
Abstract
Hedgehog (Hh) signaling pathway plays an essential role during vertebrate embryonic development and tumorigenesis. It is already known that Sonic hedgehog (Shh) pathway is important for the evolution of radio and chemo-resistance of several types of tumors. Most of the brain tumors are resistant to chemotherapeutic drugs, consequently, they have a poor prognosis. So, a better knowledge of the Shh pathway opens an opportunity for targeted therapies against brain tumors considering a multi-factorial molecular overview. Therefore, emerging studies are being conducted in order to find new inhibitors for Shh signaling pathway, which could be safely used in clinical trials. Shh can signal through a canonical and non-canonical way, and it also has important points of interaction with other pathways during brain tumorigenesis. So, a better knowledge of Shh signaling pathway opens an avenue of possibilities for the treatment of not only for brain tumors but also for other types of cancers. In this review, we will also highlight some clinical trials that use the Shh pathway as a target for treating brain cancer.
Similar content being viewed by others
Background
Hedgehog (Hh) is one of few of signaling pathways that is frequently used during development for intercellular communication. Hh is important for the organogenesis of almost all organs in mammals, as well as in regeneration and homeostasis. Further, Hh signaling is disrupted in diverse types of cancer [1, 2]. The vertebrate Hh signaling is not entirely dependent on an extremely specialized organelle, the primary cilium (PC), unlike other essential developmental signaling pathways. The PC is an organelle, microtubule-based, that emerges from the cell surface of most vertebrate cells. This organelle is important to process several cellular signals and/or extracellular environmental changes necessary for animal development, as Wingless (Wnt), Platelet-derived growth factor (PDGF), Shh, and Notch [3].
There are three mammalian Hh proteins, Shh, Indian-Hedgehog (Ihh), and Desert-Hedgehog (Dhh). Shh and Ihh have important, and sometimes coinciding, functions in several tissues. Shh has particularly marked roles in nervous system cell type specification and limbs patterning, whereas Ihh has important roles in skeletal development, mainly endochondral ossification. Dhh is restricted to the gonads including granulosa cells of ovaries and sertoli cells of testis [4,5,6]. The best-studied function of Shh, during mouse embryogenesis, is to instruct neural progenitors patterning, in which it is possible to distinguish six different cell types based on molecular markers, such as interneurons progenitors and motor neurons, that differentiate due to a gradient of Shh [7, 8].
Several evidences demonstrate that embryogenesis and tumorigenesis have common characteristics, where both processes depend on coordinated mechanisms of proliferation, differentiation and migration [9]. Vital signaling pathways for embryonic development and organogenesis are modulated in tumorigenesis. Aberrant activation of Hh signaling has been shown to be associated with the formation of brain tumors, as well as its cross talking with other pathways like transforming growth factor beta (TGFßs), Wnt, Notch and Shh [10,11,12]. Moreover, several studies have investigated the role of Hh-Gli (Gli means glioma-associated oncogene homologue) signaling in cancer initiating stem cells (CSCs) and suggested that it regulates self-renewal and tumorigenic potential [13]. This review focused on updating the role of these molecules in brain tumorigenesis as well as suggesting new therapeutic strategies/clinical trials using the Shh pathway as a potential future treatment.
Shh signaling pathway components in tumorigenesis
The canonical pathway
Activation of Shh pathway can happen in two major ways: 1. canonical signaling: by ligand-dependent interaction or through receptor-induced signaling and 2. non-canonical signaling, when there’s a mechanism of activation downstream of smoothened (Smo) (Fig. 1) [14].

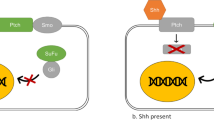
The Canonical activation of Shh pathway in vertebrates. The activation occurs by ligand-dependent interaction when Shh binds to Ptch at the cell membrane. In response to this binding, Ptch no longer inhibits Smo, which accumulates at the PC and initiates the downstream signaling pathway cascade. So, Smo regulates the Gli processing and activation at the PC. When Gli is activated, it translocates to the nucleus, where it activates target genes. (Diagram by Carballo, VC). (Adapted from Robbins et al., 2012) [54]
The Shh canonical signaling occurs when the glycoprotein Shh binds and inactivates the 12-transmembrane protein Patched (Ptch1). In the lack of the ligand Shh, the activity of the 7-transmembrane protein Smo is inhibited by Ptch1, so Shh protein binding Ptch1 regulates Smo activity [15, 16]. Smo is a GPCR-like (G protein–coupled receptor) protein, and the translocation into the cilia membrane is a requisite for Gli activation [3, 17]. In response to Shh signaling, Ptch1 inhibition of Smo at the PC is abolished, when Ptch1 is internalized and degraded [18]. So, after Ptch1 degradation, Smo accumulates at the PC where is activated and stabilized by initiating the Shh downstream signaling cascade [18]. This downstream signaling cascade results in the translocation of Gli family proteins to the nucleus that begins the transcription of target genes, including Ptch1 and Gli1, in a negative and positive feedback loop, respectively (Fig. 1) [14]. Furthermore, Gli translocation to the nucleus also induces protein modulation of Wnt and Noggin [16, 19, 20]. Patched 2 (Ptch2) is another receptor for Shh that shares approximately 54% homology with Ptch1. However, the expression and signaling of Ptch2 is different from Ptch1, having decreased ability to inhibit Smo in absence of Shh ligand [21].
The Gli1 gene was initially cloned as an amplified oncogene of a malignant glioma and then characterized as a transcription factor of the hedgehog signaling pathway [22, 23]. Three Gli proteins (Gli1, Gli2 and Gli3) are zinc-finger transcription factors and are expressed in vertebrates, in overlapping and partially redundant domains. These three proteins are Shh-dependent, where only Gli1 occurs as a full-length transcriptional activator, while Gli2 and Gli3 act as either a negative or positive regulators (Gli2A - Gli2 activated or Gli2R - Gli2 repressor and Gli3A - Gli3 activated or Gli3R - Gli3 repressor, respectively) of the pathway which is determined by post-transcriptional and post-translational processing [24, 25]. Moreover, the change of Gli3A to Gli3R form is favored with respect to Gli2. Consequently, Gli2 has mainly an activator transcriptional behavior, while Gli3 acts as a repressor [26]. It has already been demonstrated that Gli2 can accumulate in the primary cilium and controls transcriptional activation, in response to Shh ligand binding, overcoming thereby the negative regulation of Gli3 [27].
The Gli3 has also a very important function in regulating Shh signaling. Without Shh, Gli3 has a repressor form (Gli3R). When Shh binds to Ptch and activates Smo, Smo converts Gli3R into an activated form (Gli3A). So, Gli3 works as a transcriptional factor with a dual function. The ratio of Gli3R/Gli3A is directly related to the control of several processes during organogenesis, such as digit types and number [28, 29].
Shh signaling pathway can also be controlled by Supressor of Fused (SUFU) (Fig. 2) [30]. SUFU is a negative regulator of the Shh signaling pathway, acting on the Gli transcription factors. When Shh ligand is not present, SUFU binds directly the Gli proteins and inhibits their translocation to the nucleus, preventing the pathway activation [31]. However, the specific mechanisms concerning Gli inactivation by SUFU are not completely understood, but the full-length Gli proteins are converted to a C-terminal shorten repressor form: Gli-R. This truncated form of Glis is then partially degraded after subsequent phosphorylation by glycogen synthase kinase 3 beta (GSK3β), casein kinase I (CK1) and protein kinase A (PKA) [26]. Gli proteins retained at the cytoplasm by SUFU are then degraded or processed and thereby inhibiting Shh signaling [32]. When Gli-R moves to the nucleus, it represses SHH target genes including Ptch1 and Gli1 itself. When Shh pathway is activated, it is necessary that SUFU inhibition of Glis occurs by hyper-phosphorilation of SUFU [33]. Therefore, it has been previously demonstrated that several protein kinases, such as PKA and protein kinase C (PKC), CK1, mitogen activated protein kinase kinase (Mek1), GSK3, Phosphoinositide-3 kinase (PI3K), or dual specificity Yak1-related kinase (DYRK1) can modulate this pathway at several levels [33,34,35,36,37,38,39] (Fig. 2). This mechanism of regulation of the Shh pathway by ubiquitination-related posttranslational modifications of the Gli transcription factors leads to massive protein degradation or a proteasome-dependent proteolytic cleavage [40]. This process was first identified in Drosophila, but it was also demonstrated in vertebrates [41]. It is important to note that mutations in Ptch and SUFU, which are the negative regulators of Shh signaling, are linked to tumorigenesis, although the exact mechanism is unknown [42]. It was demonstrated in knockout mice, that the loss of SUFU is enough to activate the pathway without the support of the receptors [43, 44]. This constitutive Shh signaling activation in medulloblastoma (MB) is not sufficient to induce tumorigenesis, because a second tumor suppressor must be inactivated, such as p53 [45].

The non-canonical activation of Shh pathway. The non–canonical activation occurs through Gli-independent mechanisms and it can be of two types. A) Type I which modulates Ca2+ and actin cytoskeleton (left). When Shh binds the receptor Ptch, Smo is no longer inhibited and couple Gi proteins (G) and small GTPases RhoA and Rac1 activated. In addition, Smo stimulates calcium (Ca2+) release from the endoplasmic reticulum (ER) and PLC-γ-catalyzed the opening of IP3-dependent channels by the generation of IP3. B) Type II which is independent on Smo. When Shh binds Ptch, the interaction of Ptch with cyclin B1 is disrupted, leading to an increase in cell proliferation and survival (right). (Diagrams by Carballo, VC). (Adapted from Robbins et al., 2012) [54]

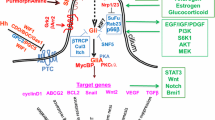
The crosstalk between Shh pathway and others. Shh signaling pathway can crosstalk with several pathways, especially EGF, Wnt and TGF-β. Here we can observe the Shh signaling pathway in blue, the EGF pathway in orange, the Wnt pathway in, and TGF-β pathway in green. The crosstalk between these pathways and Shh occurs at different moments, and it becomes more important to understand this molecular interaction in order to search for new therapeutical drugs. (Diagrams by Carballo, VC). (Adapted from Matias et al., 2017; Berg and Soreide, 2012 and https://www.mycancergenome.org/content/molecular-medicine/pathways/TGF-beta-signaling) [128, 129]
Besides ubiquitination, mainly of Gli3, to control Shh pathway, it was also demonstrated that Gli1 and Gli2 can be acetylated at lysine 518 and 757, respectively [46]. The mechanism of deacetylation of these proteins is mediated by the enzyme histone deacetylase 1 (HDAC1), which promotes transcriptional activation of the pathway. This activation is turned off by the degradation of HDAC1, which sustains a positive autoregulatory loop, when Shh is present. This degradation is mediated through an E3 ubiquitin ligase complex [46].
Shh signaling pathway is a valid therapeutic goal in a broad range of cancers, such as pancreas, prostate, breast and brain tumors. We focus here on brain tumors. The transcriptomics data on 149 clinical cases of The Cancer Genome Atlas-Glioblastoma (GBM) database showed a robust correlation between PTCH1 and GLI1 mRNA expression as an indication of the canonical Shh pathway activity in this malignancy. The expression of GLI1 mRNA varied in three orders of significance among the GBM patients of the same cohort, demonstrating a single continuous distribution different from the discrete high/low-GLI1 mRNA expressing clusters of MB [47]. Furthermore, it has already been well-established that tumor microenvironment plays an important role in controlling GBM pathology and their drug-resistance mechanisms [48]. Cells from the tumor microenvironment usually secrete inflammatory cytokines, growth factors [49,50,51] and other proteins that can activate Shh signaling in a typical or atypical manner (canonical or non-canonical) [52]. It was demonstrated that in the tumor microenvironment the endothelial cells provide Shh to activate the Hh signalling pathway in GBM cells, thereby promoting glioma stem cells (GSC) properties and tumor propagation [53].
Non-canonical Shh signaling
The “non-canonical Shh signaling” usually occurs through Gli-independent mechanisms. The Gli-independent mechanisms include two types: Type I is downstream of Smo, which modulates Ca2+ and actin cytoskeleton and type II is independent of Smo and increases cell proliferation and survival [54]. The non-canonical Shh signaling can regulate chemotaxis and cell migration through actin rearrangement. Additionally, it can stimulate cell proliferation via calcium-induced extracellular signal-regulated kinases (ERK) activation and activate Src family kinase, which is required axon guidance [54,55,56].
Some studies emerged mainly in tumor cells concerning the non-canonical Shh signaling in the ten last years. However it has not been completely elucidated how Smo selects between canonical or non-canonical routes. Usually the non-canonical route occurs when Smo couples to Gαi in vertebrates and modulates Ca2+ flux, Ras homolog gene family, member A (RhoA) and Rac activation and Warburg-like metabolism [56,57,58].
Interestingly, it was first believed that only Shh canonical signaling occurs when Smo enters the PC [59], and if Smo does not route through PC, it signals through a non-canonical pathway [17]. However, it was recently demonstrated that non-canonical Shh signaling leads to acetylation of α-tubulin via Smo-mediated calcium which increases in a primary cilia-dependent manner in mouse embryonic fibroblasts [17]. There are rare studies of this type of signaling associated with tumorigenesis and none with brain tumors. A ligand-independent Smo mutant resulted in tumors over-expressing Shh that show pronounced chromosomal instability and smoothened-independent up-regulation of Cyclin B1, a putative non-canonical branch of the Shh pathway in lung cancer. These results strongly support an autocrine, ligand-dependent model of Shh signaling in Small Cell Lung Cancer tumorigenesis and explain a new role for non-canonical Shh signaling through the induction of chromosomal instability [60]. Moreover, Hh signaling has an important role on the switch of hypoxia-induced pancreatic cancer epithelial to mesenchymal transition and invasion in a ligand-independent manner [61].
Recently, it was demonstrated that the intraflagellar transport protein 80 (IFT80) promotes Hh canonical signaling via activation of Hh-Smo-Ptch1-Gli signaling pathway during osteoblasts (OBs) differentiation. On the other hand, when this occurs, the non-canonical Hh signaling is inhibited via Hh-Smo-Gαi-RhoA-stress fibre signaling, demonstrating that non-canonical Hh signaling negatively regulates OBs differentiation [62]. Moreover, this study demonstrated that at least in OBs differentiation and bone formation, IFT80 is essential for the balance of the non-canonical and canonical Hh signaling pathways [62].
However, the researchers are still unveiling the mystery of the non-canonical Smo signaling axis, as well as how Smo selects between canonical and non-canonical routes.
Shh interaction with others pathways
It is already known that Shh signaling is very important for embryonic development and in adults, deregulation or mutation of this pathway plays an important role in both differentiation and proliferation, inducing tumorigenesis [63, 64]. Furthermore, CSCs follow the same pathways than normal stem cells such as Wnt, Shh, Notch and others and are also present during embryonic development, organogenesis and tumorigenesis [10,11,12].
Emerging evidence suggests that Shh signaling pathway can interact with other signaling components, such as TGF-β, epidermal growth factor receptor (EGFR), K-Ras, PKA, Notch, and Wnt/β-catenin (Fig. 3) [65, 66]. Furthermore, it has been suggested that more than one of these pathways are active, in different types of tumors, at the same time [16].
The Shh and Wnt pathways could interact in two ways: 1. through Gli1 and Gli2, which have been shown to regulate positively the expression of secreted frizzled-related protein-1 (sFRP-1) and thus inhibiting Wnt ligands and/or their receptors [67] and 2. through downstream GSK3β (an essential component of complexes that inhibit Shh and WNT morphogenetic pathways) [68]. GSK3β, can act as a positive regulator of Shh signaling by phosphorylating SUFU and promoting the release of SUFU from Gli, at least when the pathway is active [69]. It has already been demonstrated that in mice without normal APC function (citoplasmatic degradation and nuclear exporting of β-catenin) that SUFU negatively regulates Tcf-dependent transcription by reducing nuclear β-catenin levels [70]. So, Shh can regulate Wnt signaling. This crosstalk between Shh and Wnt has also been demonstrated in medulloblastoma cells, where the loss of SUFU activates both pathways, inducing excessive proliferation and tumorigenesis [71]. Besides, Wnt signaling can also increase Shh pathway activity, as β-catenin may potentially affect the Gli1 transcriptional activity via TCF/LEF in an independent manner [66]. Interestingly, in gastric cancer, the Shh signaling pathway activation seems to be inversely correlated with the level of Wnt pathway activation. It was observed that Gli1 overexpression suppressed Wnt transcriptional activity, nuclear β-catenin accumulation and proliferation of gastric cancer cells [72].
It is well established that aberrant RAS activation has a protagonist role in tumorigenesis, and activating RAS mutation occurs in 30% of all human cancers [73]. It has been demonstrated that activation of the RAS/MAPK pathway (KRAS), induced by divers upstream signals and converging at the level of Gli transcription factors, is important in promoting cancer development during pancreatic tumorigenesis [74]. Another pathway that has been demonstrated to interact with Shh is the ERK signaling pathway, which controls Gli transcription factor function in Shh signaling, when stimulated by exogenous ligands (like basic fibroblast growth factor -bFGF) [39].
In addition to Wnt/βcatenin and KRAS, TGF-β/TGF-βR, EGFR, and platelet-derived growth factor receptor α (PDGFRα) can also cooperate with the canonical Shh pathway [39, 66, 75,76,77,78]. There is an important increase in Gli1 and Gli2 expression induced through the activation of TGF-β/TGF-βR/Smads in pancreatic cell lines. Furthermore, these cells were resistant to Shh inhibition, but the pharmacologic blockade of TGF-β signaling leads to repression of cell proliferation accompanied by a reduction in Gli2 expression [79].
Another signaling pathway that crosstalk with Shh and contributes to tumorigenesis is EGFR signaling. The stimulation of EGFR/RAS/RAF/MEK/ERK in different cancer cell lines such as gastric, and pancreatic cancer cell lines and was able to activate the Gli transcription factor and selective transcriptional modulation of Gli target gene expression [76,77,78, 80]. However, it was observed in MB cells, a crosstalk mechanism where EGFR signaling silences proteins acting as negative regulators of Hh signaling, as an ERK and AKT-signaling independent method. Reciprocally, a high-level synergism was also observed, due to a significant and strong up-regulation of several canonical EGF-targets. Synergistic outcomes between EGFR and Hh signaling can selectively promote a shift from a canonical HH/GLI profile to a gene profile specific target modulated. It indicates that there are more diffuse, yet context-dependent (i.e. cancer-dependent) interactions, between growth factor receptors and HH/GLI signaling in human tumorigenesis [81].
So, it is becoming more and more evident that the integration of these signaling pathways, which are important for embryonic morphogenesis, may support a more malignant behavior by tumor cells and consequently maintain the tumorigenesis of diverse aggressive tumors, such as pancreas, prostate, breast and brain [78, 80]. The need to understand the role of these pathways in tumorigenesis is becoming increasingly evident, mainly the molecular crosstalk between them, as it is an important consideration for the development of HH-targeting agents and the appropriate selection of a class of inhibitors for therapeutic intervention [82]. Furthermore, it is valid to be proposed in the future treatments of Shh-dependent tumors using inhibitors of Akt, PI3K, MEK, ERK, Wnt, EGFR and TGF-β [38, 66, 78,79,80].
Hh inhibitors and clinical trials
The importance of stem cells in brain tumors
Nowadays, several studies support the hypothesis that malignant tumors are initiated and maintained by CSCs. Although the origin of the CSCs in human tumors is not fully understood, it is already well established that these cells are responsible for the chemo and radioresistance of the most malignant tumors [49, 50]. The recurrence of the tumor is usually due the existence of these cells in the tumor bulk [49, 50]. Moreover, studies have demonstrated that CSCs could de-differentiate from a more differentiated cancer cell present in the tumor mass that acquires self-renewal properties, clonal tumor initiation capacity and clonal long-term repopulation potential, perhaps as a result of epithelial-to-mesenchymal transition (EMT) [83,84,85].
The hypothesis that the existence of CSCs initiates malignant tumor came from the observation that tumor cells, like adult tissues, originate from cells that can self-renew. Furthermore, that these cells also are able to differentiate into cell forming the tumor bulk [86]. In the adult tissue, these cells are the adult stem cells that are tissue-specific and multipotent, being able to differentiate between all cell types of the tissue of origin [86].
In the adult brain, it is already well established that the existence of a neurogenic niche, which is extremely dynamic and complex microenvironment where new glial cells and neurons are generated when necessary from the stem or progenitor cells [87]. This neurogenic niche has a very important role, as it provides signals that regulate whether the stem cells should differentiate, remain quiescent, or actively divide, controlling the self-renewal properties in this way and maintaining neural stem/progenitor cell populations [87]. These neural stem cells (NSCs) are found in two main niches in adult brain, in the lateral ventricles (ventricular-subventricular zone (V-SVZ)) and in the hippocampus (subgranular zone (SGZ)), and these microenvironments ensures the self-renew and multipotent properties [88, 89].
It is interesting to note that Shh is very important for determining cell fate and patterning during embryo development, having a mitogenic effect on proliferative cells throughout development [90]. Recently it was demonstrated that in the adulthood, the level of Shh signaling pathway activation played an important role to regulate the balance between quiescent and activated NSCs. Moreover, when the Shh pathway was genetically activated the number of quiescent NSCs increased and the pool of activated NSCs decreased [91]. However, there was an initial transitory period over the short term when activated NSCs are actively proliferating, apparently when their G1 and S-G2/M phases were short [91].
Taking into account that in GBM, the Shh pathway is usually upregulated, affecting GBM CSC proliferation and self-renewal [87, 92], this discovery opened an avenue for clinical trials that managed not only to stop the tumor to growth but also the tumor to relapse after surgery.
The importance of Shh and MGMT interaction in clinical trials
Nowadays, the standard treatment for most brain tumors comprises resection of the majority of the tumor mass, followed by chemo- and radiotherapy [49, 93], being Temozolmide (TMZ) and radiotherapy being the gold standard treatment [94]. TMZ is an alkylating agent prodrug, and its effect on tumor cells is to methylate the O6 residues of guanine preventing DNA duplication during cell proliferation and inducing cell death and apoptosis [95]. However, the DNA repair enzyme O-6-methylguanine-DNA methyltransferase (MGMT) is able to reverse the effects of alkylating agents as TMZ [96,97,98]. The MGMT promoter methylation is directly related to patient’s prognosis, as low promoter methylation status induces a high MGMT expression and a shorter survival due to a remarkable chemoresistance. On the other hand, a higher promoter methylation status predicts a good response to TMZ chemotherapy, as the MGMT enzyme is downregulated, resulting in longer survival for the patients [99, 100]. Therefore, studies are being done in order to control and impairs the MGMT enzyme activity in chemoresitant tumors It is interestingly to note that many DNA repair proteins could be potential targets for inhibiting cancer cells without affecting normal cells; as they usually are upregulated in several chemorresistant cells and cancers [101].
The most malignant tumors are also highly mutated and present CSCs, which make them difficult to treat. So efforts are being made in order to bypass the chemoresitance in tumors. As written above, the Shh pathway is upregulated in CSCs [87, 102]. Moreover, these cells express also usually high levels of MGMT, and therefore they are involved in chemotherapy resistance and are responsible for tumor recurrence [103]. Emerging evidences are demonstrating that Shh signaling pathway could regulate MGMT expression and chemoresistance to TMZ in human GBM. Moreover, this regulation occurred independently from MGMT promoter methylation status, offering a probable target to reestablish chemosensitivity to TMZ in tumor that developed chemoresitance [104]. Furthermore, it is believed that Gli1 expression is also responsible for chemoresistance in gliomas and that it’s overexpression is related to tumor recurrence after treatment. So in the other hand, when Shh pathway is inhibited, [105] the sensitivity to chemotherapy improves by down-regulating many genes related to apoptosis, cell survival, multi-drug resistance, and especially MGMT [102, 106,107,108,109].
Smo-based inhibitors
Presently, there are several Hh inhibitors employed in clinical trials for different types of brain tumors (www.clinicaltrials.gov) (see Table 1). SMO is the principal target for the development of Shh-pathway inhibitors; however preclinical and clinical studies have demonstrated that the use of Smo inhibitors induces the development of mutations that lead to treatment resistance [110, 111].
The first clinical trial, targeting Smo and so using Shh pathway inhibitor as therapy, considered several patients with recurrent or metastatic basal cell carcinoma (BCC). At that time, a preliminary study was performed with cyclopamine in a topical application and cream formulation. This study has revealed that the tumors rapidly regressed in all cases without adverse effects, and the normal skin and putative stem cells exposed to cyclopamine were preserved [112]. Cyclopamine is a natural steroidal alkaloid derived from Veratrum californicum which inhibits the cellular response to Shh signaling by antagonizing the proto-oncogene SMO [113]. The histological and immunohistochemical analyses from this study have also indicated that the topical cyclopamine application resulted in an inhibition of the proliferation and induced the apoptotic death of tumor cells [112]. In 2006, Herman started a Phase III clinical trial to assess cyclopamine as a chemo-preventive agent to inhibit the recurrence of BCC following surgical resection. At that moment, neither a phase I nor a phase II clinical trials have evaluated the possible side effects of cyclopamine in human subjects, so the patients may choose not to take part in the study. It is important to note that in both clinical trials, the cyclopamine was administered topically that diminished the side effects of the drug [112, 114].
However, cyclopamine has never been used orally in clinical trials. Test using animal models demonstrated that cyclopamine besides being poorly soluble orally, at high doses, it has a potential teratogenic effect, causing many potential side effects, including weight loss, dehydration, and death [115], which limits its clinical use. Therefore, some other potent SMO inhibitors have also reached the clinical trials, such as: the orally active IPI-926, a semi-synthetic derivative of cyclopamine and different synthetic compounds, such as GDC-0449 (vismodegib), Cur61414, and NVPLDE-225 (Erismodegib or Sonidegib or Odomzo) [116,117,118,119].
Presently some ongoing and completed clinical trials used Shh inhibitors to treat brain tumors (see Tables 2 and 3). The first clinical trial performed using a Shh inhibitor to treat a brain tumor was conducted in 2008. At the time, a 26-year-old man with metastatic MB that was refractory to multiple therapies was treated with a novel Hh pathway inhibitor, GDC-0449 [117]. Interestingly, the group did molecular analyses of the patient’s tumor specimens obtained before treatment which suggested activation of Shh pathway, as there was a high expression of Hh target genes including GLI1, PTCH1, PTCH2 and sFRP1. So, the treatment resulted in rapid regression of the tumor and reduction of symptoms, but unfortunately, this effect was transient and the patient died after five months of treatment [117]. It was observed that the Hh pathway inhibition with GDC-0449 induced the malignant transformation in MB which induced the tumor regrowth and the rapid progress of the disease [117].
Only in 2012, the US Food and Drug Administration (FDA) approved GDC-0449 as a standard therapy in patients with locally advanced and metastatic BCC [120]. Then few phase I and II clinical trials emerged with the objective to define the pediatric maximum tolerated dose and the efficacy of GDC-0449 in SHH-MB. Some studies evaluated the use of GDC-0449 in combination with TMZ. The studies were performed through the Pediatric Brain Tumor Consortium. These were phase II studies which evaluated the efficacy of GDC-0449 in younger patients, as well as in adult patients with recurrent or refractory MB [121]. Many other collaborative studies using GDC-0449 are still ongoing which directs therapy based on both clinical and molecular risk stratification (see Table 2 and see www.clinicaltrials.gov). GDC-0449 has an advantage for the use in clinical trials since it has low toxicity and high specificity for the Shh pathway. Additionally, this drug may also be used together with other pathway inhibitors or chemotherapy [122]. Moreover, GDC-0449 usually is well tolerated because of a lack of Smo receptor in most normal tissues [111]. It is believed that the use of Shh pathway inhibitors in MB treatment may offer an adequate therapeutic option. However, it is important to note that, as Shh pathway is very important during development, the adverse effect of blocking Shh pathway in prepubescent children is not completely understood [123]. Recently a study demonstrated that the used of GDC-0449 in pediatric oncologic patients induces short stature and growth abnormalities as they developed physeal fusion [124]. So, the use of Hh inhibitors in skeletally immature patients should be widely discussed and may be limited to those patients whom treatment options are limited or absent.
Gli-based inhibitors
Shh-MBs as GBMs are highly mutated tumors, and it is not uncommon for those tumors to demonstrate primary resistance to SMO inhibition, as they present alterations in downstream SHH pathway genes such as SUFU, GLI2, or MYCN [125]. As described above, it is typical for those tumors to acquire secondary resistance to Shh inhibition, and in this case, a Shh inhibition monotherapy is not efficient [117]. This is why, several pharmaceutical companies such as Exelixis/Bristol-Myers Squibb, Novartis, Infinity, and Pfizer developed alternative Shh antagonists that act directly in Gli (see Table 1). Some of these inhibitors are already being tested in the brain and central nervous system tumors as coadjuvant therapy with TMZ (see Tables 2 and 3 and www.clinicaltrials.gov).
So, besides GDC-0449, NVPLDE-225 and BMS-833923 (XL139) were also tested in brain tumors. There were some phase I and phase II clinical trials completed with the purpose of testing the efficacy tolerability, pharmacokinetics, pharmaco-dynamics, and safety of these drugs orally [65].
Another drug that is being tested for gliomas in phase I and II clinical trials is the arsenic trioxide (ATO). ATO is an FDA-approved drug that has been shown to inhibit Gli-dependent growth in MB mouse model, which was first used for the treatment of patients with acute promyelocytic leukemia (APL) [126]. Recently, a study demonstrated that apparently, the treatment of patients in combination with ATO, TMZ, and radiation does not improve the overall outcome in GBM patients; however, it might have some benefit in anaplastic astrocytoma patients [127].
Most of Hh inhibitors that have entered clinical trials targeted Smo, although several mechanisms of resistance to Smo inhibitors have been identified. Therefore, the discovery of new Hh pathway inhibitors may be crucial to bypass these resistance mechanisms and control tumorigenesis.
Conclusions
The Shh pathway is a well-established pillar of neural development and cancer cells use this mechanism to resist therapy and recur. The Shh pathway is thought to be very simple, as it usually signals canonically through Gil proteins; however, the shh pathway can be very complex, as demonstrated by the emerging evidence. Moreover, this pathway can not only be controlled through several mechanisms and molecules, such as Gli2R and Gli3R, SUFU and Ptch, which are components of the pathway, but also through posttranslational modifications, such as ubiquitination and acetylation. Several reports demonstrated that Shh could also signal through a non-canonical route; however, it is still a mystery how the cells select between canonical and non-canonical routes. Shh pathway can also interact with other signaling components that are important during embryonic development and tumorigenesis, such as TGF-β, EGFR, and Wnt. Cross-talking between these pathways and Shh signaling plays a pivotal role in the presevation of CSCs postulated to have intrinsic resistance to chemotherapy. So, a better understanding of the mechanisms is involved in the interaction between Shh pathway, and these pathways open a huge window of opportunities for the development of new therapeutic drugs for multiple cancers. Moreover, the inhibition of Shh signaling components may prove to be key to resistance and potential therapeutic targets to GBM and MB. The CSC hypothesis provides an explanation for the heterogeneity and recurrence of these tumors, and the Shh signaling pathway plays an important role in the maintenance of these cells. However, we believe that the best way to control the turmor recurrence is combining Shh antagonist with convetional therapies that are actually used in the clinic. Nowadays, the primary target used for development of Shh-pathway inhibitors in clinical trials is SMO, and there are several clinical trials for different types of brain tumors ongoing. So, current clinical trials offer a great outlook to overcome brain tumor. But, we still believe that more researchers must be conducted, as unfortunately we did not reach the cure for most of the cancers, such as GBMs and MBs, that are very aggressive.
Abbreviations
- APL:
-
Promyelocytic leukemia
- ATO:
-
Arsenic trioxide
- BCC:
-
Basal cell carcinoma
- bFGF:
-
Basic fibroblast growth factor
- CK1:
-
Casein kinase 1
- CSCs:
-
Cancer initiating stem cells
- Dhh:
-
Desert-Hedgehog
- DYRK1:
-
Dual specificity Yak1-related kinase
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-to-mesenchymal transition
- ERK:
-
Extracellular signal-regulated kinases
- FDA:
-
Food and Drug Administration
- GBM:
-
Glioblastoma
- GDC-0449:
-
Vismodegib
- Gli:
-
Glioma-associated oncogene homologue
- Gli2A:
-
Gli2 activated
- Gli2R:
-
Gli2 repressor
- Gli3A:
-
Gli3 activated
- Gli3R:
-
Gli3 repressor
- GPCR:
-
G protein–coupled receptor
- GSC:
-
Glioma stem cells
- GSK3:
-
Glycogen synthase kinase 3
- HDAC1:
-
Histone deacetylase 1
- Hh:
-
Hedgehog
- IFT80:
-
Intraflagellar transport protein 8
- Ihh:
-
Indian-Hedgehog
- MB:
-
Medulloblastoma
- Mek1:
-
Mitogen activated protein kinase kinase
- MGMT:
-
O-6-methylguanine-DNA methyltransferase
- NSCs:
-
Neural stem cells
- NVPLDE-225:
-
Erismodegib or Sonidegib or Odomzo
- Obs:
-
Osteoblasts
- PC:
-
primary cilium
- PDGF:
-
Platelet-derived growth factor
- PDGFRα:
-
Platelet-derived growth factor receptor α
- PI3K:
-
Phosphoinositide-3 kinase
- PKA:
-
Protein kinase A
- PKC:
-
Protein kinase C
- Ptch1 and 2:
-
Patched 1 and Patched 2
- RhoA:
-
Ras homolog gene family, member A
- sFRP-1:
-
Secreted frizzled-related protein-1
- SGZ:
-
Subgranular zone
- Shh:
-
Sonic-Hedgehog
- Smo:
-
Smoothened
- SUFU:
-
Supressor of Fused
- TGFß:
-
Transforming growth factor beta
- TMZ:
-
Temozolamide
- V-SVZ:
-
Ventricular-subventricular zone
- Wnt:
-
Wingless
References
Davis FG, Kupelian V, Freels S, McCarthy B, Surawicz T. Prevalence estimates for primary brain tumors in the United States by behavior and major histology groups. Neuro-Oncology. 2001;3:152–8.
Bangs F, Anderson KV. Primary cilia and mammalian hedgehog signaling. Cold Spring Harb Perspect Biol. 2017;9
Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–44.
Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by desert hedgehog regulates the male germline. Curr Biol. 1996;6:298–304.
Yao HH, Whoriskey W, Capel B. Desert hedgehog/patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002;16:1433–40.
Wijgerde M, Ooms M, Hoogerbrugge JW, Grootegoed JA. Hedgehog signaling in mouse ovary: Indian hedgehog and desert hedgehog from granulosa cells induce target gene expression in developing theca cells. Endocrinology. 2005;146:3558–66.
Briscoe J, Ericson J. Specification of neuronal fates in the ventral neural tube. Curr Opin Neurobiol. 2001;11:43–9.
Dessaud E, McMahon AP, Briscoe J. Pattern formation in the vertebrate neural tube: a sonic hedgehog morphogen-regulated transcriptional network. Development. 2008;135:2489–503.
Ma Y, Zhang P, Wang F, Yang J, Yang Z, Qin H. The relationship between early embryo development and tumourigenesis. J Cell Mol Med. 2010;14:2697–701.
Roth W, Wild-Bode C, Platten M, Grimmel C, Melkonyan HS, Dichgans J, Weller M. Secreted frizzled-related proteins inhibit motility and promote growth of human malignant glioma cells. Oncogene. 2000;19:4210–20.
Golestaneh N, Mishra B. TGF-beta, neuronal stem cells and glioblastoma. Oncogene. 2005;24:5722–30.
Lima FR, Kahn SA, Soletti RC, Biasoli D, Alves T, da Fonseca AC, Garcia C, Romao L, Brito J, Holanda-Afonso R, et al. Glioblastoma: therapeutic challenges, what lies ahead. Biochim Biophys Acta. 2012;1826:338–49.
Cochrane CR, Szczepny A, Watkins DN, Cain JE. Hedgehog signaling in the maintenance of cancer stem cells. Cancers. 2015;7:1554–85.
Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, Foresta U, Mitsiades CS, Rossi M, Todoerti K, et al. Canonical and noncanonical hedgehog pathway in the pathogenesis of multiple myeloma. Blood. 2012;120:5002–13.
Choudhry Z, Rikani AA, Choudhry AM, Tariq S, Zakaria F, Asghar MW, Sarfraz MK, Haider K, Shafiq AA, Mobassarah NJ. Sonic hedgehog signalling pathway: a complex network. Ann Neurosci. 2014;21:28–31.
Rimkus TK, Carpenter RL, Qasem S, Chan M, Lo HW. Targeting the sonic hedgehog signaling pathway: review of smoothened and GLI inhibitors. Cancers. 2016;8
Lee H, Ko HW. Ciliary smoothened-mediated noncanonical hedgehog signaling promotes tubulin acetylation. Biochem Biophys Res Commun. 2016;480:574–9.
Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102:521–31.
Dahmane N, Ruiz i, Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–100.
Kandel ER, Schwartz JH, Jessell TM. Principles of neural science. 4th ed. New York: McGraw-Hill, Health Professions Division; 2000.
Rahnama F, Toftgard R, Zaphiropoulos PG. Distinct roles of PTCH2 splice variants in hedgehog signalling. Biochem J. 2004;378:325–34.
Kinzler KW, Bigner SH, Bigner DD, Trent JM, Law ML, O'Brien SJ, Wong AJ, Vogelstein B. Identification of an amplified, highly expressed gene in a human glioma. Science. 1987;236:70–3.
Ruiz i, Altaba A, Mas C, Stecca B. The Gli code: an information nexus regulating cell fate, stemness and cancer. Trends Cell Biol. 2007;17:438–47.
Ruiz i, Altaba A. Catching a Gli-mpse of hedgehog. Cell. 1997;90:193–6.
Sasaki H, Nishizaki Y, Hui C, Nakafuku M, Kondoh H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development. 1999;126:3915–24.
Teglund S, Toftgard R. Hedgehog beyond medulloblastoma and basal cell carcinoma. Biochim Biophys Acta. 2010;1805:181–208.
Kim J, Kato M, Beachy PA. Gli2 trafficking links hedgehog-dependent activation of smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci U S A. 2009;106:21666–71.
Litingtung Y, Dahn RD, Li Y, Fallon JF, Chiang C. Shh and Gli3 are dispensable for limb skeleton formation but regulate digit number and identity. Nature. 2002;418:979–83.
Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. 2011;27:513–37.
Gonnissen A, Isebaert S, Haustermans K. Targeting the hedgehog signaling pathway in cancer: beyond smoothened. Oncotarget. 2015;6:13899–913.
Kogerman P, Grimm T, Kogerman L, Krause D, Unden AB, Sandstedt B, Toftgard R, Zaphiropoulos PG. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. 1999;1:312–9.
Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R. The output of hedgehog signaling is controlled by the dynamic association between suppressor of fused and the Gli proteins. Genes Dev. 2010;24:670–82.
Lauth M, Bergstrom A, Shimokawa T, Toftgard R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc Natl Acad Sci U S A. 2007;104:8455–60.
Kaesler S, Luscher B, Ruther U. Transcriptional activity of GLI1 is negatively regulated by protein kinase a. Biol Chem. 2000;381:545–51.
Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–34.
Mao J, Maye P, Kogerman P, Tejedor FJ, Toftgard R, Xie W, Wu G, Wu D. Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J Biol Chem. 2002;277:35156–61.
Pan Y, Bai CB, Joyner AL, Wang B. Sonic hedgehog signaling regulates Gli2 transcriptional activity by suppressing its processing and degradation. Mol Cell Biol. 2006;26:3365–77.
Riobo NA, Haines GM, Emerson CP, Jr.: Protein kinase C-delta and mitogen-activated protein/extracellular signal-regulated kinase-1 control GLI activation in hedgehog signaling. Cancer Res 2006, 66:839–845.
Riobo NA, Lu K, Ai X, Haines GM, Emerson CP, Jr.: Phosphoinositide 3-kinase and Akt are essential for sonic hedgehog signaling. Proc Natl Acad Sci U S A 2006, 103:4505–4510.
Welchman RL, Gordon C, Mayer RJ. Ubiquitin and ubiquitin-like proteins as multifunctional signals. Nat Rev Mol Cell Biol. 2005;6:599–609.
Gulino A, Di Marcotullio L, Canettieri G, De Smaele E, Screpanti I. Hedgehog/Gli control by ubiquitination/acetylation interplay. Vitam Horm. 2012;88:211–27.
Archer TC, Weeraratne SD, Pomeroy SL. Hedgehog-GLI pathway in medulloblastoma. Journal of clinical oncology. 2012;30:2154–6.
Cooper AF, Yu KP, Brueckner M, Brailey LL, Johnson L, McGrath JM, Bale AE. Cardiac and CNS defects in a mouse with targeted disruption of suppressor of fused. Development. 2005;132:4407–17.
Svard J, Heby-Henricson K, Persson-Lek M, Rozell B, Lauth M, Bergstrom A, Ericson J, Toftgard R, Teglund S. Genetic elimination of suppressor of fused reveals an essential repressor function in the mammalian hedgehog signaling pathway. Dev Cell. 2006;10:187–97.
Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, McKinnon PJ. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–7.
Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, Pietrosanti L, De Smaele E, Ferretti E, Miele E, et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–42.
Chandra V, Das T, Gulati P, Biswas NK, Rote S, Chatterjee U, Ghosh SN, Deb S, Saha SK, Chowdhury AK, et al. Hedgehog signaling pathway is active in GBM with GLI1 mRNA expression showing a single continuous distribution rather than discrete high/low clusters. PLoS One. 2015;10:e0116390.
Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2012;60:502–14.
Dubois LG, Campanati L, Righy C, D'Andrea-Meira I, Spohr TC, Porto-Carreiro I, Pereira CM, Balca-Silva J, Kahn SA, DosSantos MF, et al. Gliomas and the vascular fragility of the blood brain barrier. Front Cell Neurosci. 2014;8:418.
Moura Neto V, Campanati L, Pereira CM, Freitas C, Coelho-Aguiar J, Tavares AL, SPOHR TCLS, Aguiar D, Kahn SA, Matias D, et al: Glioblastoma: a hidden side of the astrocyte. In In: Glioma Cell Biology (Mentlein ASaR ed., vol. 1. Vienna: Springer Netherlends Vienna; 2014.
Oliveira-Nunes MC, Assad Kahn S, de Oliveira Barbeitas AL, TC ES, Dubois LG, Ventura Matioszek GM, Querido W, Campanati L, de Brito Neto JM, Lima FR, et al. The availability of the embryonic TGF-beta protein nodal is dynamically regulated during glioblastoma multiforme tumorigenesis. Cancer Cell Int. 2016;16:46.
Shevde LA, Samant RS. Nonclassical hedgehog-GLI signaling and its clinical implications. Int J Cancer. 2014;135:1–6.
Yan GN, Yang L, Lv YF, Shi Y, Shen LL, Yao XH, Guo QN, Zhang P, Cui YH, Zhang X, et al. Endothelial cells promote stem-like phenotype of glioma cells through activating the hedgehog pathway. J Pathol. 2014;234:11–22.
Robbins DJ, Fei DL, Riobo NA. The hedgehog signal transduction network. Sci Signal. 2012;5:re6.
Yam PT, Langlois SD, Morin S, Charron F. Sonic hedgehog guides axons through a noncanonical, Src-family-kinase-dependent signaling pathway. Neuron. 2009;62:349–62.
Polizio AH, Chinchilla P, Chen X, Kim S, Manning DR, Riobo NA. Heterotrimeric Gi proteins link hedgehog signaling to activation of rho small GTPases to promote fibroblast migration. J Biol Chem. 2011;286:19589–96.
Belgacem YH, Borodinsky LN. Sonic hedgehog signaling is decoded by calcium spike activity in the developing spinal cord. Proc Natl Acad Sci U S A. 2011;108:4482–7.
Teperino R, Amann S, Bayer M, McGee SL, Loipetzberger A, Connor T, Jaeger C, Kammerer B, Winter L, Wiche G, et al. Hedgehog partial agonism drives Warburg-like metabolism in muscle and brown fat. Cell. 2012;151:414–26.
Arensdorf AM, Marada S, Ogden SK. Smoothened regulation: a tale of two signals. Trends Pharmacol Sci. 2016;37:62–72.
Szczepny A, Rogers S, Jayasekara WSN, Park K, McCloy RA, Cochrane CR, Ganju V, Cooper WA, Sage J, Peacock CD, et al: The role of canonical and non-canonical hedgehog signaling in tumor progression in a mouse model of small cell lung cancer. Oncogene 2017;36(39):5544-50.
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, Duan W, Sun Q, Xu J, Wu Z, Wu E. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer. 2013;12:66.
Yuan X, Cao J, He X, Serra R, Qu J, Cao X, Yang S. Ciliary IFT80 balances canonical versus non-canonical hedgehog signalling for osteoblast differentiation. Nat Commun. 2016;7:11024.
Kimura H, Stephen D, Joyner A, Curran T. Gli1 is important for medulloblastoma formation in Ptc1+/− mice. Oncogene. 2005;24:4026–36.
Dahmane N, Lee J, Robins P, Heller P, Ruiz i, Altaba A. Activation of the transcription factor Gli1 and the sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389:876–81.
Santoni M, Burattini L, Nabissi M, Morelli MB, Berardi R, Santoni G, Cascinu S. Essential role of Gli proteins in glioblastoma multiforme. Curr Protein Pept Sci. 2013;14:133–40.
Maeda O, Kondo M, Fujita T, Usami N, Fukui T, Shimokata K, Ando T, Goto H, Sekido Y. Enhancement of GLI1-transcriptional activity by beta-catenin in human cancer cells. Oncol Rep. 2006;16:91–6.
He J, Sheng T, Stelter AA, Li C, Zhang X, Sinha M, Luxon BA, Xie J. Suppressing Wnt signaling by the hedgehog pathway through sFRP-1. J Biol Chem. 2006;281:35598–602.
Bijlsma MF, Spek CA, Peppelenbosch MP. Hedgehog: an unusual signal transducer. BioEssays. 2004;26:387–94.
Takenaka K, Kise Y, Miki H. GSK3beta positively regulates hedgehog signaling through Sufu in mammalian cells. Biochem Biophys Res Commun. 2007;353:501–8.
Meng X, Poon R, Zhang X, Cheah A, Ding Q, Hui CC, Alman B. Suppressor of fused negatively regulates beta-catenin signaling. J Biol Chem. 2001;276:40113–9.
Taylor MD, Zhang X, Liu L, Hui CC, Mainprize TG, Scherer SW, Wainwright B, Hogg D, Rutka JT. Failure of a medulloblastoma-derived mutant of SUFU to suppress WNT signaling. Oncogene. 2004;23:4577–83.
Yanai K, Nakamura M, Akiyoshi T, Nagai S, Wada J, Koga K, Noshiro H, Nagai E, Tsuneyoshi M, Tanaka M, Katano M. Crosstalk of hedgehog and Wnt pathways in gastric cancer. Cancer Lett. 2008;263:145–56.
Bos JL. Ras oncogenes in human cancer: a review. Cancer Res. 1989;49:4682–9.
Ji Z, Mei FC, Xie J, Cheng X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem. 2007;282:14048–55.
Xie J, Aszterbaum M, Zhang X, Bonifas JM, Zachary C, Epstein E, McCormick F. A role of PDGFRalpha in basal cell carcinoma proliferation. Proc Natl Acad Sci U S A. 2001;98:9255–9.
Bigelow RL, Jen EY, Delehedde M, Chari NS, McDonnell TJ. Sonic hedgehog induces epidermal growth factor dependent matrix infiltration in HaCaT keratinocytes. The Journal of investigative dermatology. 2005;124:457–65.
Kasper M, Schnidar H, Neill GW, Hanneder M, Klingler S, Blaas L, Schmid C, Hauser-Kronberger C, Regl G, Philpott MP, Aberger F. Selective modulation of hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol Cell Biol. 2006;26:6283–98.
Schnidar H, Eberl M, Klingler S, Mangelberger D, Kasper M, Hauser-Kronberger C, Regl G, Kroismayr R, Moriggl R, Sibilia M, Aberger F. Epidermal growth factor receptor signaling synergizes with hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009;69:1284–92.
Dennler S, Andre J, Alexaki I, Li A, Magnaldo T, ten Dijke P, Wang XJ, Verrecchia F, Mauviel A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007;67:6981–6.
Seto M, Ohta M, Asaoka Y, Ikenoue T, Tada M, Miyabayashi K, Mohri D, Tanaka Y, Ijichi H, Tateishi K, et al. Regulation of the hedgehog signaling by the mitogen-activated protein kinase cascade in gastric cancer. Mol Carcinog. 2009;48:703–12.
Gotschel F, Berg D, Gruber W, Bender C, Eberl M, Friedel M, Sonntag J, Rungeler E, Hache H, Wierling C, et al. Synergism between hedgehog-GLI and EGFR signaling in hedgehog-responsive human medulloblastoma cells induces downregulation of canonical hedgehog-target genes and stabilized expression of GLI1. PLoS One. 2013;8:e65403.
Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, Yang SX, Ivy SP. Targeting notch, hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. 2015;12:445–64.
Espinoza I, Miele L. Deadly crosstalk: notch signaling a 41 t the intersection of EMT and cancer stem cells. Cancer Lett. 2013;341:–45.
Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells--perspectives on current status and future directions: AACR workshop on cancer stem cells. Cancer Res. 2006;66:9339–44.
Nguyen LV, Vanner R, Dirks P, Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012;12:133–43.
Tan BT, Park CY, Ailles LE, Weissman IL. The cancer stem cell hypothesis: a work in progress. Lab Investig. 2006;86:1203–7.
Yabut OR, Pleasure SJ. The crossroads of neural stem cell development and tumorigenesis. Opera Med Physiol. 2016;2:181–7.
Decimo I, Bifari F, Krampera M, Fumagalli G. Neural stem cell niches in health and diseases. Curr Pharm Des. 2012;18:1755–83.
Massirer KB, Carromeu C, Griesi-Oliveira K, Muotri AR. Maintenance and differentiation of neural stem cells. Wiley Interdiscip Rev Syst Biol Med. 2011;3:107–14.
Sousa VH, Fishell G. Sonic hedgehog functions through dynamic changes in temporal competence in the developing forebrain. Curr Opin Genet Dev. 2010;20:391–9.
Daynac M, Tirou L, Faure H, Mouthon MA, Gauthier LR, Hahn H, Boussin FD, Ruat M. Hedgehog controls quiescence and activation of neural stem cells in the adult ventricular-subventricular zone. Stem Cell Reports. 2016;7:735–48.
Bar EE, Chaudhry A, Farah MH, Eberhart CG. Hedgehog signaling promotes medulloblastoma survival via Bc/II. Am J Pathol. 2007;170:347–55.
Alves TR, Lima FR, Kahn SA, Lobo D, Dubois LG, Soletti R, Borges H, Neto VM. Glioblastoma cells: a heterogeneous and fatal tumor interacting with the parenchyma. Life Sci. 2011;89:532–9.
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–96.
Roos WP, Batista LF, Naumann SC, Wick W, Weller M, Menck CF, Kaina B. Apoptosis in malignant glioma cells triggered by the temozolomide-induced DNA lesion O6-methylguanine. Oncogene. 2007;26:186–97.
Belinsky SA, Klinge DM, Liechty KC, March TH, Kang T, Gilliland FD, Sotnic N, Adamova G, Rusinova G, Telnov V. Plutonium targets the p16 gene for inactivation by promoter hypermethylation in human lung adenocarcinoma. Carcinogenesis. 2004;25:1063–7.
Srivenugopal KS, Ali-Osman F. The DNA repair protein, O(6)-methylguanine-DNA methyltransferase is a proteolytic target for the E6 human papillomavirus oncoprotein. Oncogene. 2002;21:5940–5.
Chen L, Wang Y, Liu F, Xu L, Peng F, Zhao N, Fu B, Zhu Z, Shi Y, Liu J, et al. A systematic review and meta-analysis: association between MGMT hypermethylation and the clinicopathological characteristics of non-small-cell lung carcinoma. Sci Rep. 2018;8:1439.
Dunn J, Baborie A, Alam F, Joyce K, Moxham M, Sibson R, Crooks D, Husband D, Shenoy A, Brodbelt A, et al. Extent of MGMT promoter methylation correlates with outcome in glioblastomas given temozolomide and radiotherapy. Br J Cancer. 2009;101:124–31.
van Nifterik KA, van den Berg J, van der Meide WF, Ameziane N, Wedekind LE, Steenbergen RD, Leenstra S, Lafleur MV, Slotman BJ, Stalpers LJ, Sminia P. Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide. Br J Cancer. 2010;103:29–35.
Bhattacharjee S, Nandi S. Synthetic lethality in DNA repair network: a novel avenue in targeted cancer therapy and combination therapeutics. IUBMB Life. 2017;69:929–37.
Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, Piccirillo S, Vescovi AL, DiMeco F, Olivi A, Eberhart CG. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. 2007;25:2524–33.
Pistollato F, Abbadi S, Rampazzo E, Persano L, Della Puppa A, Frasson C, Sarto E, Scienza R, D'Avella D, Basso G. Intratumoral hypoxic gradient drives stem cells distribution and MGMT expression in glioblastoma. Stem Cells. 2010;28:851–62.
Wang K, Chen D, Qian Z, Cui D, Gao L, Lou M. Hedgehog/Gli1 signaling pathway regulates MGMT expression and chemoresistance to temozolomide in human glioblastoma. Cancer Cell Int. 2017;17:117.
Cui D, Xu Q, Wang K, Che X. Gli1 is a potential target for alleviating multidrug resistance of gliomas. J Neurol Sci. 2010;288:156–66.
Sims-Mourtada J, Izzo JG, Ajani J, Chao KS. Sonic hedgehog promotes multiple drug resistance by regulation of drug transport. Oncogene. 2007;26:5674–9.
Sims-Mourtada J, Izzo JG, Apisarnthanarax S, Wu TT, Malhotra U, Luthra R, Liao Z, Komaki R, van der Kogel A, Ajani J, Chao KS. Hedgehog: an attribute to tumor regrowth after chemoradiotherapy and a target to improve radiation response. Clin Cancer Res. 2006;12:6565–72.
Chen X, Horiuchi A, Kikuchi N, Osada R, Yoshida J, Shiozawa T, Konishi I. Hedgehog signal pathway is activated in ovarian carcinomas, correlating with cell proliferation: it's inhibition leads to growth suppression and apoptosis. Cancer Sci. 2007;98:68–76.
Yoon JW, Gilbertson R, Iannaccone S, Iannaccone P, Walterhouse D. Defining a role for sonic hedgehog pathway activation in desmoplastic medulloblastoma by identifying GLI1 target genes. Int J Cancer. 2009;124:109–19.
Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, Burdick D, Goldsmith R, Robarge K, Sutherlin D, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–44.
Laukkanen MO, Castellone MD. Hijacking the hedgehog pathway in cancer therapy. Anti Cancer Agents Med Chem. 2016;16:309–17.
Tas S, Avci O. Rapid clearance of psoriatic skin lesions induced by topical cyclopamine. A preliminary proof of concept study. Dermatology. 2004;209:126–31.
Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development. 1998;125:3553–62.
Herman S. A Phase III, Double-Blind, Placebo-Controlled Clinical Trial to Assess Cyclopamine as a Chemopreventive Agent Inhibiting Recurrence of Basal Cell Carcinoma following Surgical Resection. DORIS DUKE MEDICAL STUDENTS’ JOURNAL. 2006;V:29–35.
Mimeault M, Batra SK. Frequent deregulations in the hedgehog signaling network and cross-talks with the epidermal growth factor receptor pathway involved in cancer progression and targeted therapies. Pharmacol Rev. 2010;62:497–524.
Molckovsky A, Siu LL. First-in-class, first-in-human phase I results of targeted agents: highlights of the 2008 American society of clinical oncology meeting. J Hematol Oncol. 2008;1:20.
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–8.
Von Hoff DD, LoRusso PM, Rudin CM, Reddy JC, Yauch RL, Tibes R, Weiss GJ, Borad MJ, Hann CL, Brahmer JR, et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N Engl J Med. 2009;361:1164–72.
Stanton BZ, Peng LF. Small-molecule modulators of the sonic hedgehog signaling pathway. Mol BioSyst. 2010;6:44–54.
Gould SE, Low JA, Marsters JC Jr, Robarge K, Rubin LL, de Sauvage FJ, Sutherlin DP, Wong H, Yauch RL. Discovery and preclinical development of vismodegib. Expert Opin Drug Discov. 2014;9:969–84.
Gajjar A, Stewart CF, Ellison DW, Kaste S, Kun LE, Packer RJ, Goldman S, Chintagumpala M, Wallace D, Takebe N, et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: a pediatric brain tumor consortium study. Clin Cancer Res. 2013;19:6305–12.
Li H, Li J, Feng L. Hedgehog signaling pathway as a therapeutic target for ovarian cancer. Cancer Epidemiol. 2016;40:152–7.
Gupta S, Takebe N, Lorusso P. Targeting the hedgehog pathway in cancer. Ther Adv Med Oncol. 2010;2:237–50.
Robinson GW, Kaste SC, Chemaitilly W, Bowers DC, Laughton S, Smith A, Gottardo NG, Partap S, Bendel A, Wright KD, et al. Irreversible growth plate fusions in children with medulloblastoma treated with a targeted hedgehog pathway inhibitor. Oncotarget. 2017;8:69295–302.
Lee DY, Deng Z, Wang CH, Yang BB. MicroRNA-378 promotes cell survival, tumor growth, and angiogenesis by targeting SuFu and Fus-1 expression. Proc Natl Acad Sci U S A. 2007;104:20350–5.
Beauchamp EM, Ringer L, Bulut G, Sajwan KP, Hall MD, Lee YC, Peaceman D, Ozdemirli M, Rodriguez O, Macdonald TJ, et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking hedgehog/GLI pathway. J Clin Invest. 2011;121:148–60.
Kumthekar P, Grimm S, Chandler J, Mehta M, Marymont M, Levy R, Muro K, Helenowski I, McCarthy K, Fountas L, Raizer J. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J Neuro-Oncol. 2017;133:589–94.
Matias D, Predes D, Niemeyer Filho P, Lopes MC, Abreu JG, Lima FRS, Moura Neto V. Microglia-glioblastoma interactions: new role for Wnt signaling. Biochim Biophys Acta. 2017;1868:333–40.
Berg M, Soreide K. EGFR and downstream genetic alterations in KRAS/BRAF and PI3K/AKT pathways in colorectal cancer: implications for targeted therapy. Discov Med. 2012;14:207–14.
Acknowledgements
We thank to Professor Vivaldo Moura Neto for careful reading the manuscript.
Funding
This work was supported by the National Institute for Translational Neuroscience (INNT) of the Ministry of Science and Technology; Brazilian Federal Agency for the Support and Evaluation of Graduate Education (CAPES) of the Ministry of Education; National Council for Scientific and Technological Development (CNPq); Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro Carlos Chagas Filho Research Support Foundation (FAPERJ); Ary Frauzino Foundation for Cancer Research and Pró-Saúde Associação Beneficiente de Assistência Social e Hospitalar.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
All Authors participated in the elaboration of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Carballo, G.B., Honorato, J.R., de Lopes, G.P. et al. A highlight on Sonic hedgehog pathway. Cell Commun Signal 16, 11 (2018). https://doi.org/10.1186/s12964-018-0220-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-018-0220-7