
Abstract
Background
FBLIM1 gene has been recently demonstrated to be involved in the pathogenesis of bone sterile inflammation. The aim of the study is to evaluate the prevalence of FBLIM1 gene variants in a cohort of 80 Italian patients with Chronic Non-bacterial Osteomyelitis (CNO).
Methods
The coding regions of FBLIM1 gene were sequenced in a cohort of 80 patients with CNO using DNA extracted from blood lymphocytes, and PCR products were sequenced. Only rare (global MAF < 2%), coding variants detected were considered. Clinical evaluation of patients with rare variants and those without was performed. Fisher’s exact test was used to compare categorical and ordinal data, and Student’s t-test was used to analyze continuous data.
Results
Eighteen out of 80 patients (~ 22%) presented at least one rare coding variant in FBLIM1. Eight patients presented a variant never associated before with CNO. All patients presented classical features of CNO and no statistical difference between patients with presence of FBLMI1 variants and those without were found in terms of clinical manifestation, treatment, and outcome.
Conclusion
Considering the high frequency of rare variants in our CNO cohort, our data seem to confirm a possible role of FBLIM1 in the pathogenesis of CNO suggesting that CNO is a disorder of chronic inflammation and imbalanced bone remodeling.
Similar content being viewed by others


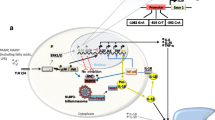
Background
Chronic Non-bacterial Osteomyelitis (CNO) is a rare inflammatory disorder that is characterized by onset of pain, local bone expansion and radiological findings suggestive of osteomyelitis, usually at multiple sites, not related to an infectious disease [1]. Although its pathogenesis remains still unknown, there is consensus about the hypothesis that CNO might be a genetic disease within the spectrum of autoinflammatory disorders [2,3,4]. Although the existence of genes contributing to sporadic CNO has been proposed, their identification is still missing [5].
Cox et al have recently demonstrated that FBLIM1, a gene that codes for a protein involved in the regulation of bone remodeling, could be involved in the pathogenesis of sterile bone inflammation. The authors, via whole-exome sequencing, detected a homozygous mutation in the filamin-binding domain of FBLIM1 in an affected child with consanguineous parents. They also sequenced FBLIM1 in 96 subjects with CNO and found a second patient with a distinct frameshift variant [6].
On this basis, we sequenced the FBLIM1 gene in a cohort of Italian patients with CNO and correlated the results with clinical manifestations.
Methods
This is a multicenter observational study. In the absence of validated international diagnostic criteria, we used the proposed Jansson criteria for CNO diagnosis [7]. The clinical and radiological data were entered into a customized and anonymized database and considered as variables for correlation analysis. Blood samples were collected from patients affected by CNO from 7 Italian rheumatology centers: IRCCS Burlo Garofolo, Trieste; Spedali Civili, Brescia; Anna Meyer Children’s Hospital, Florence; Bambino Gesù Children’s Hospital, Rome; G. Di Cristina Children’s Hospital, Palermo; “Giovanni XXIII” Pediatric Hospital, Bari and IRCCS Giannina Gaslini, Genoa. Written informed parental consent, according to the approved protocol of the IRCCS “Burlo Garofolo” ethics committee (n°27/14) was obtained for genetic analysis. Blood collected for DNA analysis was taken specifically for this study during a routine blood sampling. After standard genomic isolation, DNA was used for Sanger sequencing on entire coding and flanking regions of FBLIM1. All sequences were analyzed with codon code aligner software (V.7.1.1 version). Only rare (global MAF < 2%), coding variants detected were considered. To predict if each variation could be harmful for the protein function, we used the following softwares: Polyphen2 (http://genetics.bwh.harvard.edu/pph2/); SIFT (http://sift.jcvi.org); MUTATION ASSESSOR (http://mutationassessor.org/r3/); Human Splicing Finder (http://www.umd.be/HSF/) and TRANSFAC (http://gene-regulation.com/pub/databases.html#transfac). Controls were taken from the gnomAD, a worldwide database of hundreds of thousands of individuals with no manifested pathologies, stratified by regions of origin.
Clinical evaluation between patients with and without rare variants was performed. Fisher’ s exact test was used to compare categorical and ordinal data, and Student’s t test was used to analyze continuous data.
Results
Eighty patients diagnosed with CNO were enrolled, 52 females (65%) and 28 males (35%). Bone pain was the most common clinical complaint; median age at onset was 9 years (range 4–14 years). We found a median number of six bone localizations per patient; 9 patients had a single lesion. Most lesions were located to the long bones (65%), pelvis (48%) and spine (37%). Three patients presented skull lesions (they underwent bone biopsy to exclude malignancy). Five patients (5.8%) had a first-degree relative affected by an autoimmune disorder. Thirty patients (35%) had a comorbidity; among these, 18 (22.5%) had skin manifestations such as psoriasis, pustulosis or severe acne, 4 (5%) had inflammatory bowel disease. No consanguinity nor family history for CNO was reported.
Sanger sequencing was conducted in all patients. 18 out of 80 (22,5%) patients presented at least one rare (≤ 0.02) variant in FBLIM1 gene. In particular, 15 patients presented 1 rare variant; 2 patients presented 2 rare variants and 1 patient had 5 rare variants. In total, 11 rare variants were found (7 out of 11 variants presented a MAF ≤ 0.01).
Table 1 displays the variants /SNPs identified. 5 out of 11 variants were in the coding regions and 6 were intronic. Among the SNP in the coding region 3 are missense and 2 synonymous variants. Two of the missense (Arg38Gln and Gly311Arg) and one of the intronic variants (c.250 + 32 C > A) have been previously reported in CNO by Cox et al [9]. The other eight variants have never been associated with CNO.
Patient P18 harbored 5 FBLIM1 variants: two missense variants (rs146575757 Arg38Gln and rs114077715 Gly311Arg), two intronic (rs201006671 and rs76050903) and one synonymous (rs61733331) variant. By parental sequencing we verified that the child inherited from his mother the allele with the missense Arg38Gln, the two intronic and the synonymous variant while he inherited from his father the allele with the only Gly311Arg missense variant, found also in heterozygosis in P16. In patient P13 we identified in homozygosis the intronic variant rs187479896 and in heterozygosis the rs140170023, a synonymous variant present also in heterozygosis in other 9 patients (P2, P3, P4, P5, P6, P8, P9, P10, P13 and P15). In patient P3, in addition to the synonymous variant, the SNPs rs41310367 was also detected in heterozygosis. An intronic heterozygous variant was identified also in P11. Finally, P12 carried the missense variant Arg22His (rs540511146), P7 the variant rs766409425 and patients P1, P14, and P17 the intronic variant rs144567113.
All the 11 identified variants, especially those never described before and so orphaned of functional studies were analyzed “in silico” using specific software; results are shown in Table 2. Moreover, according to dbSNP, all the variants identified are extremely rare in healthy controls examined (supplementary table), thus enforcing their possible pathogenic roles.
All patients presented classical features of CNO and no statistical differences between patients with or without FBLIM1 variants were found in terms of gender prevalence, positive family history, age at onset, number of sites involved, presence of fever, arthritis and skin involvement as well as remission at the end of follow-up (Table 3).
Discussion
CNO is a rare pediatric autoinflammatory bone disease and several factors such as family pedigree and syndromic monogenic forms of CNO suggest a strong genetic component. Moreover, the association between the presence of polymorphisms of the IL-10 promoter with CNO pathogenesis has been reported [10]. It has been also demonstrated that mutation of the pstpip2 gene in mice results in an autoinflammatory disease very similar to human CNO [8]. However, candidate genes including PSTPIP1, CARD15/NOD2, and IL1RN, were not associated with CNO in humans when analyzed in small cohorts [11, 12].
Cox et al. recently showed that recessive mutations in FBLIM1 contribute to the pathogenesis of CNO [9]. FBLIM1 codes for Filamin-binding LIM protein 1 (FBLP1 or migfilin), a filamin-binding protein involved in the regulation of bone remodeling [13].
FBLP1 is a key regulator of the cytoskeleton, as it is recruited to cell-matrix contacts in response to adhesion and colocalizes with beta-catenin at cell-cell junctions in epithelial and endothelial cells. Through interactions with multiple binding partners, including filamin, FBLP1 links the cell adhesion structures to the actin cytoskeleton [14]. FBLP1 competes with integrin β for filamin binding to promote integrin activation in neutrophils as well as bone homeostasis [15]. Therefore, a mutation in the filamin-binding domain of FBLP1 may disrupt FBLP1-FLN binding, resulting in aberrant integrin activation in neutrophils and leading to sterile inflammation.
In our cohort of 80 CNO patients we found a rare coding variant of FBLIM1 in 18 patients. Three of these variants (rs146575757, rs41310367 and rs114077715) were previously described by Cox et al. [6]. The missense variant Arg38Gln (rs146575757) is in the filamin-binding domain (exon 3), so it may be causative of neutrophil activation. The second variant (rs41310367) is centrally located in the middle of an enhancer, in a STAT3 binding region and in an NR4A2 recognition site, reported to be active in several cell lines, including osteoblasts, so it may disrupt balance between osteoclasts and osteoblasts activity leading to bone remodeling. The other missense variant Gly311Arg on the last exon is localized in the third LIM domain of the protein, a small protein -protein interaction domain, containing two zinc fingers.
For the further eight variants identified in our study no literature data are available about their association with the disease or their functional impact. These variants are all rare (MAF < 2%) in the general population, and extremely rare in the case of rs540511146 (MAF = 0.00011). The high prevalence of FBLM1 rare variants in our CNO cohort may support a role in the pathogenesis of the disease.
Considering the intronic variants, they were predicted to possibly alter binding sites for splicing factors, in particular for the serine/arginine-rich protein (SR) specific binding sites. We observed by Human Splicing Finder a prediction in which the substitution of a single nucleotide in that specific genomic region leads to a loss and/or possible new sequence recognition by these factors and a possible creation a sequence for a new splice site. The in silico analysis and its predictions could address the research towards functional studies to verify the pathogenicity of the variants.
No statistical association was found between patients with and without FBLIM1 gene variants, suggesting that FBLIM1 might be considered a non-specific predisposing factor to CNO in a subgroup of patients.
Conclusions
CNO remains a not completely understood disease but it probably belongs to the family of autoinflammatory diseases. A unique causing gene was not found yet. However, our data seem may support the fact that some FBLIM1 variants might increase the susceptibility to the CNO pathogenesis.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- CNO:
-
Chronic Non-bacterial Osteomyelitis
References
Taddio A, Ferrara G, Insalaco A, Pardeo M, Gregori M, Finetti M, et al. Dealing with Chronic Non-Bacterial Osteomyelitis: a practical approach. Pediatr Rheumatol Online J. 2017;15:87.
Majeed HA, Kalaawi M, Mohanty D, Teebi AS, Tunjekar MF, al-Gharbawy F, et al. Congenital dyserythropoietic anemia and chronic recurrent multifocal osteomyelitis in three related children and the association with sweet syndrome in two siblings. J Pediatr. 1989;115:730–4.
Caorsi R, Picco P, Buoncompagni A, Martini A, Gattorno M. Osteolytic lesion in PAPA syndrome responding to anti-interleukin 1 treatment. J Rheumatol. 2014;41:2333–4.
Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med. 2009;360:2426–37.
Golla A, Jansson A, Ramser J, Hellebrand H, Zahn R, Meitinger T, et al. Chronic recurrent multifocal osteomyelitis (CRMO): evidence for a susceptibility gene located on chromosome 18q21.3-18q22. Eur J Hum Genet. 2002;10:217–21.
Cox AJ, Darbro BW, Laxer RM, Velez G, Bing X, Finer AL, et al. Recessive coding and regulatory mutations in FBLIM1 underlie the pathogenesis of chronic recurrent multifocal osteomyelitis (CRMO). PLoS One. 2017;12:e0169687.
Jansson AF, Muller TH, Gliera L, Ankerst DP, Wintergest U, Belohradsky BH, et al. Clinical score for nonbacterial osteitis in children and adults. Arthritis Rheum. 2009;60:1152–9.
Ferguson PJ, Bing X, Vasef MA, Ochoa LA, Mahgoub A, Waldschmidt TJ, et al. A missense mutation in pstpip2 is associated with the murine autoinflammatory disorder chronic multifocal osteomyelitis. Bone. 2006;38:41–7.
Cox AJ, Ferguson PJ. Update on the genetics of nonbacterial osteomyelitis in humans. Curr Opin Rheumatol. 2018;30:521–5.
Hamel J, Paul D, Gahr M, Hedrich CM. Pilot study: possible association of IL10 promoter polymorphisms with CRMO. Rheumatol Int. 2012;32:555–6.
Beck C, Girschick HJ, Morbach H, Schwarz T, Yimam T, Frenkel J, et al. Mutation screening of the IL-1 receptor antagonist gene in chronic non-bacterial osteomyelitis of childhood and adolescence. Clin Exp Rheumatol. 2011;29:1040–3.
Huber AM, Lam PY, Duffy CM, Yeung RS, Ditchfield M, Laxer D, et al. Chronic recurrent multifocal osteomyelitis: clinical outcomes after more than five years of follow-up. J Pediatr. 2002;141:198–203.
Xiao G, Cheng H, Cao H, Chen K, Tu Y, Yu S, et al. Critical role of filamin-binding LIM protein 1 (FBLP-1)/migfilin in regulation of bone remodeling. J Biol Chem. 2012;287:21450–60.
Wu C. Migfilin and its binding partners: from cell biology to human diseases. J Cell Sci. 2005;118:659–64.
Das M, Ithychanda SS, Qin J, Plow EF. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PLoS One. 2011;6:e26355.
Acknowledgements
Not applicable.
Funding
The manuscript was supported by Institute for Maternal and Child Health - IRCCS “Burlo Garofolo” (grant number RC 27/14).
Author information
Authors and Affiliations
Consortia
Contributions
Andrea Taddio, Adamo Pio d’Adamo, Giovanna Ferrara, Rolando Cimaz and Marco Gattorno: conceptualized and designed the study, drafted the initial manuscript, and approved the final manuscript as submitted. Anna Monica Bianco and Martina La Bianca performed the Sanger analysis and the evaluation of the variants. Serena Pastore, Alberto Tommasini, Marco Cattalini, Gabriele Simonini, Virginia Messia, Antonella Insalaco, Clotilde Alizzi, Manuela Pardeo, Martina Finetti and Francesco La Torre carried out the initial analyses, reviewed and revised the manuscript, and approved the final manuscript as submitted. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study protocol and genetic tests were approved by IRCCS “Burlo Garofolo” ethics committee (n°27/14).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Supplementary table
. Frequencies (Global, European, South Asian, East Asian and African) of FBLIM1 gene variants in healthy control people (from GnomAD v2.1.1 controls dataset).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
d’Adamo, A.P., Bianco, A.M., Ferrara, G. et al. High prevalence of rare FBLIM1 gene variants in an Italian cohort of patients with Chronic Non-bacterial Osteomyelitis (CNO). Pediatr Rheumatol 18, 55 (2020). https://doi.org/10.1186/s12969-020-00447-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12969-020-00447-4