 3′UTR sequencing, the RNA was extracted directly from serum and mosquitoes macerate, previously detected by RT-PCR. For the full genome sequencing of the DENV-3 strain BR73354/01, the RNA was extracted from the first passage in cell culture.
RT –PCR (Reverse transcriptase- polymerase chain reaction)
RT—PCR for detecting and typing DENV was performed as described previously[35].
Sequencing and phylogenetic analysis
PCR products were sequenced in both directions using the BigDye Dideoxy Terminator sequencing kit (Applied Biosystems). The mosquitoes’ DENV-3 full-length genome sequence and 3′UTR sequences obtained in this study were deposited in GenBank (http://www.ncbi.nlm.nih.gov) and are described on Table1. Sequence and similarity identity analysis was performed using BioEdit software (http://www.mbio.ncsu.edu/bioedit/bioedit.htmL). The multiple alignments were performed using CLUSTAL W (http://www.ebi.ac.uk/clustalw/) and the phylogenetic analysis by MEGA 5 software (http://www.megasoftware.net), using the Neighbor-joining method, according to the Tamura-Nei model, with a bootstrap of 1,000 replications for the analysis of the complete genome. For the 3′UTR analysis, the Maximum likelihood method, according to the Kimura-2 model was chosen as determined by the best-fit substitution model provided by MEGA 5. Strains representative from the five genotypes available in GenBank (http://www.ncbi.nlm.nih.gov) were used for the comparison, DENV-1 (GenBank accession number #AF513110), DENV-2 (#AF489932) and DENV-4 (# AF326573) strains were used as outgroup to root the tree.
Secondary structure analysis
The predicted secondary structures were generated by MFOLD web server (http://mfold.rna.albany.edu/?q=mfold/RNA-Folding-Form) with default folding parameters and folding predictions at 37oC on the VR from the 3’ UTR of DENV-3.
Results and discussion
The DENV-3 was re-introduced in Latin America in 1994, after an absence of 17 years, being initially isolated in Panama and spreading throughout Central America[36, 37] to Caribbean[38–40] and South America[6, 28, 41–43]. However, some phylogenetic studies point to its introduction through Mexico[44] a few years earlier[13]. This introduction caused by the genotype III of DENV-3, originally from the Southeast Asia and characterized by an increased virulence, coincided with the occurrence of a higher number of severe and DHF cases[37, 45–47].
In Brazil, the first DENV-3 was isolated in December of 2000 in Nova Iguaçu, RJ[5] when the Ae aegypti infestation level was 8.1% and, 58% of those mosquitoes were resistant to temephos at that time[8, 48]. Due to the role of the city of Nova Iguaçu in dengue epidemiology, after the DENV-1 introduction in 1986[4], field studies were conducted for detection of DENV in field-caught vectors[8, 49]. The potential emergence of strains associated with severe disease highlights the need for the surveillance of DENV in human host and vectors, as the detection of DENV in infected field-caught vectors is considered a useful tool for the early prediction of epidemics and detection of new serotypes/genotypes introductions[50, 51].
The entomological surveillance performed in the first semester of 2001 in Nova Iguaçu, RJ, resulted in the isolation of three DENV-3 strains from the districts of Santa Efigênia (BR73354/01), California (BR73356/01) and Morro Agudo (BR73636/01) isolated from three pools (9 mosquitoes/pool) of naturally infected Ae. aegypti females[8]. In January 2006, one DENV-3 strain (BR81200/06) was isolated from one Ae. aegypti pool composed of three females, collected indoors in the Vargem Pequena neighborhood, west region of RJ.
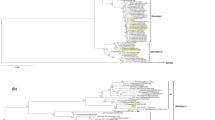
In order to access the differences among Brazilian DENV-3, we sequenced and deposited on Genbank (FJ177308) the entire genome sequence of one virus isolated from naturally infected Ae. aegypti (BR73354/01) and compared to the Brazilian strain 74886/02 (AY679147), isolated from the liver of a fatal case during the DENV-3 epidemic occurred in 2002[31]. The nucleotide similarity was 99.3% and the phylogeny based on the analysis of the complete coding region characterized the Brazilian strain as belonging to genotype III (Indian Subcontinent), Figure1.

Neighbor-joining phylogenetic analysis of the complete genome sequence from DENV-3 isolated from naturally infected mosquitoes in Brazil, 2001. Black circle represent DENV-3 sequence generated in this study. Strains representative from the four genotypes available in Genbank (http://www.ncbi.nlm.nih.gov) were used for the comparison, DENV-1, DENV-2 and DENV-4 strains were used as outgroup to root the tree. The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches. DENV strains used were named as follows: GenBank accession number/country/year
Amino acid substitutions were observed throughout the entire coding region, when the Brazilian DENV-3 strains were compared to the prototype PHIL/H87/1956 and strains representative of the other genotypes. Some substitutions were exclusive to the Brazilian DENV-3 strains analyzed (Table2) and some were shared among the Brazilian strains and the strain isolated from Ae. aegypti in Taiwan in 1998 (TAIWAN/TWmosq/1998), Table2. Exclusive substitutions to the DENV-3 Brazilian strains analyzed in this study were observed on the capsid, prM and envelope genes. Besides those others observed were also shared by other genotype III strains previously analyzed[11].
On the NS1 gene only one residue substitution was observed, and substitution on NS2A and NS2B were also reported. On the NS3 gene, a substitution on only NS3115 was exclusive to the Brazilian DENV-3. On NS4B, the same substitution was observed on positions NS4B21 and NS4B138. However, on the NS5 gene, besides the ten substitutions exclusive to the Brazilian DENV-3, substitutions exclusive to the Brazilian DENV-3 isolated from Ae. agypti (BR73354/2001) were observed almost consecutively, on NS597 and NS598. On NS5101, the substitution resulted in a stop codon and other substitutions throughout NS5 were also observed. Despite this, the original macerate was re-inoculated in C6/36 cells culture and the DENV −3 infection confirmed by the presence of cytopathic effect and a positive RT-PCR. The presence of genome-defective DENV-3 containing either stop codons or deletions in vivo has been reported previously[52].
Despite the use of the E gene for DENV phylogenetic and evolutionary studies[9, 12, 28, 53–62] due to its biological properties and selective pressure imposed by the host immune response, the role of sequences heterogeneity in other genomic regions which includes the non-structural genes and the genome UTRs cannot be excluded[53].
Previous studies have suggested that the sequence and secondary structures of the 5′ and 3′UTR of flaviviruses play an important role in viral replication and differences in these regions may influence viral virulence[24, 26, 27, 63]. Mutations and deletions within these regions may alter infectivity and reduce efficiency of viral replication[22, 23]. Furthermore, the domain I from the DENV 3′UTR is considered the most variable region (VR) from the 3′UTR[18] and can serve as a good marker for DENV evolution[19–21].
The analysis of the 3′UTR of the strain BR73354/01 genome showed an 8- nucleotides deletion within the 11-nucleotides insertion on the VR, previously observed for the Brazilian DENV-3 strain isolated from humans[31] and common to genotype III DENV-3 strains from the Latin America/Caribbean and Sri Lanka regions[27, 64]. Nucleotides substitutions exclusive to the BR73354/01 were observed on positions 10,383 and 10,391 from the 3′UTR. One substitution on the RCS2 and one on the CS2 were shared by all Brazilian DENV-3 when compared to the prototype.
We additionally analyzed and deposited on GenBank the 3′UTR sequences from other three DENV-3 strains isolated from naturally infected Ae. aegypti isolated in 2001 and 2006 in RJ (BR73356/01, BR73636/01 and BR81200/06) and from ten DENV-3 isolated from humans from 2001 to 2008. The strain BR73356/01 presented the same 8-nucleotides deletion observed for the strain BR73354/01. However, the other two strains also isolated from mosquitoes in RJ (BR73636/01 and BR81200/06) presented the 11-nucleotides insertion common to the human strains. The analysis of the 3′UTR from DENV-3 isolates from humans showed that nine out of ten strains also presented the 11-nucleotides previously described. However, one of the strains isolated in Rio de Janeiro in 2002 (BR74792/02) showed the same 8-nucleotides deletion observed on the mosquito strains (Figure2). Previous studies have shown deletions and nucleotide variations in the VR within a same serotype[28, 29]. Despite those observations, it was also shown a high conservation on the 3′UTR RCS2, CS1 and CS2 conserved regions (Figure2, gray-shadowed areas) among all the Brazilian strains analyzed. This was quite expected as domain II is of moderate conservation and domain III, the most conserved region of all[18].Therefore, we focused on the VR of the 3′UTR, aiming to better characterize those mutations by predicting the secondary structures of that region. Not all strains presenting the 11-nucleotides insertion were similar in structure (Figure3A and3B). In fact, despite the 11-nucleotides insertion, the strain BR72/2008 presented a unique secondary structure (Figure3B). The only difference from the latter is a nucleotide substitution (G →A) on the 11-nucleotides insertion region, when compared to all other Brazilian sequences analyzed (Figure2). The strain BR80996/2006, also showed a unique secondary structure due to the nucleotides substitution presented in the VR, despite the presence of the insertion shared with the other strains. The slight difference presented by the strain BR83904/2007, was due to a substitution (C→T) exclusive to this sequence. Furthermore, all three sequences with the 8-nucleotides deletion (BR73354mosq/2001, BR73356mosq/2001 and BR74792/2002) presented the same secondary structure (Figure3C).

Multiple nucleotide sequence alignment of the Brazilian DENV-3 3′UTR from additional strains isolated from mosquitoes Ae. aegypti ( n= 4) and humans ( n= 10) from 2001 to 2008. Dots (.) indicate identity among strains based on the Brazilian strain BR74886/02, characterized by the 11 nucleotides insertion on the variable region (VR). Dashes (−) indicate gaps in the alignment. The 8 nucleotides deletion characteristic to strains isolated from mosquitoes BR73354mosq/2001, BR73356mosq/2001 and from a strain isolated in human (BR74792/2002) are marked by black squares. Eleven nucleotide insertion and conserved sequence regions (RCS2, CS2 and CS1) are gray-shadowed

On (A) the predicted secondary structure of the variable region (VR) from Brazilian DENV-3 strains isolated from humans and mosquitoes Ae. Aegypti (nucleotides 1 to 108) presenting the 11-nucleotides insertion and compared to the prototype PHIL/87/1956 (nucleotides 1–104). On (B) the predicted secondary structures from the Brazilian strains (nucleotides 1 to108) presenting the 11-nucleotides insertion, but with nucleotides substitutions differing from the strains on (A) and on (C) the structures from the sequences (nucleotides 1 to 100) with the 8-nucleotides deletion
Phylogenetic studies based on the 5′and 3′UTR have shown to be very useful for molecular epidemiological studies[19, 21, 27, 28, 65]. The Maximum-Likelihood phylogenetic tree of Brazilian DENV-3 strains isolates from naturally infected Ae. aegypti mosquitoes and humans based on the 3′UTR sequence analysis places those strains as belonging to genotype III, corroborating the findings of the full-length genome analysis (data not shown).
Conclusions
Here, we analyzed the coding region and the 3′UTR of DENV-3 from both human host and mosquitoes and described insertions, deletions and a substitution leading to stop codon formation. The majority of DENV-3 in this study was characterized by the 11-nucleotide insertion in the 3′UTR, despite the observation of strains carrying the 8-nucleotide deletion. In spite the presence of distinct viral variants, it is suggested that the major variant is transmitted. However, how those distinct viral populations are maintained or transmitted is not fully understood, therefore the availability of viruses isolated from both hosts are crucial for the better comprehension of the vector-virus-human host interactions and for quasispecies investigations. Furthermore, the analysis of those distinct viral populations in experimentally infected mosquitoes may help to elucidate those observations.
Financial support
CNPq, CAPES, FIOCRUZ, FIOCRUZ/PAPES V and FAPERJ. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
Lindenbach B, Rice C, PH KDH: Flaviviridae: The viruses and their replication. In Fields Virology. Philadelphia: Lippincott Williams and Wilkins; 2001:991-1041.
Whitehead S, Blaney J, Durbin A, Murphy B: Prospects for a dengue virus vaccine. Nat Rev Microbiol 2007, 5: 518-528. 10.1038/nrmicro1690
Kyle J, Harris E: Global spread and persistence of dengue. Annu Rev Microbiol 2008, 62: 71-92. 10.1146/annurev.micro.62.081307.163005
Schatzmayr HG, Nogueira RM, da Rosa AP T: An outbreak of dengue virus at Rio de Janeiro--1986. Mem Inst Oswaldo Cruz 1986, 81: 245-246. 10.1590/S0074-02761986000200019
Nogueira RM, Miagostovich MP, de Filippis AM, Pereira MA, Schatzmayr HG: Dengue virus type 3 in Rio de Janeiro. Brazil. Mem Inst Oswaldo Cruz 2001, 96: 925-926. 10.1590/S0074-02762001000700007
Nogueira RM, Schatzmayr HG, de Filippis AM, dos Santos FB, da Cunha RV, Coelho JO, de Souza LJ, Guimarães FR, de Araújo ES, De Simone TS, et al.: Dengue virus type 3, Brazil, 2002. Emerg Infect Dis 2005, 11: 1376-1381. 10.3201/eid1109.041043
Miagostovich MP, dos Santos FB, de Simone TS, Costa EV, Filippis AM, Schatzmayr HG, Nogueira RM: Genetic characterization of dengue virus type 3 isolates in the State of Rio de Janeiro, 2001. Braz J Med Biol Res 2002, 35: 869-872. 10.1590/S0100-879X2002000800002
Lourenco-de-Oliveira R, Honorio NA, Castro MG, Schatzmayr HG, Miagostovich MP, Alves JC, Silva WC, Leite PJ, Nogueira RM: Dengue virus type 3 isolation from Aedes aegypti in the municipality of Nova Iguaçu, State of Rio de Janeiro. Mem Inst Oswaldo Cruz 2002, 97: 799-800. 10.1590/S0074-02762002000600009
Lanciotti RS, Lewis JG, Gubler DJ, Trent DW: Molecular evolution and epidemiology of dengue-3 viruses. J Gen Virol 1994,75(Pt 1):65-75.
Wittke V, Robb TE, Thu HM, Nisalak A, Nimmannitya S, Kalayanrooj S, Vaughn DW, Endy TP, Holmes EC, Aaskov JG: Extinction and rapid emergence of strains of dengue 3 virus during an interepidemic period. Virology 2002, 301: 148-156. 10.1006/viro.2002.1549
King C, Chao D, Chien L, Chang G, Lin T, Wu Y, Huang J: Comparative analysis of full genomic sequences among different genotypes of dengue virus type 3. Virol J 2008, 5: 63. 10.1186/1743-422X-5-63
Araújo JM, Bello G, Schatzmayr HG, Santos FB, Nogueira RM: Dengue virus type 3 in Brazil: a phylogenetic perspective. Mem Inst Oswaldo Cruz 2009, 104: 526-529. 10.1590/S0074-02762009000300021
Araújo J, Nogueira R, Schatzmayr H, Zanotto P, Bello G: Phylogeography and evolutionary history of dengue virus type 3. Infect Genet Evol 2009, 9: 716-725. 10.1016/j.meegid.2008.10.005
Brinton MA, Fernandez AV, Dispoto JH: The 3'-nucleotides of flavivirus genomic RNA form a conserved secondary structure. Virology 1986, 153: 113-121. 10.1016/0042-6822(86)90012-7
Proutski V, Gould EA, Holmes EC: Secondary structure of the 3' untranslated region of flaviviruses: similarities and differences. Nucleic Acids Res 1997, 25: 1194-1202. 10.1093/nar/25.6.1194
Markoff L: 5'- and 3'-noncoding regions in flavivirus RNA. Adv Virus Res 2003, 59: 177-228.
Alvarez D, De Lella Ezcurra A, Fucito S, Gamarnik A: Role of RNA structures present at the 3'UTR of dengue virus on translation, RNA synthesis, and viral replication. Virology 2005, 339: 200-212. 10.1016/j.virol.2005.06.009
Iglesias NG, Gamarnik AV: Dynamic RNA structures in the dengue virus genome. RNA Biol 2011, 8: 249-257. 10.4161/rna.8.2.14992
Shurtleff AC, Beasley DW, Chen JJ, Ni H, Suderman MT, Wang H, Xu R, Wang E, Weaver SC, Watts DM, et al.: Genetic variation in the 3' non-coding region of dengue viruses. Virology 2001, 281: 75-87. 10.1006/viro.2000.0748
Klungthong C, Putnak R, Mammen M, Li T, Zhang C: Molecular genotyping of dengue viruses by phylogenetic analysis of the sequences of individual genes. J Virol Methods 2008, 154: 175-181. 10.1016/j.jviromet.2008.07.021
Pankhong P, Weiner D, Ramanathan M, Nisalak A, Kalayanarooj S, Nimmannitya S, Attatippaholkun W: Molecular Genetic Relationship of the 3' Untranslated Region Among Thai Dengue-3 Virus, Bangkok Isolates, During 1973–2000. DNA Cell Biol 2009,28(10):481-491. 10.1089/dna.2008.0835
Men R, Bray M, Clark D, Chanock RM, Lai CJ: Dengue type 4 virus mutants containing deletions in the 3' noncoding region of the RNA genome: analysis of growth restriction in cell culture and altered viremia pattern and immunogenicity in rhesus monkeys. J Virol 1996, 70: 3930-3937.
Mandl CW, Holzmann H, Meixner T, Rauscher S, Stadler PF, Allison SL, Heinz FX: Spontaneous and engineered deletions in the 3' noncoding region of tick-borne encephalitis virus: construction of highly attenuated mutants of a flavivirus. J Virol 1998, 72: 2132-2140.
Leitmeyer KC, Vaughn DW, Watts DM, Salas R, Villalobos I, de C, Ramos C, Rico-Hesse R: Dengue virus structural differences that correlate with pathogenesis. J Virol 1999, 73: 4738-4747.
Cologna R, Rico-Hesse R: American genotype structures decrease dengue virus output from human monocytes and dendritic cells. J Virol 2003, 77: 3929-3938. 10.1128/JVI.77.7.3929-3938.2003
Clyde K, Kyle J, Harris E: Recent advances in deciphering viral and host determinants of dengue virus replication and pathogenesis. J Virol 2006, 80: 11418-11431. 10.1128/JVI.01257-06
Silva R, de Silva A, Harris E, MacDonald G: Genetic analysis of Dengue 3 virus subtype III 5' and 3' non-coding regions. Virus Res 2008, 135: 320-325. 10.1016/j.virusres.2008.03.007
Aquino V, Anatriello E, Gonçalves P, DA Silva E, Vasconcelos P, Vieira D, Batista W, Bobadilla M, Vazquez C, Moran M, Figueiredo L: Molecular epidemiology of dengue type 3 virus in Brazil and Paraguay, 2002–2004. Am J Trop Med Hyg 2006, 75: 710-715.
Roche C, Cassar O, Laille M, Murgue B: Dengue-3 virus genomic differences that correlate with in vitro phenotype on a human cell line but not with disease severity. Microbes Infect 2007, 9: 63-69. 10.1016/j.micinf.2006.10.010
Vasilakis N, Weaver S: The history and evolution of human dengue emergence. Adv Virus Res 2008, 72: 1-76.
Miagostovich M, dos Santos F, Fumian T, Guimarães F, da Costa E, Tavares F, Coelho J, Nogueira R: Complete genetic characterization of a Brazilian dengue virus type 3 strain isolated from a fatal outcome. Mem Inst Oswaldo Cruz 2006, 101: 307-313.
Consoli R, Lourenço-de-Oliveira R: Principais mosquitos de importância sanitária do Brasil. Rio de Janeiro: Fiocruz; 1994.
Igarashi A: Isolation of a Singh's Aedes albopictus cell clone sensitive to Dengue and Chikungunya viruses. J Gen Virol 1978, 40: 531-544. 10.1099/0022-1317-40-3-531
Gubler DJ, Kuno G, Sather GE, Velez M, Oliver A: Mosquito cell cultures and specific monoclonal antibodies in surveillance for dengue viruses. AmJTrop Med Hyg 1984, 33: 158-165.
Lanciotti RS, Calisher CH, Gubler DJ, Chang GJ, Vorndam AV: Rapid detection and typing of dengue viruses from clinical samples by using reverse transcriptase-polymerase chain reaction. J Clin Microbiol 1992, 30: 545-551.
Harris E, Sandoval E, Xet-Mull AM, Johnson M, Riley LW: Rapid subtyping of dengue viruses by restriction site-specific (RSS)-PCR. Virology 1999, 253: 86-95. 10.1006/viro.1998.9481
Usuku S, Castillo L, Sugimoto C, Noguchi Y, Yogo Y, Kobayashi N: Phylogenetic analysis of dengue-3 viruses prevalent in Guatemala during 1996–1998. Arch Virol 2001, 146: 1381-1390. 10.1007/s007050170098
Rigau-Pérez JG, Ayala-López A, García-Rivera EJ, Hudson SM, Vorndam V, Reiter P, Cano MP, Clark GG: The reappearance of dengue-3 and a subsequent dengue-4 and dengue-1 epidemic in Puerto Rico in 1998. AmJTrop Med Hyg 2002, 67: 355-362.
Peyrefitte C, Pastorino B, Bessaud M, Gravier P, Tock F, Couissinier-Paris P, Martial J, Huc-Anais P, Césaire R, Grandadam M, Tolou H: Dengue type 3 virus, Saint Martin, 2003–2004. Emerg Infect Dis 2005, 11: 757-761. 10.3201/eid1105.040959
Rodriguez-Roche R, Alvarez M, Holmes EC, Bernardo L, Kouri G, Gould EA, Halstead S, Guzman MG: Dengue virus type 3, Cuba, 2000–2002. Emerg Infect Dis 2005, 11: 773-774.
Uzcategui NY, Camacho D, Comach G, Cuello de Uzcategui R, Holmes EC, Gould EA: Molecular epidemiology of dengue type 2 virus in Venezuela: evidence for in situ virus evolution and recombination. J Gen Virol 2001, 82: 2945-2953.
Ocazionez R, Cortés F, Villar L, Gómez S: Temporal distribution of dengue virus serotypes in Colombian endemic area and dengue incidence. Re-introduction of dengue-3 associated to mild febrile illness and primary infection. Mem Inst Oswaldo Cruz 2006, 101: 725-731.
Barrero P, Mistchenko A: Genetic analysis of dengue virus type 3 isolated in Buenos Aires. Argentina. Virus Res 2008, 135: 83-88.
Briseño-García B, Gómez-Dantés H, Argott-Ramírez E, Montesano R, Vázquez-Martínez AL, Ibáñez-Bernal S, Madrigal-Ayala G, Ruíz-Matus C, Flisser A, Tapia-Conyer R: Potential risk for dengue hemorrhagic fever: the isolation of serotype dengue-3 in Mexico. Emerg Infect Dis 1996, 2: 133-135. 10.3201/eid0202.960210
Istúriz RE, Gubler DJ, BreadelCastillo J: Dengue and dengue hemorrhagic fever in Latin America and the Caribbean. Infect Dis Clin North Am 2000, 14: 121-140. 10.1016/S0891-5520(05)70221-X
Messer WB, Vitarana UT, Sivananthan K, Elvtigala J, Preethimala LD, Ramesh R, Withana N, Gubler DJ, De Silva AM: Epidemiology of dengue in Sri Lanka before and after the emergence of epidemic dengue hemorrhagic fever. AmJTrop Med Hyg 2002, 66: 765-773.
Dash P, Parida M, Saxena P, Abhyankar A, Singh C, Tewari K, Jana A, Sekhar K, Rao P: Reemergence of dengue virus type-3 (subtype-III) in India: implications for increased incidence of DHF & DSS. Virol J 2006, 3: 55. 10.1186/1743-422X-3-55
Braga IA, Lima JB, Soares SS, Valle D: Aedes aegypti resistance to temephos during 2001 in several municipalities in the states of Rio de Janeiro, Sergipe, and Alagoas, Brazil. Mem Inst Oswaldo Cruz 2004, 99: 199-203.
Honório NA, Lourenço-De-Oliveira R: [Frequency of Aedes aegypti and Aedes albopictus larvae and pupae in traps, Brazil]. Rev Saude Publica 2001, 35: 385-391. 10.1590/S0034-89102001000400009
Chow VT, Chan YC, Yong R, Lee KM, Lim LK, Chung YK, Lam-Phua SG, Tan BT: Monitoring of dengue viruses in field-caught Aedes aegypti and Aedes albopictus mosquitoes by a type-specific polymerase chain reaction and cycle sequencing. AmJTrop Med Hyg 1998, 58: 578-586.
Kow CY, Koon LL, Yin PF: Detection of dengue viruses in field caught male Aedes aegypti and Aedes albopictus (Diptera: Culicidae) in Singapore by type-specific PCR. J Med Entomol 2001, 38: 475-479. 10.1603/0022-2585-38.4.475
Wang WK, Lin SR, Lee CM, King CC, Chang SC: Dengue type 3 virus in plasma is a population of closely related genomes: quasispecies. J Virol 2002, 76: 4662-4665. 10.1128/JVI.76.9.4662-4665.2002
Chao D, King C, Wang W, Chen W, Wu H, Chang G: Strategically examining the full-genome of dengue virus type 3 in clinical isolates reveals its mutation spectra. Virol J 2005, 2: 72. 10.1186/1743-422X-2-72
Podder G, Breiman R, Azim T, Thu H, Velathanthiri N, Mai Q, Lowry K, Aaskov J: Origin of dengue type 3 viruses associated with the dengue outbreak in Dhaka, Bangladesh, in 2000 and 2001. AmJTrop Med Hyg 2006, 74: 263-265.
Kochel T, Aguilar P, Felices V, Comach G, Cruz C, Alava A, Vargas J, Olson J, Blair P: Molecular epidemiology of dengue virus type 3 in Northern South America: 2000–2005. Infect Genet Evol 2008, 8: 682-688. 10.1016/j.meegid.2008.06.008
Amarilla A, de Almeida F, Jorge D, Alfonso H, de Castro-Jorge L, Nogueira N, Figueiredo L, Aquino V: Genetic diversity of the E protein of dengue type 3 virus. Virol J 2009, 6: 113. 10.1186/1743-422X-6-113
Villabona-Arenas C, Miranda-Esquivel D, Jimenez R: Phylogeny of dengue virus type 3 circulating in Colombia between 2001 and 2007. Trop Med Int Health 2009.
de Mora D, Andrea L, Alvarez M, Regato M, Fajardo A, Recarey R, Colina R, Khan B, Cristina J: Evidence of diversification of dengue virus type 3 genotype III in the South American region. Arch Virol 2009, 154: 699-707. 10.1007/s00705-009-0343-7
Ramírez A, Fajardo A, Moros Z, Gerder M, Caraballo G, Camacho D, Comach G, Alarcón V, Zambrano J, Hernández R, et al.: Evolution of dengue virus type 3 genotype III in Venezuela: diversification, rates and population dynamics. Virol J 2010, 7: 329. 10.1186/1743-422X-7-329
Moi ML, Takasaki T, Kotaki A, Tajima S, Lim CK, Sakamoto M, Iwagoe H, Kobayashi K, Kurane I: Importation of dengue virus type 3 to Japan from Tanzania and Cote d'Ivoire. Emerg Infect Dis 2010, 16: 1770-1772. 10.3201/eid1611.101061
Ospina MC, Diaz FJ, Osorio JE: Prolonged co-circulation of two distinct Dengue virus Type 3 lineages in the hyperendemic area of Medellin. Colombia. Am J Trop Med Hyg 2010, 83: 672-678. 10.4269/ajtmh.2010.09-0766
Chen R, Vasilakis N: Dengue–quo tu et quo vadis? Viruses 2011, 3: 1562-1608. 10.3390/v3091562
Cologna R, Armstrong PM, Rico-Hesse R: Selection for virulent dengue viruses occurs in humans and mosquitoes. J Virol 2005, 79: 853-859. 10.1128/JVI.79.2.853-859.2005
Peyrefitte CN, Couissinier-Paris P, Mercier-Perennec V, Bessaud M, Martial J, Kenane N, Durand JP, Tolou HJ: Genetic characterization of newly reintroduced dengue virus type 3 in Martinique (French West Indies). J Clin Microbiol 2003, 41: 5195-5198. 10.1128/JCM.41.11.5195-5198.2003
Aquino J, Tang W, Ishii R, Ono T, Eshita Y, Aono H, Makino Y: Molecular epidemiology of dengue virus serotypes 2 and 3 in Paraguay during 2001–2006: the association of viral clade introductions with shifting serotype dominance. Virus Res 2008, 137: 266-270. 10.1016/j.virusres.2008.07.011
Acknowledgements
We thank to the field staff of the Sanitary District of Nova Iguaçu/Vector Control Program for its help on the vectors collection. To Mauro Menezes, Wellington Silva, Anielly Ferreira, José da Costa Farias Filho, Jaqueline Simoes and Clarice R Santos for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors have no conflict of interest.
Authors’ contributions
FBS, RMRN and RLO designed the study. MGC, FNB performed the experiments, MGC, FBS and RLO wrote the paper. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
de Castro, M.G., de Nogueira, F.B., Nogueira, R.M.R. et al. Genetic variation in the 3’ untranslated region of dengue virus serotype 3 strains isolated from mosquitoes and humans in Brazil. Virol J 10, 3 (2013). https://doi.org/10.1186/1743-422X-10-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-10-3