
Abstract
Background
Astrocyte activation is one of the earliest findings in the brain of methamphetamine (Meth) abusers. Our goal in this study was to identify the characteristics of the astrocytic acute response to the drug, which may be critical in pathogenic outcomes secondary to the use.
Methods
We developed an integrated analysis of gene expression data to study the acute gene changes caused by the direct exposure to Meth treatment of astrocytes in vitro, and to better understand how astrocytes respond, what are the early molecular markers associated with this response. We examined the literature in search of similar changes in gene signatures that are found in central nervous system disorders.
Results
We identified overexpressed gene networks represented by genes of an inflammatory and immune nature and that are implicated in neuroactive ligand-receptor interactions. The overexpressed networks are linked to molecules that were highly upregulated in astrocytes by all doses of methamphetamine tested and that could play a role in the central nervous system. The strongest overexpressed signatures were the upregulation of MAP2K5, GPR65, and CXCL5, and the gene networks individually associated with these molecules. Pathway analysis revealed that these networks are involved both in neuroprotection and in neuropathology. We have validated several targets associated to these genes.
Conclusions
Gene signatures for the astrocytic response to Meth were identified among the upregulated gene pool, using an in vitro system. The identified markers may participate in dysfunctions of the central nervous system but could also provide acute protection to the drug exposure. Further in vivo studies are necessary to establish the role of these gene networks in drug abuse pathogenesis.
Similar content being viewed by others


Background
Astrocytes are glial cells that are involved in numerous brain functions, including interacting with neurons and maintaining brain structure [1, 2], synthesizing cholesterol [3], and controlling synaptogenesis and neuronal plasticity [4–6]. In addition, together with microglia cells, astrocytes are first responders to both systemic and central nervous system (CNS) localized insults [7, 8]. These cells can initiate an inflammatory response, which can interestingly both promote tissue healing and neuronal loss and damage, as well as control the permeability of the blood-brain barrier (BBB) [9–11].
Drug abuse is one of the factors that can induce the activation of astrocytes [12]. One such drug is methamphetamine (Meth), which is widely used due to its strong effects as a psychotropic stimulant and its low price. Meth abusers develop an enormous number of degenerative symptoms, particularly in the CNS, manifested by cognitive deficits and motor dysfunction [13]. Astrocytic activation is one of the most common findings in the brain of Meth abusers, as well as in various models of Meth exposure [14–17], often associated with neurotoxicity. The effects of Meth on astrocytes could be potentially direct and have been attributed to its binding to sigma1 receptors [18].
In the present studies, we tested the hypothesis that astrocytes, as important first responders, may develop phenotypic changes that can contribute and be associated to neurological decline and multiple disorders commonly found in Meth abusers. We evaluated whether Meth can affect astrocytes by producing changes in gene expression and whether such changes can be critical attributes in the development of CNS disorders. We examined genes that were upregulated following Meth exposure, with an in vitro approach using primary cortical astrocytic cultures. Using systems biology, we have integrated the expression changes caused by Meth and have identified important astrocytic patterns, fingerprints, and pathways among genes that are transcriptionally enriched by the exposure to Meth. Although the analysis is limited to upregulated genes, these changes could have important implications in the development of neurological symptoms commonly associated with drug abuse.
Methods
Primary rat astrocyte cultures
E18 embryonic Sprague Dawley rat cortical astrocytes (BrainBits LLC, Springfield, IL) were cultured on poly-D-lysine-coated coverslips and were maintained in Neuro basal medium (Invitrogen, Carlsbad, CA) with 10% horse serum and 3 mM glutamine (Invitrogen), until confluence was reached (day 12).
Methamphetamine treatment
(+)-Methamphetamine hydrochloride (Sigma-Aldrich, Saint Louis, MO) was added to the confluent astrocytic cultures, at the final concentrations of 1, 10, and 100 μM, diluted in PBS. The drug was maintained in the cultures for 24 h prior to the harvesting of the cells. Control cultures were incubated with PBS vehicle. All results derive from three independent experiments, each one performed in duplicate.
Apoptosis detection
The rat astrocytes were tested for development of apoptosis 24 h after Meth treatment, using the terminal deoxynucleotidyl transferase dUTP (TdT) in situ TACS Blue (R&D systems), following the manufacturer’s instructions. Counterstaining was performed with Gill’s hematoxylin (Sigma-Aldrich, St. Louis, MO). Coverslips were applied over Cytoseal 60 mounting media (EMS, Hatfield, PA), and cells were inspected in light microscope.
Cell harvest and RNA extraction
Total RNA was isolated from the cells using Trizol reagent (Thermofisher, Waltham, MA), according to the manufacturer’s instructions. Total RNA concentration was measured using the Nanodrop spectrophotometer and then used for reverse transcription and for gene array (below).
Gene expression array
The integrity of total RNAs was examined in an Agilent Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Total RNA concentration was measured using the Nanodrop spectrophotometer. The mouse Agilent microarray service was performed by Phalanx Biotech (San Diego, CA). A total of 4 μg Cy5-labeled RNA targets were hybridized to Gene Expression v2 4x44K Microarrays (Agilent Technologies, Santa Clara, CA), according to the manufacturer’s protocol. The data were analyzed using the provided manufacturer’s protocol. Following the hybridization, fluorescent signals were scanned using an Axon 4000 (Molecular Devices, Sunnyvale, CA, USA). Three replicates per condition were used. Microarray signal intensity of each spot was analyzed using the GenePix 4.1 software (Molecular Devices, Sunnyvale, CA, USA). Each signal value was normalized using the R program in the limma linear models package (Bioconductor 3.2, https://bioconductor.org).
Gene expression analysis
Raw data was loaded into ArrayStudio (Omicsoft Corporation, Cary, NC) and first filtered based on a built-in ANOVA, as well as a t test, applied to fold changes between experimental and control conditions. Significant changes had a p value <0.05. In addition, maximum least-squares (Max LS) mean ≥ 6 and a false discovery rate by the Benjamini-Hochberg correction (FDR_BH) <0.01 were applied. Using this method, we found many genes with raw p values <0.05, but if the FDR_BH did not reach <0.01, they were discarded. In this particular analysis set, the genes were further filtered to express a robust fold change above 4 between control and experimental cultures. These filters allowed the identification of significant, above background gene expression changes. The list of genes that were significantly upregulated by Meth in astrocytes, following the described criteria, were loaded into Cytoscape 3.3 (http://cytoscape.org), using GeneMania [19], to identify significantly changed interaction networks of genes and relevant pathways. Pathway enrichment was examined using iPathwayGuide (Advaita Bioinformatics, Plymouth, MI) platform [20] and DAVID Bioinformatics database [21] (https://david.ncifcrf.gov), which utilize the Kyoto Encyclopedia of Genes and Genomes (KEGG) (www.genome.jp/kegg) and in Gene Ontology (GO) terms (http://geneontology.org/page/go-enrichment-analysis). Diseases associated to the changed profiles and genes were identified in KEGG.
RT-PCR
Validation of gene array data was performed both in the same samples and in two additional independent experiments. RNA was reverse transcribed using SuperScript III Reverse Transcriptase (Invitrogen, Waltham, MA). Most primers were purchased from Qiagen (Valencia, CA). PCRs were performed using RT2 SYBR Green ROX FAST Mastermix (Qiagen), in a 7900HT Fast Real-Time PCR System with Fast 96-Well Block Module (Applied Biosystems, Foster City, CA) with a SDS Plate utility v2.2 software (Applied Biosystems). The results were normalized to the expression of GAPDH.
Protein extraction and western blots
Following a wash with ice-cold PBS, protein from cell cultures was extracted by lysis in radio-immunoprecipitation assay buffer (RIPA—Thermo Fisher Scientific, Waltham, MA) in the presence of Complete protease inhibitor cocktail tablets (Roche Molecular Biochemicals, Indianapolis, IN). The cells were scraped and transferred to a microfuge tube and span at 10,000 rpm at 4 °C for 10 min. The supernatant was transferred to a new tube and protein concentration was measured using a Bradford Reagent (BioRad, Hercules, CA). Protein was stored in −20 °C until use. Ten micrograms of protein were loaded into each lane of SDS-PAGE electrophoresis gels (BioRad) in 4–20% gradient gels under reducing conditions. Transfer and immunodetection were performed as previously described [22]. Nonspecific antibody binding was blocked using 5% nonfat dried milk for 1 h at room temperature. Immunoblotting was carried out with antibodies against MEK5/MAP2K5 (PA5-29236, Thermo Fisher Scientific), TDAG8/GPR65 (BS-7668R, Bioss, Inc./VWR, Radnor, PA), and b-actin (Cell Signaling, Danvers, MA), followed by secondary antibody HRP-conjugated anti-rabbit IgG (GE Healthcare, Little Chalfont, UK). Blots were developed in film (Kodak) with 1:1 solution of Super Signal West Pico Chemiluminescent Substrate and Luminol/Enhancer (Thermo Fisher Scientific, Rockford, IL). Bands were scanned and band intensities were calculated in ImageJ 1.43u (National Institute of Health, Bethesda, MD). Experimental bands were normalized to the intensity of b-actin bands in each sample.
Immunocytochemistry
Cells were cultured on poly-L-lysine (Sigma-Aldrich)-treated 8-well glass chamber slides (Thermo Scientific), fixed with 4% paraformaldehyde for 20 min in the dark, and then washed with PBS. Wells were then incubated with PBS containing 0.1% Triton X-100 for 15 min at room temperature, rinsed 3 times with PBS, and then blocked with 5 g/l Casein (Sigma-Aldrich) in PBS, containing 0.5 g/l Thimerosal (Sigma-Aldrich) for 1 h at room temperature. The primary antibodies against MEK5/MAP2K5 (PA5-29236, Thermo Fisher Scientific), TDAG8/GPR65 (BS-7668R, Bioss, Inc./VWR, Radnor, PA), and CXCL5 (BS-2549R, Bioss Inc./VWR, Radnor, PA) were diluted in blocking solution and placed in the wells for 2 h at room temperature. Then, cells were rinsed 3 times for 10 min with 1% blocking solution in PBS, followed by incubation with a secondary Alexa594-labeled donkey anti-rabbit IgG (Thermo Fisher Scientific) for 2 h at room temperature, in the dark. After rinsing, 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) was diluted to 300 ng/ml in 1% blocking solution for 10 min, in the dark. Cells were rinsed and maintained in PBS and observed in a Nikon A1R laser-scanning confocal mounted onto a Nikon-inverted Ti-E scope (Nikon, Melville, NY), and with a 20× PlanApo objective, 0.8NA (Nikon) and images were acquired using a NIS-Elements C software (Nikon). Fluorescence intensity was normalized against background (secondary antibody only) and calculated in ImageJ 1.43u (National Institute of Health, Bethesda, MD).
Statistical analysis
Group comparisons for individual genes across different culture conditions were performed using one-way ANOVA, followed by Bonferroni’s post hoc tests. The difference between the means was considered significant at p < 0.05. Tests were performed using Prism software (GraphPad Software, San Diego, CA, USA) for Macintosh.
Results
Astrogliosis is among the earliest consequences of Meth use to the CNS. We investigated the early effects of Meth exposure on primary cortical astrocytes, by focusing on genes that were upregulated to above a conservative 4-fold threshold, which corresponded to more than 62% of the total changes in gene expression caused by the drug at any concentration over 24 h of exposure, as determined by gene array. Importantly, over the course of the experiment, we did not observe significant decrease of cell viability due to the exposure to different concentrations of Meth, as assessed by TdT detection by in situ hybridization (not shown).
We found that 411 genes were increased when the astrocytic cultures were exposed to Meth at concentrations of 10 or 100 μM, in comparison to controls, while in treatments with 1 μM of Meth, 180 genes were significantly increased. All together, 179 genes were significantly upregulated to above 4-fold by all the three doses of Meth compared to vehicle-treated astrocytes (Table 1). The correlation coefficient confirms that for the majority of the genes, Meth induced a dose-dependent response pattern (Table 1), which is visualized in Fig. 1. Figure 1a shows fold change of all the upregulated genes at the different doses of Meth. All genes that increased to above 4-fold at 10 μM of Meth compared to control were also significantly increased with 100 μM, but only 32% of those were also increased by the 1-μM condition. The calculation of the average fold change showed a significant overall dose-response effect (Fig. 1b), with a Pearson coefficient equal to 0.89 and a p value ≤0.0001. However, the examination of a correlation coefficient in all the individual upregulated genes showed that 28% of all genes exhibited a flat response that was equal in all 3 doses, while 22% of the genes showed a negative correlation coefficient, suggesting an inverse dose-response effect.

Changes in astrocytic gene expression pattern following exposure to different doses of Meth for 24 h. a Three doses of Meth were used, 1 μM (blue diamonds), 10 μM (red squares), and 100 μM (green triangles). The genes that showed a robust 4-fold increase in all Meth treatments were plotted as fold change in Meth-treated cells compared to controls. b The average fold change of all the genes was plotted for each one of the doses of Meth, and the Pearson coefficient was calculated (r 2 = 0.89). One-way ANOVA p value ≤0.0001
We examined the top 30 most upregulated genes in each of the three doses of Meth used to stimulate astrocytes (Table 2). Surprisingly, the majority of the genes differed between the three doses. However, a few genes appeared among the 30 most upregulated genes by all conditions, compared to controls. These genes, which appear in Table 1 as italics, were the mitogen-activated protein kinase kinase 5 (MAP2K5), the G protein-coupled receptor 65 (GPR65), ectodysplasin A (ED1), the neuron navigator 3 (NAV3), and CXCL5, which were chosen for a deeper analysis of the behavior of genes in network with them and pathway changes resulting from Meth exposure.
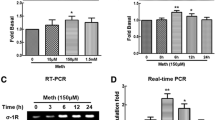
MAP2K5, GPR65, and CXCL5 were further validated by PCR and also at the protein levels. The qPCR validation results confirmed the potential importance of these three molecules in the direct response of astrocytes to Meth (Fig. 2a). The PCRs were performed in the same samples used for gene array and also in two independent experiments. MAP2K5 and GRP65 were significantly upregulated by all three doses, and CXCL5 was significantly upregulated in 10 and 100 μM treatments (Fig. 2a). We confirmed the relevance of these findings at the protein level, using specific antibodies against MEK5, the protein transcribed by the MAP2K5 gene, and GRP65, by western blot (Fig. 2b, c). The enrichment of these proteins was also confirmed by imaging, using specific antibodies against MEK5, GRP65, and CXCL5 (Fig. 2c, d). The increase in these markers first identified using systems biology tools suggests the value of the approach and its power for the identification of changes in genes that depend on or are associated with MAP2K5, GRP65, and CXCL5. We again utilized systems biology to search for gene networks associated to these genes, as well as to ED1 and NAV3, exhibiting synchronic behaviors in response to Meth in astrocytes.

Validation of selected upregulated genes. a Transcriptional levels of MAP2K5, GRP65, and CXCL5 in astrocytes stimulated with 1, 10, and 100 μM of Meth for 24 h, examined by SyBrGreen qRT-PCR. Results are the Mean ± SEM of three independent experiments performed in duplicate. b Representative western blots for detection of MEK5, the protein encoded by MAP2K5 gene, GPR65, and normalizing b-actin, in protein extracts from astrocytes stimulated with 1, 10, and 100 μM of Meth for 24 h. c Normalized band intensity was calculated in ImageJ software (NIH). d Confocal imaging showing representative astrocytic cultures stained with specific antibodies for detection of MEK5, GPR65, and CXCL5 in 10 and 100 μM Meth treatments. e Fluorescence intensity of the expression of MEK5, GPR65, and CXCL5 was calculated in ImageJ (NIH) for cultures stimulated with 1, 10, and 100 μM, as well as controls. *p < 0.05 in one-way ANOVA followed by Bonferroni’s post hoc comparison against control conditions
For the examination of the behavior of genes in network with MAP2K5, GPR65, CXCL5, ED1, and NAV3, we used GeneMania in Cytoscape, and for that, we focused on the 10 μM dose, which represents levels of drug reaching the brain in Meth users [23]. The analysis of the changes was conducted using a protocol utilized in our lab, to determine the behavior of genes associated to the ones we chose to prioritize, based on pathway, physical and genetic interactions, shared protein domains, or coexpression, in order to predict molecular networks with which astrocytes might respond to acute drug abuse. Using GeneMania and JActiveModules in Cytoscape [24–27], we identified such gene node clusters. The highest score node contained 141 genes, which were all upregulated, and which clustered with a coefficient of 0.174, suggesting that Meth has a strong effect on astrocytic gene networks. Ninety of those genes (63.8%) showed multi-edged node pairs, suggesting a strong interaction between molecular changes and processes triggered by Meth in astrocytes (Additional file 1: Figure S1). MAP2K5, GPR65, and ED1 (EDA or the rat homolog of CD68), which were consistently among the 30 genes most upregulated by all three doses of Meth (Table 2—italic letters), were also represented in this large gene cluster. A literature examination suggests that these genes could be a link between the acute response of astrocytes to Meth and the potential development of CNS alterations. For instance, ED1 is a microglial marker but it can be found expressed on tumoral astrocytes [28]. Interestingly, MAP2K5 is also a characteristic of tumorigenesis [29]. GPR65, on the other hand, is a proton- and acid-sensing G-protein receptor that plays an important role in cell survival and is also known as TDAG8 [30, 31].
We examined subfamilies of genes assembled as child nodules, by connecting first neighbors of MAP2K5 (Fig. 3a) and GPR65 (Fig. 3b), which led to subnetworks respectively assigned to neuronal support and inflammation. For instance, the genes that appeared in connection with MAP2K5 (Fig. 3a) were annotated to MAPK signaling (p = 0.0022), gap junction (p = 0.0051), the GnRH signaling pathway (p = 0.0062), and also neuroactive ligand-receptor interactions (p = 0.008), suggesting association to neurological outcomes. The genes connected to GPR65 (Fig. 3b) were associated to pathways involving cytokine-cytokine interaction (p = 0.00027), chemokine signaling pathway (p = 0.04), B cell receptor signaling pathway (p = 0.003), Fc-gamma R-mediated phagocytosis (p = 0.0048), and systemic lupus erythematous (p = 0.0052), suggesting a role in potential inflammatory outcomes.

Connectivity of genes that were upregulated in astrocytes by Meth. JActiveModules was applied to the whole data set, using a significant (p < =0.05) minimum of a 4-fold upregulation, in genes identified in astrocytes that were treated with 10 μM Meth for 24 h, for identification of hotspots. This approach identified a (a) cluster of MAP2K5-first neighbor genes, a (b) cluster of GPR65-first neighbor genes, and a (c) cluster of ED1-first neighbor genes. Shades of red represent the level of upregulation of each gene, where light red is less upregulated and darker red is more upregulated. Genes that are connected by a green line are in the same pathway, whereas orange lines connect genes that are colocalized, yellow lines mean coexpression, gray lines mean genetic interactions, blue lines mean shared protein domains, and red lines mean physical interactions
ED1 was not represented in either one of these subnetworks, but a cluster analysis centered on first neighbors of this gene (Fig. 3c), resulted in a group of genes functionally assigned as glycoproteins (p = 0.0018), and functionally annotated to immune response (p = 0.029) and cell adhesion (p = 0.03).
Other genes among the 30 most upregulated ones by all three doses were NAV3 and CXCL5 (Table 2). NAV3 was represented within subnetworks associated to GPR65 (Fig. 3b), as well as to ED1 (Fig. 3c). CXCL5, on the other hand, segregated on a network (Fig. 4) that was heavily associated to chemokine signaling pathway (p = 8.7E−7), cytokine-cytokine receptor interaction (p = 6E−5), neuroactive ligand-receptor interaction (p = 0.0037), and calcium signaling pathways (p = 0.01).

CXCL5-centered gene network upregulated in astrocytes in response to Meth exposure. Shades of red represent the level of upregulation of each gene, where light red is less upregulated and darker red is more upregulated. Genes that are connected by a green line are in the same pathway, whereas orange lines connect genes that are colocalized, yellow lines mean coexpression, gray lines mean genetic interactions, blue lines mean shared protein domains, and red lines mean physical interactions
We further dissected the highest score node, to produce subnetworks that were derived by the introduction of a display restriction connecting genes only through pathway and physical associations. This restrictive approach generated one network (Fig. 5), in which MAP2K5 appeared as the strongest upregulated gene, in correlation with other genes described in neurological processes, metabolism, and inflammation (Fig. 5). Importantly, as a control, the cortical astrocyte marker ErbB3 [32] was represented in this network.

MAP2K5-centered gene clusters exclusively based on pathway and physical interactions. The data was filtered by eliminating colocalization, coexpression, genetic interactions, and shared protein domains from the clustering criteria, but maintaining physical and pathway interactions. Shades of red represent the level of upregulation, and green represents downregulation of each gene. Red lines represent pathway associations, and green lines represent associations based on protein physical interactions. A group attributes layout was applied to discriminate p values and scores. Scores can be also identified by the size of individual nodes. Genes grouped on the left had lower p values, and highest scores than genes on the right. Genes in the center of the left group were the ones with highest scores, or highest number of connectors
We performed pathway enrichment analysis on the set of genes that were upregulated by Meth in astrocytes, using the DAVID Bioinformatics Database (KEGG_PATHWAY) (Table 3) and the iPathwayGuide (Fig. 6). We found that Meth treatment on astrocytes caused an important enrichment of genes that are relevant in neuroactive ligand-receptor interactions, immunity, and metabolic outcomes, as shown in Table 3.

Pathway enrichment analysis on genes upregulated by Meth treatment of astrocytes. iPathwayGuide derived analysis showing molecular pathways segregated according to total perturbation accumulation (pACC) and gene overrepresentation within pathways (pORA). Red/Yellow dots represent pathways modified with a significant p value
Further analysis was conducted to examine the number of gene perturbation accumulation (pACC) versus overrepresentation p value (pORA) within pathways (Fig. 6) and that also indicated that CNS- and pathology-relevant genes were strongly represented. For instance, genes involved in neuroactive ligand-receptor interaction (p = 0.001) and circadian rhythm (p = 0.012) led to an important contribution to changes induced by Meth in astrocytes. A substantial, but not statistically significant, representation of genes involved in chemokine signaling pathways was also observed (p = 0.056). Interestingly, Meth also triggered genes that are involved in the resistance to infections (Fig. 6). The strong representation of pathways that may be involved in neurological outcomes prompted an analysis of individual genes.
Of the genes annotated to the neuroactive ligand-receptor interaction pathway (Fig. 7), a few were interesting for being associated to neurological disorders in other systems, for instance, the Period Circadian Clock 2 (PER2) [33, 34]. Others have been also described to have essential inflammatory roles, for instance, prostanoid receptors, such as the prostaglandin E receptor 3 (PTGER3), the thromboxane A2 receptor (TBXA2R), and the prostaglandin D2 receptor (PTGDR), which play important roles in inflammatory reactions and hyperalgesia [35].

Genes mapped to the neuroactive ligand-receptor pathway, identified by gene array in astrocytes that were treated with Meth. Data corresponds to significant fold change in 10 μM Meth-treated astrocytes compared to vehicle-treated controls in the gene expression array
The other genes upregulated by Meth treatment of astrocytes that could be mentioned for their potential connection with CNS-related syndromes were the following: the inositol 1,4,5-triphosphate receptor, type 1 (ITPR1, 9.24-fold upregulation) is involved in spinocerebellar ataxia [36]; the cold autoinflammatory syndrome 1 (CIAS1, 10.94-fold) in cryopirin-associated periodic syndrome [37]; the RET proto-oncogene (7.4-fold) in congenital central hypoventilation syndrome [38, 39]; PER2 (4-fold) in familial advanced sleep phase syndrome [40]; nucleoporin 62 kDa (NUP62, 4.72-fold) in infantile bilateral striatal necrosis [41]; ubiquitin-activating E1 (UBE1,10–45-fold) [42] and ankyrin repeat domain 2 (stretch responsive muscle, ANKRD2, 9.06-fold) in paralytic syndromes and cerebral palsy; interleukin 1 receptor antagonist (IL1RN, 6.43-fold), the interleukin 2 receptor common gamma chain (IL2RG, 10.02-fold), the ATP-transporter 2 (TAP2, 20.47-fold), PTGDR (11.97-fold), PTGER3 (27.56-fold), TBXA2R (6.3-fold), and the Complement Component 2 (C2, 5.8-fold) in CNS inflammation; the AarF domain containing kinase 4 (ADCK4, 11.04-fold) in coenzyme Q deficiency [43]; the zinc finger protein 41 (ZNF41, 5.39-fold) and the angiotensin II receptor, type 2 (AGTR2, 10.44-fold) in fragile X syndrome, cerebral ataxia, mental retardation, and disequilibrium syndrome [44, 45]; laminin, alpha 2 (LAMA2, 4.31-fold) in congenital muscular dystrophies and control of the blood-brain barrier [46]; and the cholinergic receptor, nicotinic, alpha 1 (CHRNA1, 7.73-fold) involved in congenital myasthenic syndrome and considered as an important potential drug target [47].
Using qRT-PCR, we confirmed the transcriptional upregulation of several of these genes, prioritized by their presence in the two most strongly represented pathways, the neuroactive-ligand and the cytokine-cytokine receptor interaction pathways (Table 4), in all three doses of Meth in vitro. These genes were the IL1RN, IL2RG, as well as the prostanoid receptors PTGDR, PTGER3, and TBXA2R (Fig. 8 and Table 4). These genes play important functions in inflammatory processes in the brain and elsewhere. We also examined other upregulated genes that have been described as molecules that could potentially influence the immune environment in the CNS, TAP2, and C2 (Fig. 8). All these validated genes have been suggested to play important roles in the CNS and neurological disorders [23, 48–53]. Our validation by PCR confirmed that the Meth treatment has the capacity to directly stimulate the upregulation of these genes, which have an involvement in inflammatory processes in the brain. Among them, TAP2 and PTGDR were upregulated by 10 and 100 μM Meth but not by 1 μM, while the other genes were transcriptionally increased by all the doses, validating the gene array data.

Transcriptional validation of inflammatory gene changes induced by Meth in astrocytes. Gene expression was examined by SyBrGreen qRT-PCR on mRNA extracts from primary astrocytes treated with 1 μM, 10 μM, and 100 μM of Meth, as well as controls, upon GAPDH normalization. Results are from one representative experiment, confirmed by two independent biological replicates. *p < 0.05 in one-way ANOVA followed by Bonferroni’s post hoc test, in comparisons indicated by lines
Although our analysis of changes in astrocytic gene expression was focused on enrichments, a few of the genes highlighted in our study have showed network connections with genes downregulated by the Meth treatment. The increase in MAP2K5, for instance, was associated with the decrease on serine/threonine protein kinase 2 (PAK2) (0.6-fold, p = 0.041) (Fig. 5). Other molecule that showed several downregulated connectors was IL2RG (Fig. 9). A gene cluster centered on IL2RG was associated with several components of the Jak-Stat signaling pathway (p = 0.0002, Benjamini = 0.002), primary immunodeficiency (p = 0.008, Benjamini = 0.03), and cytokine-cytokine receptor interaction (p = 0.006, Benjamini = 0.06) pathways. Of the genes in the IL2RG interactive subnetwork, several were significantly downregulated, such as the inducible T cell costimulator ligand (ICOSLG) (0.3-fold, p = 0.006) or the cytochrome P450 family member CYP2B6 (0.1-fold, p = 0.0012), suggesting that the upregulated genes may interfere with, or become affected by, transcriptional suppressions. The role of the downregulated genes in the development of astrocytic changes caused by Meth must be examined in the future.

IL2RG-centered gene subnetwork. Network shows connected genes based on pathway (red lines) and physical interactions (brown). The data was filtered by eliminating colocalization, coexpression, genetic interactions, and shared protein domains from the clustering criteria, but maintaining physical and pathway interactions. Shades of red represent the level of upregulation, and green represents downregulation of each gene
Discussion
We examined the hypothesis that astrocytes respond to Meth exposure by developing gene expression signatures and upregulation of genes that may participate in pathogenesis. For that, we used an in vitro approach, where primary cortical astrocyte cultures were directly exposed to Meth for 24 h. Our system-wide approach combined experimental and computational methods to systematically identify and integrate the important characteristics of astrocytes following the direct exposure of Meth, without taking into account downregulated genes. This approach was chosen in order to address a gap in the knowledge of dopamine-independent effects of drug abuse, which may play an important role in the development of response patterns in glial cells. With a focus on significantly upregulated genes, we identified signature patterns that were validated by qPCR and by changes at the protein level. A review of the literature revealed that several of these markers could have implications to CNS pathogenesis.
The validated changes in MAP2K5 (MEK5) levels and the detection of changes in many genes associated with this kinase were particularly remarkable and indicated a pathway of interest. MAP2K5 is a component of the MAPK family intracellular signaling pathway in the brain. In the context on cell-cell communication, it is highly responsive to extracellular growth factors such as brain-derived neurotrophic factor (BDNF), nerve growth factor (NGF), insulin-like growth factor 2 (IGF2) [54], granulocyte colony-stimulating factor [55], and epidermal growth factor [56]. MAP2K5 is particularly responsive to oxidative stress, for instance, in muscle differentiation where it activates ERK5 [57]. In the brain, its upregulation could play a role in neuroprotection of dopaminergic neurons [58], suggesting that the acute response of astrocytes to drug abuse could provide survival signals. On the other hand, in isolated astrocyte cultures, the MAP2K5 gene clusters with other genes that suffered similar changes, which have been described to be involved in CNS disorders. One of these genes is LAMA2, which is associated with muscular dystrophies and with the blood-brain barrier control [46]. LAMA2 also regulates other genes, such as the ladybird homeobox corepressor 1 (LBXCOR1), which is a corepressor of transcription playing a role in GABAergic phenotype of interneurons in some areas of the brain, associated for instance to the susceptibility to restless legs syndrome [59]. Another one of these genes is MDM2, which is described as an important regulator of tumorgenesis in astroglioma models [60].
GPR65, which was also validated, is also known as T cell death-associated gene 8 (TDAG8), an acidosis-sensing molecule [61, 62] and that has been described in association with susceptibility to autoimmunity, including in the CNS. Given that one of the adverse effects of Meth abuse is metabolic acidosis [63], this finding could be of relevance. On the other hand, GPR65 is a negative regulator of inflammation [64, 65]. Its upregulation may partially protect the brain environment in the response to acute Meth. In astrocytes exposed to Meth, CXCL5 was one of the genes highly upregulated by all Meth doses, with a strong connection to inflammatory pathways that can potentially lead to important changes in the context of the brain environment [66]. Importantly, CXCL5 has been reported as a potential marker of ischemic brain injury [67]. Conversely, the most enriched molecular pathways corresponded to Neuroactive ligand-receptor interactions and cytokine-cytokine receptor interactions, which respectively include MAP2K5 and GPR65, as well as CXCL5 genes.
We found that Meth caused the upregulation of several prostanoid receptors, including TBXA2R, PTGDR, and PTGER3, on astrocytes. These characteristics in the context of the brain could contribute to the inflammatory pathophysiology seen in the CNS of Meth abusers. These targets were validated by quantitative real-time polymerase chain reaction (qRT-PCR). Interestingly, the expression of PTGER3 was particularly sensitive to Meth, in a dose-dependent manner. In the context of pathology, PTGER3 has been found to be upregulated in aged patients with schizophrenia, suggesting the participation of the eicosanoid signaling in mental disorders [68]. In addition, the prostanoid receptors could affect major signaling pathways, such as the MAP kinase pathway [69], providing a potential link between the inflammatory and neuroactive aspects of the astrocytic acute response to the drug.
There was an intriguing induction of circadian rhythm-associated molecules in astrocytes that were treated with Meth. This supports findings in the literature where mammalian astrocytes display circadian functional profiles, particularly regarding the expression of the clock genes such as PER2, as well as regarding ATP release [33, 34]. It remains to be determined whether the expression of this gene specifically in astrocytes can play a role in vivo, since it has been shown that PER2 does modify circadian sleep cycle during sleep disruptions [70] and also in substance abuse [71, 72].
The genes that promote inflammation are of particular interest, because when applied to the context of the brain, they could play a role both in neuroprotection and neurodegeneration [73, 74]. In Meth abuse, it is known that neuroinflammation plays a critical role in the development of neurological decline [75–78]. Our results indicate that the acute, direct response of astrocytes to the drug could contribute to the inflammatory pathogenesis. Therefore, the genes with pro-inflammatory roles in the two most enriched pathways, which were the neuroactive ligand-receptor and the cytokine-cytokine receptor interactions, were prioritized for validation. These genes were the IL2RG, TAP2, and IL1RN, in addition to the prostanoid receptors. The ability of Meth to upregulate them was confirmed by qRT-PCR. The induction of IL2RG by Meth in astrocytes could be a factor modulating phenotype of other glial cells in vivo, as previously described by us in the context of HIV/Meth comorbidities [23]. TAP2, on the other hand, is a molecule that is involved in the expression of class I major histocompatibility complex molecules, which affects antigen presentation [79–82]. The role of the class I-mediated cytotoxic response in the HIV and in the HIV/Meth comorbidities has also been described by us [80, 81, 83, 84]. We also validated the upregulation of IL1RN by Meth, which could contribute to controlling astrocytosis, as suggested in the prion disease model [85]. IL1RN has been also suggested to participate in the regulation of glutamate uptake by astrocytes [86]. These genes are therefore important in a context of cellular interactions in the brain, and our results demonstrate that the acute exposure to the drug can cause their upregulation in a direct and dopamine-independent manner.
Several of the genes acutely induced by Meth in astrocytes have been described in association with neurological and inflammatory functions and with CNS disorders that are linked to motor dysfunction [87, 88]. Interestingly, astrocytes have been previously suggested as important cellular targets in CNS motor disorders [87, 88], although specific molecular targets are not clear. In the context of Meth abuse, movement disorders are very common sequels and comorbidities [89]. On the other hand, the genes upregulated by Meth in astrocytes also suggest a potential acute neuroprotective response. Interestingly, the use of low doses of Meth has been previously suggested to be beneficial immediately following severe traumatic brain injury [90, 91]. However, whether the character of the astrocytic response favors protection or neuronal damage and disorder, acutely or during chronic exposure remains to be investigated in vivo.
An important aspect of some upregulated genes examined here was their connection with downregulated network components. That was the case for genes associated with the increased expression of MAP2K5 and also IL2RG. The molecules in connection with the expression of IL2RG were of particular interest, given the role of the IL2RG system components in brain inflammatory outcomes and modulation of microglial phenotypes [92]. The IL2RG subnetwork was associated with the increase of important pro-inflammatory molecules such as IL21 [93] but also with the decrease of molecules such as the inducible T cell costimulator ligand (ICOSLG), which is a pattern recognition element that may impact immune response [94], or the cytochrome P450 family member CYP2B6, which plays a role in mood disorders and depression [95]. The interaction between genes that are up- and downmodulated suggests the importance of extending the analysis to genes that are decreased by Meth, for understanding the full spectrum and implications of changes caused by the direct exposure of astrocytes to that drug.
Our data analysis has suggested that acute Meth exposure drives the development of response patterns in astrocytes that may cause these cells to play essential roles in vivo, through dopamine-independent mechanisms. There are limitations in the single-cell type system, as in vivo factors derived from the other cells may further modulate the effects of Meth on astrocytes, and the response patterns may not be exclusively a result of direct stimulation. Therefore, given that the genes that are increased directly by Meth can play critical roles influencing the brain cell network, the changes in astrocyte gene expression do need further examination, using in vivo models of drug abuse. The focus on upregulated genes may limit the interpretation of the data but may accelerate pre-clinical approaches, since upregulated molecules are favored as targets for therapeutics. The gene signatures identified among upregulated genes are highly relevant to inflammation and to CNS disorders but also suggest that Meth may trigger neuroprotection pathways. These deviations in the astrocytic response pattern may point to important targets to be further investigated, for preventing and ultimately reverting deleterious consequences and neurological sequels of drug abuse.
Conclusions
Astrocytes modify their molecular signatures in response to direct exposure to methamphetamine. By applying a systems biology analysis approach with a focus on upregulated molecular markers, we have identified overexpressed gene networks represented by genes of an inflammatory and immune nature and that are implicated in neuroactive ligand-receptor interactions. MAP2K5, GPR65, and CXCL5 were molecules situated in the core of the highest score gene networks and were linked to markers associated with both neuroprotection and neuropathology. We have validated several targets and discussed their potential association with human neurological disease. Further in vivo studies are necessary to examine the role of these gene networks in drug abuse pathogenesis and their potential as biomarkers.
Abbreviations
- ADCK4:
-
AarF domain containing kinase 4
- AGTR2:
-
Angiotensin II receptor, type 2
- ANKRD2:
-
Ankyrin repeat domain 2
- ANOVA:
-
Analysis of variance
- BBB:
-
Blood-brain barrier
- BDNF:
-
Brain-derived neurotrophic factor
- C2:
-
Complement component 2
- CHRNA1:
-
Cholinergic receptor, nicotinic, alpha 1
- CIAS1:
-
Cold autoinflammatory syndrome 1
- CNS:
-
Central nervous system
- CXCL5:
-
C-X-C chemokine ligand 5
- DAVID:
-
Database for annotation, visualization, and integrated discovery
- ED1:
-
Ectodysplasin A
- FDR_BH:
-
False discovery rate by the Benjamini-Hochberg correction
- GPR65:
-
G protein-coupled receptor 65
- IGF2:
-
Insulin-like growth factor 2
- IL1RN:
-
Interleukin 1 receptor antagonist
- IL2RG:
-
interleukin 2 receptor common gamma chain
- ITPR1:
-
Inositol 1,4,5-triphosphate receptor, type 1
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LAMA2:
-
Laminin, alpha 2 (, 4.31-fold)
- LBXCOR1:
-
Ladybird homeobox corepressor 1
- MA2K5:
-
Mitogen-activated protein kinase kinase 5
- Max LS:
-
Maximum least-squares
- Meth:
-
Methamphetamine
- NAV3:
-
Neuron navigator 3
- NGF:
-
Nerve growth factor
- pACC:
-
Gene perturbation accumulation
- PER2:
-
Period Circadian Clock 2
- pORA:
-
Overrepresentation p value
- PTGDR:
-
Prostaglandin D2 receptor
- PTGER3:
-
Prostaglandin E receptor 3
- TAP2:
-
ATP-transporter 2
- TBXA2R:
-
Thromboxane A2 receptor
- UBE1:
-
Ubiquitin-activating E1
- ZNF41:
-
Zinc finger protein 41
References
Barres BA. Neuronal-glial interactions. A new form of transmission? Nature. 1989;339:343–4.
Seth P, Koul N. Astrocyte, the star avatar: redefined. J Biosci. 2008;33:405–21.
Mulder M. Sterols in the central nervous system. Curr Opin Clin Nutr Metab Care. 2009;12:152–8.
Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–55.
Barker AJ, Ullian EM. New roles for astrocytes in developing synaptic circuits. Commun Integr Biol. 2008;1:207–11.
Barker AJ, Ullian EM. Astrocytes and synaptic plasticity. Neuroscientist. 2010;16:40–50.
Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson’s disease. Mov Disord. 2011;26:6–17.
Sofroniew MV. Reactive astrocytes in neural repair and protection. Neuroscientist. 2005;11:400–7.
Sofroniew MV. Multiple roles for astrocytes as effectors of cytokines and inflammatory mediators. Neuroscientist. 2014;20:160–72.
Abbott NJ. Astrocyte-endothelial interactions and blood-brain barrier permeability. J Anat. 2002;200:629–38.
Abbott NJ, Ronnback L, Hansson E. Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci. 2006;7:41–53.
Miguel-Hidalgo JJ. The role of glial cells in drug abuse. Curr Drug Abuse Rev. 2009;2:76–82.
Scott JC, Woods SP, Matt GE, Meyer RA, Heaton RK, Atkinson JH, Grant I. Neurocognitive effects of methamphetamine: a critical review and meta-analysis. Neuropsychol Rev. 2007;17:275–97.
Narita M, Suzuki M, Kuzumaki N, Miyatake M, Suzuki T. Implication of activated astrocytes in the development of drug dependence: differences between methamphetamine and morphine. Ann N Y Acad Sci. 2008;1141:96–104.
Cadet JL, Krasnova IN, Jayanthi S, Lyles J. Neurotoxicity of substituted amphetamines: molecular and cellular mechanisms. Neurotox Res. 2007;11:183–202.
Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker Jr W, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–80.
Pu C, Vorhees CV. Developmental dissociation of methamphetamine-induced depletion of dopaminergic terminals and astrocyte reaction in rat striatum. Brain Res Dev Brain Res. 1993;72:325–8.
Zhang Y, Lv X, Bai Y, Zhu X, Wu X, Chao J, Duan M, Buch S, Chen L, Yao H. Involvement of sigma-1 receptor in astrocyte activation induced by methamphetamine via up-regulation of its own expression. J Neuroinflammation. 2015;12:29.
Warde-Farley D, Donaldson SL, Comes O, Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT, et al. The GeneMANIA prediction server: biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010;38:W214–20.
Draghici S, Khatri P, Tarca AL, Amin K, Done A, Voichita C, Georgescu C, Romero R. A systems biology approach for pathway level analysis. Genome Res. 2007;17:1537–45.
Da Huang W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Alirezaei M, Kiosses WB, Flynn CT, Brady NR, Fox HS. Disruption of neuronal autophagy by infected microglia results in neurodegeneration. PLoS ONE. 2008;3, e2906.
Madden L, Flynn CT, Zandonatti M, May M, Parsons LH, Katner SH, Henriksen SJ, Fox HS. Modeling Human Methamphetamine Exposure in Nonhuman Primates: Chronic Dosing in the Rhesus Macaque Leads to Behavioral and Physiological Abnormalities. Neuropsychopharmacology. 2005;30:350–59.
Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL, Morris Q, Bader GD. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–8.
Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat Protoc. 2007;2:2366–82.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Ideker T, Ozier O, Schwikowski B, Siegel AF. Discovering regulatory and signalling circuits in molecular interaction networks. Bioinformatics. 2002;18 Suppl 1:S233–40.
Strojnik T, Kavalar R, Zajc I, Diamandis EP, Oikonomopoulou K, Lah TT. Prognostic impact of CD68 and kallikrein 6 in human glioma. Anticancer Res. 2009;29:3269–79.
Berezovsky AD, Poisson LM, Cherba D, Webb CP, Transou AD, Lemke NW, Hong X, Hasselbach LA, Irtenkauf SM, Mikkelsen T, DeCarvalho AC. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia. 2014;16:193–206. 206 e119-125.
Rosko AE, McColl KS, Zhong F, Ryder CB, Chang MJ, Sattar A, Caimi PF, Hill BT, Al-Harbi S, Almasan A, Distelhorst CW. Acidosis sensing receptor GPR65 correlates with anti-apoptotic Bcl-2 family member expression in CLL cells: potential implications for the CLL microenvironment. J Leuk (Los Angel). 2014;2:5.
Ryder C, McColl K, Zhong F, Distelhorst CW. Acidosis promotes Bcl-2 family-mediated evasion of apoptosis: involvement of acid-sensing G protein-coupled receptor Gpr65 signaling to Mek/Erk. J Biol Chem. 2012;287:27863–75.
Sharif A, Duhem-Tonnelle V, Allet C, Baroncini M, Loyens A, Kerr-Conte J, Collier F, Blond S, Ojeda SR, Junier MP, Prevot V. Differential erbB signaling in astrocytes from the cerebral cortex and the hypothalamus of the human brain. Glia. 2009;57:362–79.
Beaule C, Swanstrom A, Leone MJ, Herzog ED. Circadian modulation of gene expression, but not glutamate uptake, in mouse and rat cortical astrocytes. PLoS ONE. 2009;4, e7476.
Marpegan L, Swanstrom AE, Chung K, Simon T, Haydon PG, Khan SK, Liu AC, Herzog ED, Beaule C. Circadian regulation of ATP release in astrocytes. J Neurosci. 2011;31:8342–50.
Bley KR, Hunter JC, Eglen RM, Smith JA. The role of IP prostanoid receptors in inflammatory pain. Trends Pharmacol Sci. 1998;19:141–7.
van de Leemput J, Chandran J, Knight MA, Holtzclaw LA, Scholz S, Cookson MR, Houlden H, Gwinn-Hardy K, Fung HC, Lin X, et al. Deletion at ITPR1 underlies ataxia in mice and spinocerebellar ataxia 15 in humans. PLoS Genet. 2007;3, e108.
Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, Teillac-Hamel D, Fischer A, De Saint BG. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet. 2002;71:198–203.
Amiel J, Laudier B, Attie-Bitach T, Trang H, De Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simonneau M, et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–61.
Bolk S, Angrist M, Schwartz S, Silvestri JM, Weese-Mayer DE, Chakravarti A. Congenital central hypoventilation syndrome: mutation analysis of the receptor tyrosine kinase RET. Am J Med Genet. 1996;63:603–9.
Vanselow K, Vanselow JT, Westermark PO, Reischl S, Maier B, Korte T, Herrmann A, Herzel H, Schlosser A, Kramer A. Differential effects of PER2 phosphorylation: molecular basis for the human familial advanced sleep phase syndrome (FASPS). Genes Dev. 2006;20:2660–72.
Basel-Vanagaite L, Muncher L, Straussberg R, Pasmanik-Chor M, Yahav M, Rainshtein L, Walsh CA, Magal N, Taub E, Drasinover V, et al. Mutated nup62 causes autosomal recessive infantile bilateral striatal necrosis. Ann Neurol. 2006;60:214–22.
Ramser J, Ahearn ME, Lenski C, Yariz KO, Hellebrand H, Von Rhein M, Clark RD, Schmutzler RK, Lichtner P, Hoffman EP, et al. Rare missense and synonymous variants in UBE1 are associated with X-linked infantile spinal muscular atrophy. Am J Hum Genet. 2008;82:188–93.
Ashraf S, Gee HY, Woerner S, Xie LX, Vega-Warner V, Lovric S, Fang H, Song X, Cattran DC, Avila-Casado C, et al. ADCK4 mutations promote steroid-resistant nephrotic syndrome through CoQ10 biosynthesis disruption. J Clin Invest. 2013;123:5179–89.
Sun H, Wu H, Yu X, Zhang G, Zhang R, Zhan S, Wang H, Bu N, Ma X, Li Y. Angiotensin II and its receptor in activated microglia enhanced neuronal loss and cognitive impairment following pilocarpine-induced status epilepticus. Mol Cell Neurosci. 2015;65:58–67.
Raymond FL, Tarpey P. The genetics of mental retardation. Hum Mol Genet. 2006;15:R110–6.
Menezes MJ, McClenahan FK, Leiton CV, Aranmolate A, Shan X, Colognato H. The extracellular matrix protein laminin alpha2 regulates the maturation and function of the blood-brain barrier. J Neurosci. 2014;34:15260–80.
Tei S, Ishii HT, Mitsuhashi H, Ishiura S. Antisense oligonucleotide-mediated exon skipping of CHRNA1 pre-mRNA as potential therapy for congenital myasthenic syndromes. Biochem Biophys Res Commun. 2015;461:481–6.
Rafiei A, Hosseini SH, Taheri M, Hosseni-khah Z, Hajilooi M, Mazaheri Z. Influence of IL-1RN intron 2 variable number of tandem repeats (VNTR) polymorphism on bipolar disorder. Neuropsychobiology. 2013;67:116–21.
Yagami T, Koma H, Yamamoto Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the central nervous system. Mol Neurobiol. 2015.
Yagami T, Nakazato H, Ueda K, Asakura K, Kuroda T, Hata S, Sakaeda T, Sakaguchi G, Itoh N, Hashimoto Y, et al. Prostaglandin E2 rescues cortical neurons from amyloid beta protein-induced apoptosis. Brain Res. 2003;959:328–35.
Sanchez-Mut JV, Aso E, Panayotis N, Lott I, Dierssen M, Rabano A, Urdinguio RG, Fernandez AF, Astudillo A, Martin-Subero JI, et al. DNA methylation map of mouse and human brain identifies target genes in Alzheimer’s disease. Brain. 2013;136:3018–27.
Goddard CA, Butts DA, Shatz CJ. Regulation of CNS synapses by neuronal MHC class I. Proc Natl Acad Sci U S A. 2007;104:6828–33.
Stephan AH, Barres BA, Stevens B. The complement system: an unexpected role in synaptic pruning during development and disease. Annu Rev Neurosci. 2012;35:369–89.
Carter EJ, Cosgrove RA, Gonzalez I, Eisemann JH, Lovett FA, Cobb LJ, Pell JM. MEK5 and ERK5 are mediators of the pro-myogenic actions of IGF-2. J Cell Sci. 2009;122:3104–12.
Dong F, Gutkind JS, Larner AC. Granulocyte colony-stimulating factor induces ERK5 activation, which is differentially regulated by protein-tyrosine kinases and protein kinase C. Regulation of cell proliferation and survival. J Biol Chem. 2001;276:10811–6.
Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD. Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998;395:713–6.
Dinev D, Jordan BW, Neufeld B, Lee JD, Lindemann D, Rapp UR, Ludwig S. Extracellular signal regulated kinase 5 (ERK5) is required for the differentiation of muscle cells. EMBO Rep. 2001;2:829–34.
Cavanaugh JE, Jaumotte JD, Lakoski JM, Zigmond MJ. Neuroprotective role of ERK1/2 and ERK5 in a dopaminergic cell line under basal conditions and in response to oxidative stress. J Neurosci Res. 2006;84:1367–75.
Mignot E. A step forward for restless legs syndrome. Nat Genet. 2007;39:938–9.
Ichimura K, Bolin MB, Goike HM, Schmidt EE, Moshref A, Collins VP. Deregulation of the p14ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with G1-S transition control gene abnormalities. Cancer Res. 2000;60:417–24.
Ihara Y, Kihara Y, Hamano F, Yanagida K, Morishita Y, Kunita A, Yamori T, Fukayama M, Aburatani H, Shimizu T, Ishii S. The G protein-coupled receptor T-cell death-associated gene 8 (TDAG8) facilitates tumor development by serving as an extracellular pH sensor. Proc Natl Acad Sci U S A. 2010;107:17309–14.
Ishii S, Kihara Y, Shimizu T. Identification of T cell death-associated gene 8 (TDAG8) as a novel acid sensing G-protein-coupled receptor. J Biol Chem. 2005;280:9083–7.
Kashiwagi M, Hara K, Liu Z, Kageura M, Matsusue A, Sugimura T, Kubo S. Diagnostic approach to drug-screening tests for fatal diabetic ketoacidosis: forensic autopsy of a methamphetamine abuser. Leg Med (Tokyo). 2010;12:192–4.
Onozawa Y, Fujita Y, Kuwabara H, Nagasaki M, Komai T, Oda T. Activation of T cell death-associated gene 8 regulates the cytokine production of T cells and macrophages in vitro. Eur J Pharmacol. 2012;683:325–31.
Onozawa Y, Komai T, Oda T. Activation of T cell death-associated gene 8 attenuates inflammation by negatively regulating the function of inflammatory cells. Eur J Pharmacol. 2011;654:315–9.
Wang LY, Tu YF, Lin YC, Huang CC. CXCL5 signaling is a shared pathway of neuroinflammation and blood-brain barrier injury contributing to white matter injury in the immature brain. J Neuroinflammation. 2016;13:6.
Zaremba J, Skrobanski P, Losy J. The level of chemokine CXCL5 in the cerebrospinal fluid is increased during the first 24 hours of ischaemic stroke and correlates with the size of early brain damage. Folia Morphol (Warsz). 2006;65:1–5.
Tang B, Capitao C, Dean B, Thomas EA. Differential age- and disease-related effects on the expression of genes related to the arachidonic acid signaling pathway in schizophrenia. Psychiatry Res. 2012;196:201–6.
Bos CL, Richel DJ, Ritsema T, Peppelenbosch MP, Versteeg HH. Prostanoids and prostanoid receptors in signal transduction. Int J Biochem Cell Biol. 2004;36:1187–205.
Curie T, Maret S, Emmenegger Y, Franken P. In vivo imaging of the central and peripheral effects of sleep deprivation and suprachiasmatic nuclei lesion on PERIOD-2 protein in mice. Sleep. 2015;38:1381–94.
Comasco E, Nordquist N, Gokturk C, Aslund C, Hallman J, Oreland L, Nilsson KW. The clock gene PER2 and sleep problems: association with alcohol consumption among Swedish adolescents. Ups J Med Sci. 2010;115:41–8.
Mohawk JA, Pezuk P, Menaker M. Methamphetamine and dopamine receptor D1 regulate entrainment of murine circadian oscillators. PLoS ONE. 2013;8, e62463.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34.
Wyss-Coray T, Mucke L. Inflammation in neurodegenerative disease—a double-edged sword. Neuron. 2002;35:419–32.
Goncalves J, Baptista S, Martins T, Milhazes N, Borges F, Ribeiro CF, Malva JO, Silva AP. Methamphetamine-induced neuroinflammation and neuronal dysfunction in the mice hippocampus: preventive effect of indomethacin. Eur J Neurosci. 2010;31:315–26.
McConnell SE, O’Banion MK, Cory-Slechta DA, Olschowka JA, Opanashuk LA. Characterization of binge-dosed methamphetamine-induced neurotoxicity and neuroinflammation. Neurotoxicology. 2015;50:131–41.
O’Shea E, Urrutia A, Green AR, Colado MI. Current preclinical studies on neuroinflammation and changes in blood-brain barrier integrity by MDMA and methamphetamine. Neuropharmacology. 2014;87:125–34.
Najera JA, Bustamante EA, Bortell N, Morsey B, Fox HS, Ravasi T, Marcondes MC. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol. 2016;17:7.
Liu Y, Komohara Y, Domenick N, Ohno M, Ikeura M, Hamilton RL, Horbinski C, Wang X, Ferrone S, Okada H. Expression of antigen processing and presenting molecules in brain metastasis of breast cancer. Cancer Immunol Immunother. 2012;61:789–801.
Marcondes MC, Burudi EM, Huitron-Resendiz S, Sanchez-Alavez M, Watry D, Zandonatti M, Henriksen SJ, Fox HS. Highly activated CD8(+) T cells in the brain correlate with early central nervous system dysfunction in simian immunodeficiency virus infection. J Immunol. 2001;167:5429–38.
Marcondes MC, Morsey B, Emanuel K, Lamberty BG, Flynn CT, Fox HS. CD8+ T cells maintain suppression of simian immunodeficiency virus in the central nervous system. J Infect Dis. 2015;211:40–4.
Kimura T, Griffin DE. The role of CD8(+) T cells and major histocompatibility complex class I expression in the central nervous system of mice infected with neurovirulent Sindbis virus. J Virol. 2000;74:6117–25.
Marcondes MC, Burdo TH, Sopper S, Huitron-Resendiz S, Lanigan C, Watry D, Flynn C, Zandonatti M, Fox HS. Enrichment and persistence of virus-specific CTL in the brain of simian immunodeficiency virus-infected monkeys is associated with a unique cytokine environment. J Immunol. 2007;178:5812–9.
Marcondes MC, Flynn C, Watry DD, Zandonatti M, Fox HS. Methamphetamine increases brain viral load and activates natural killer cells in simian immunodeficiency virus-infected monkeys. Am J Pathol. 2010;177:355–61.
Carroll JA, Striebel JF, Race B, Phillips K, Chesebro B. Prion infection of mouse brain reveals multiple new upregulated genes involved in neuroinflammation or signal transduction. J Virol. 2015;89:2388–404.
Hu S, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Cytokine effects on glutamate uptake by human astrocytes. Neuroimmunomodulation. 2000;7:153–9.
Baker DJ, Blackburn DJ, Keatinge M, Sokhi D, Viskaitis P, Heath PR, Ferraiuolo L, Kirby J, Shaw PJ. Lysosomal and phagocytic activity is increased in astrocytes during disease progression in the SOD1 (G93A) mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci. 2015;9:410.
Blackburn D, Sargsyan S, Monk PN, Shaw PJ. Astrocyte function and role in motor neuron disease: a future therapeutic target? Glia. 2009;57:1251–64.
Deik A, Saunders-Pullman R, Luciano MS. Substance abuse and movement disorders: complex interactions and comorbidities. Curr Drug Abuse Rev. 2012;5:243–53.
Rau TF, Kothiwal AS, Rova AR, Brooks DM, Poulsen DJ. Treatment with low-dose methamphetamine improves behavioral and cognitive function after severe traumatic brain injury. J Trauma Acute Care Surg. 2012;73:S165–72.
Rau TF, Kothiwal AS, Rova AR, Brooks DM, Rhoderick JF, Poulsen AJ, Hutchinson J, Poulsen DJ. Administration of low dose methamphetamine 12 h after a severe traumatic brain injury prevents neurological dysfunction and cognitive impairment in rats. Exp Neurol. 2014;253:31–40.
Bortell NM B, Basova L, Fox HS, Marcondes MC. Phenotypic changes in the brain of SIV-infected macaques exposed to methamphetamine parallel macrophage activation patterns induced by the common gamma-chain cytokine system. Front Microbiol. 2015;6:900.
Spolski R, Leonard WJ. Interleukin-21: a double-edged sword with therapeutic potential. Nat Rev Drug Discov. 2014;13:379–95.
Pan F, Yang TL, Chen XD, Chen Y, Gao G, Liu YZ, Pei YF, Sha BY, Jiang Y, Xu C, et al. Impact of female cigarette smoking on circulating B cells in vivo: the suppressed ICOSLG, TCF3, and VCAM1 gene functional network may inhibit normal cell function. Immunogenetics. 2010;62:237–51.
Wang H, Tompkins LM. CYP2B6: new insights into a historically overlooked cytochrome P450 isozyme. Curr Drug Metab. 2008;9:598–610.
Acknowledgements
The authors want to thank Dr. Antoine Chaillon (Pathology Department, University of California School of Medicine, San Diego, CA) for the discussions and for critically reading the manuscript and to Dr. Kathryn Sue Spencer (Dorris Neurosciences Center, The Scripps Research Institute, San Diego, CA), for helping with the confocal imaging. The authors also want to thank Lorraine Lathrop and Kim Masuda for the administrative assistance. This is the manuscript number 29349 of The Scripps Research Institute.
Funding
TR is supported by KAUST. This work was performed with funding from the NIH/NIDA R01DA036164 to MCGM.
Availability of data and materials
The dataset generated during and analyzed here is available in the Tables, and further information can be obtained from the corresponding author, upon reasonable request.
Authors’ contributions
NB carried the cell cultures, performed the cell stimulations, prepared the cells for gene expression profiling, participated in the discussions, and helped write the manuscript. SS and HSF participated in the conception of the experiments, in the analysis, and in the active discussions, with a focus on the translational significance of findings, and also revised the manuscript. TR was instrumental at providing the instructions and overseeing the systems biology analysis, participated in the discussions, and helped writing the manuscript. MCGM conceived the study, performed the data analysis, organized the discussions, obtained the funding, and wrote the manuscript. All authors read and approved the final manuscript.
Competing interest
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval
All experiments were performed using commercially available primary cells and stimulations were all performed in vitro with no requirement for TSRI Institutional Animal Care and Use Committee oversight review.
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1:
Visualization of gene changes and networks for identifying over-expression patterns and for initializing the analysis of astrocytic gene network behaviors upon Meth exposure. Genes were connected based on pathway, physical and genetic interactions, shared protein domains, or co-expression, using GeneMania and JActiveModules in Cytoscape platform. Highest score nodes were grouped by circular layout. (PNG 6917 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Bortell, N., Basova, L., Semenova, S. et al. Astrocyte-specific overexpressed gene signatures in response to methamphetamine exposure in vitro. J Neuroinflammation 14, 49 (2017). https://doi.org/10.1186/s12974-017-0825-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12974-017-0825-6