
Abstract
Background
Storage conditions during transportation of explanted ovaries are a critical step in setting up fertility preservation protocols in both animal and human fields. Here, we evaluated the effects of ovary storage at 4 °C on the preservation of preantral follicles and oocytes retrieved from antral follicles using the domestic cat as model.
Methods
Ovaries were harvested from fifty-five healthy domestic queens during ovariectomy and stored at 4 °C for 0 (control), 24, 48, 72 and 96 h. In Experiment 1, the effects of the storage period at 4 °C on the morphology, cytoskeleton (α/β tubulin) and DNA integrity (phosphorylation of histone H2AX) of preantral follicles were investigated. In Experiment 2, oocytes recovered from antral follicles were matured and fertilized in vitro to evaluate their meiotic and developmental competence. Reactive oxygen species (ROS), glutathione (GSH) and lipid peroxidation were measured in matured oocytes.
Results
The results showed that: a) storage up to 24 h did not affect the morphology and the DNA integrity of preantral follicles; b) extended storage times caused progressive morphological abnormalities, disassembling of microtubules and DNA damage; c) storage up to 48 h did not influence in vitro meiotic maturation of oocytes nor cleavage after in vitro fertilization. However, only oocytes stored within the ovary for 24 h produced blastocysts in a percentage similar to control oocytes; d) GSH levels of in vitro matured oocytes did not change at any time during ovary storage; a progressive increase in ROS levels was detected from 48 h associated with elevated lipid peroxidation at 72 and 96 h of storage.
Conclusions
Storage of cat ovaries for up to 24 h caused minimal alteration of preantral follicles and oocytes. The extension of the storage period beyond 24 h progressively impaired the structure of follicles, and modified the oxidative status of in vitro matured oocytes and their developmental competence after in vitro fertilization. This information may help when setting up programs for fertility conservation, especially for wild feline species which die in geographic areas located far away from ARTs centers.
Similar content being viewed by others


Background
Most of the 36 living species of wild cats are formally listed as threatened by extinction mainly due to poaching, habitat destruction and infectious diseases [1]. Assisted Reproductive Technologies (ARTs) contribute to the preservation of endangered feline species. In domestic felids considerable progress has been made in the development of modern methodologies of ARTs including in vitro embryo production and banking of genetic resources. Some of these techniques have been included in programs for the conservation of feline wildlife species [2, 3].
The possibility of rescuing germplasm from feline females after sudden death in their natural habitat or zoos or after medical ovariohysterectomy may at times require the shipment of ovaries for long distances, from the retrieval site to the ART centres. Therefore, storage conditions (temperature, duration, medium) during transportation of explanted ovaries are a critical step in setting up of fertility preservation protocols.
In this context, studies conducted to establish the appropriate transportation temperature for ovaries provided evidence that feline oocytes, in contrast to other species, have a unique tolerance to cold storage at 4 °C [4,5,6]. A pivotal study by Johnston et al. [7] found that oocytes from rare wild species could be matured and fertilized in vitro following ovary storage for as long as 36 h in cold (4 °C) phosphate buffer saline (PBS). Histological evaluation of cold stored ovaries of domestic cats further supported the concept that immature oocytes have some degree of cold tolerance [8]. Other studies reported unaffected in vitro maturation (IVM) rates when domestic cat ovaries were stored at 4 °C up to 72 h. However, only oocytes stored within the ovary for 24 h could produce blastocysts after in vitro fertilization [9] and retained competence to develop into live offspring after transfer into recipients [10]. More recently, findings by Luu et al. [11] indicated that the addition of relaxin to the IVM medium improved the rate of blastocyst development of cat oocytes from ovaries stored for 24 h at 4 °C. Furthermore, superoxide dismutase (SOD) supplementation to ovary transportation medium enhanced in vitro embryo production from oocytes of domestic cat ovaries preserved at 4 °C for up to 72 h [12].
Most studies on the cold storage of feline ovaries have focused on the preservation of oocytes from antral follicles. This is a limitation as an ovary contains a large amount of preantral follicles, which represents the female’s ovarian reserve and includes more than 90% of the follicular population [13]. Therefore, the pool of preantral follicles constitutes a potentially rich source of genetic material which could be used in fertility preservation programs for genetically valuable animals, including females of endangered species that die before reaching sexual maturity [14]. In fact, the ovarian cortex can be cryopreserved and utilized for xenotrasplantation or, alternatively, preantral follicles can be isolated and cultured in vitro until maturity. Slow freezing of cat ovarian cortex in conjunction with xenotransplantation was first reported by Bosch et al. [15]. Moreover, mature oocytes have been obtained after long-term transplantation of ovarian cortex from female lions into immunodeficient mice [16]. In several animal species, including cats, attempts have been made to develop in vitro culture systems for the growth of preantral follicles with variable results [17,18,19,20]. Full term culture of primordial follicles to a mature stage and birth of viable offspring after in vitro fertilization and embryo transfer have only been achieved in mice and were first described by Eppig and O’Brien [21].
Nowadays, to the best of our knowledge, there is little information about the impact of cold storage on the health status of preantral follicles of feline ovaries and on the maximal tolerable storage duration [22]. Evidence from other animal models indicates that cold temperature favors the preservation of preantral follicles [23,24,25,26,27].
The storage of ovarian tissue during transportation has great importance not only for rare and endangered species, but also for humans. Indeed, transportation of human ovarian tissue from cancer patients to a single central national center is conducted in several countries [28]. In Denmark [29, 30] Germany, Switzerland, Austria [31, 32] and in the United States [33,34,35,36] ovarian tissue is cryostored only in specific centers and finding the optimal transportation conditions will probably also be a benefit to such centers.
In the present study, the domestic cat was used as an experimental model to evaluate the preservation of preantral follicles and of oocytes retrieved from antral follicles in ovaries stored at 4 °C for different time periods. Therefore, we analyzed: a) morphology, microtubular cytoskeleton and DNA damage in preantral follicles and, b) developmental competence of oocytes after in vitro maturation and fertilization as well as oxidative status in matured oocytes.
Methods
Chemicals
All chemicals, in this study, were purchased from Sigma Chemical CO. (St. Louis, MO, USA) unless stated otherwise.
Ovary collection and storage
Ovaries were harvested from fifty five healthy, domestic queens (Felis catus, 8 months-2 years of age) at random stages of their oestrus cycles during routine ovariectomy at the Veterinary Teaching Hospital of the University of Sassari (Italy). The ovaries were placed in sterile 15-ml tubes containing Phosphate Buffered Saline (PBS, cat n°18,912–014, Gibco, Life Technologies, Carlsbad, California, USA) with penicillin (100 mg/L), streptomycin (100 mg/L) [8, 12]., Calcium Dichloride Hydrate (CaCl2H2O, 133 mg/L) and Magnesium Chloride Hexahydrate (MgCl2•6H2O, 100 mg/L). The divalent cations, calcium and magnesium, were added to the PBS solution in order to preserve cellular adhesion and tissue integrity in the ovary during storage at 4 °C [37, 38]. The ovaries were immediately transported to the laboratory at room temperature (22–25 °C) and randomly divided in two groups: fresh ovaries (0 h, control group) and ovaries to be stored at 4 °C (stored group). The ovaries from each storage group were transferred under sterile conditions to a 15-ml tube containing 4 ml of fresh pre-cooled PBS and held at 4 °C in a refrigerator for different lengths of times (24, 48, 72, 96 h). Control and stored ovaries were processed according to the following experimental design.
Experimental design
The experimental design includes two experiments. Experiment 1 was performed to evaluate the effect of different storage times (0, 24, 48, 72 and 96 h) at 4 °C on the morphology, organization of microtubular cytoskeleton and DNA integrity of preantral follicles.
In Experiment 2, the in vitro meiotic and developmental competence of oocytes collected from antral follicles of control and stored ovaries were evaluated. Moreover, the oxidative status of in vitro matured oocytes from each experimental group was analyzed by quantifying the intracellular levels of Reactive Oxygen Species (ROS), Glutathione (GSH) and lipid peroxidation.
Experiment 1
Morphological evaluation of preantral follicles
Both control and stored ovaries were fixed in Bouin’s solution for 12 h, embedded in paraffin, serially sectioned (5 μm), and mounted onto microscope slides. The sections were either stained with Hematoxylin and Eosin (HE) or used for immunohistochemistry, as described below. In order to evaluate follicular morphology, three sections from each sample were examined. Preantral follicles were classified according to their developmental status as: i) primordial with one layer of flattened cells; ii) primary with one layer of cuboidal cells; iii) secondary with two or more layers of cuboidal cells and a theca cell layer [39]. Moreover, they were graded according to their morphology: structurally normal follicles presented a spherical oocyte with a non-pyknotic nucleus surrounded by well-arranged granulosa cells, while abnormal follicles were characterized by contraction and clumping of oocyte chromatin surrounded by disorganized granulosa cells. To avoid double counting in adjacent sections, the follicles were only analyzed when an oocyte nucleus was present.
Fluorescent immunoistochemistry
The histological sections were immersed for 20 min in a 98 °C preheated solution (WCAP, citrate pH 6, BiOptica, Milan, Italy) which simultaneously allows dewaxing, rehydration and antigen unmasking. Slides were mounted in a sequenza chamber (Shandon, Runcorn, UK) before performing immunostaining.
Assessment of microtubular cytoskeleton organization
Microtubules are ubiquitous cytoskeletal structures that are formed by the polymerization of α and β tubulin heterodimers [40]. Accordingly, we used monoclonal anti α and β tubulin antibodies to detect the effect of cold storage on the microtubular cytoskeleton organization of ovarian tissue.
Ovarian sections were incubated overnight at 4 °C with a mixture of mouse monoclonal anti α-tubulin (1:1000, Sigma Chemical CO, St. Louis, MO, USA; Cat n° T5168) and mouse monoclonal anti β-tubulin (1:100; Sigma Chemical CO, Cat n° T-7941) antibodies then labeled with donkey anti-mouse Alexa Fluor 488 (1:100, Cat n° A21202, Life Technologies, Invitrogen, Carlsbad, California, USA) for 1 h at room temperature. These specific primary antibodies were chosen for immunofluorescence due to their high affinity to α and β tubulin as indicated by the manufacturer and other Authors [41,42,43,44].
The nuclei were counter-stained with Hoechst 33,342 (10 μg/ml; Cat n° B2261) in 1:1 (v/v) glycerol/PBS solution. The sections were covered with coverslips, sealed with nail polish, and kept at 4 °C in the dark until observation. Images were acquired by a laser-scanning confocal fluorescence microscope (Leica TCS SP5), equipped with 543 nm HeNe, 488 nm Argon and 405 nm 405-diode laser using an oil immersion 40× objective (NA = 1,25). The parameters related to fluorescence intensity (laser energy, gain, offset and pinhole size) were maintained at constant values during all image acquisitions.
Detection of histone H2AX phosphorylation
We investigated whether cold storage induced DNA damage in preantral follicles, examining histone H2AX phosphorylation (γH2AX), a marker of DNA double-strand breaks (DSBs) [45]. The ovary sections were incubated overnight at 4 °C with mouse monoclonal anti-γH2AX (phospho S139) antibody (Cat n° ab26350, Abcam, Cambridge; UK) and labeled with donkey anti-mouse Alexa Fluor 488 (1:100) for 1 h at room temperature. The nuclei were counter-stained with propidium iodide (1 μg/ml, Cat n°4170) in 1:1 (v/v) glycerol/PBS solution. Images were acquired as described above.
Experiment 2
Oocyte recovery and in vitro maturation
Ovaries from control and stored groups were sliced with a scalpel blade to release the cumulus-oocyte complexes (COCs). The COCs were collected in sterile Petri dishes in dissection medium (DM; 25 mM Hepes-buffered TCM 199) supplemented with 0.1% (w/v) polyvinyl alcohol (PVA) and antibiotics (100 μg/ml penicillin and streptomycin). Only the oocytes with darkly pigmented ooplasm and completely surrounded by at least one layer of cumulus cells were selected for in vitro maturation (IVM). The COCs were matured in groups of 25–35 in 650 μl of TCM 199 supplemented 0.36 mM pyruvate, 2 mM glutamine, 2.2 mM calcium lactate, 1.2 mM cysteine, 4 mg/ml BSA fatty acid free and FSH 1 IU / ml and LH 1 IU / ml (Pluset; Bio98, Milan, Italy) under mineral oil, in 4-well dishes (Nunc Cell Culture, Thermo Fisher Scientific, Waltham, Massachusetts, USA) in a humidified atmosphere of 5% CO2, at 38.5 °C for 24 h [46].
Assessment of oocyte nuclear maturation
After IVM, groups of oocytes were completely denuded of granulosa cells via gentle pipetting with a fine bore glass pipette in DM, stained with Hoechst 33,342 (10 μg/ml) in 1:1 (v/v) glycerol/PBS solution, placed on a slide and overlaid with a coverslip supported by four droplets of vaseline. The nuclear configuration was classified under an epifluorescent microscope (Olympus IX 70, Italy) as germinal vesicle (GV), germinal vesicle breakdown (GVBD), metaphase I (MI), or metaphase II (MII). The oocytes with the diffusely stained cytoplasm typical of nonviable cells and those with unidentifiable or invisible chromatin were classified as degenerated (Deg).
Measurement of intracellular ROS and GSH levels
Groups of matured oocytes (MII) from all experimental groups were selected on the basis of the presence of the first polar body and sampled for intracellular Reactive Oxygen Species (ROS) and Glutathione (GSH) level measurement [47].
Briefly, 2′7′-dichlorodihydrofluorescein diacetate (H2DCFDA, Cat n° D 6883) and 4-chloromethyl-6,8-difluoro-7-hydroxycoumarin (CMF2HC, Cell Tracker Blue, Cat n° C12881, Molecular Probes, Oregon, USA) were used to detect intracellular ROS as green fluorescence and GSH level as blue fluorescence. A total of 30–35 oocytes from each treatment group were incubated in the dark for 30 min at 38.5 °C in 5% CO2 in air in PBS-PVA containing 10 μM H2DCFDA or 10 μM CellTracker Blue. After incubation, the oocytes were washed with PBS-PVA, placed in 50 μL droplets, and observed using an epifluorescence microscope (Olympus IX 70, Italy) with UV filters (460 nm for ROS and 370 nm for GSH). The oocytes were positioned in the plane of focus, and the area of measurement was adapted to the size of the oocyte. Microscope adjustments and photomultiplier settings were kept constant for all experiments. The data of emission intensity/oocyte were reduced by compensation for the background fluorescence. The fluorescent images were saved as graphic files in. TIFF format. Fluorescence intensitiy was analyzed with Image J software (version 1.40; National Institute of health, Bethesda, MD). Results were normalized to control values for each experiment. The experiments were performed in triplicate.
Lipid peroxidation assay
Lipid peroxidation assay was performed with 4, 4-difluoro-5- (4- phenyl-1, 3-buttadienyl)-4-bora 3a, 4a-diaza-s-indacene-3-undecanoic acid 581/591C11 (BODIPY D3861, Life Technologies, Invitrogen, Carlsbad, California, USA) [48]. Briefly, a total of 30–35 MII oocytes recovered from the different groups were incubated with 10 mM BODIPY stain for 30 min at 38.5 °C in 5% CO2 in air. The dye loaded oocytes were then washed twice in DM and mounted on slides. Green and red fluorescence of BODIPY were determined using a confocal laser scanning microscope equipped with Argon and Helium/Neon lasers at excitation wavelengths of 488 and 543 nm and emission spectra of 500–530 nm (green) and greater than 560 nm (red). Levels of lipid peroxidation were measured by the ratio of green to red fluorescence. Results were normalized to control values for each experiment. The experiments were performed in triplicate.
In vitro fertilization and embryo culture
Spermatozoa were collected from epididymides of cats following routine orchiectomy [49, 50] and frozen according to the procedure described by Tsutsui et al. [51]. Before IVF, straws of semen were thawed by immersion in a 37 °C water bath for 30s and centrifuged at 620 x g for 5 min. The supernatant was discarded and the pellet was re-suspended in synthetic oviductal fluid (SOF) containing 6 mg/ml BSA, 50 μg/ml gentamicin (IVF medium). After maturation, the COCs were co-incubated with 1 × 106 motile spermatozoa/mL in IVF medium at 38.5 C in 5% CO2 in air with maximum humidity [52].
After 22 h, the presumptive zygotes were washed and cultured in SOF containing 4 mg/ml BSA, 100 IU/ml penicillin and 1% MEM non-Essential amino acids. On day 3 after IVF (day = 0) the embryos were transferred to SOF supplemented with 10% fetal calf serum and 2% MEM essential amino acids [52]. On day 2 and day 7 of IVC, respectively, the number of embryos cleaved and developing to blastocyst stage was determined. Blastocysts were stained with Hoechst 33,342 (10 μg/mL), mounted on a glass slide and examined under epifluorescent microscope (Olympus IX70, Italy). A digital image of each embryo was taken, and the number of nuclei was counted.
Statistical analysis
All statistical analyses were performed using Stata/IC 11.2 (StataCorp LP, USA). In continuous data, normal distribution was checked by Shapiro-Wilk test. When the assumption of normality was met (follicles expressing > 3 γH2AX foci, ROS and GSH, blastocyst cell number) the data were analysed by one way ANOVA using Bonferroni as post-hoc test. When the data were not normally distributed (normal preantral follicles) a non-parametric Kruskal-Wallis test was used. Categorical data of meiotic progression, cleavage and development to blastocyst stage were analyzed by chi-square test. The level of statistical significance was set at P < 0.05.
Results
Experiment 1
Morphology of preantral follicles after ovary storage at 4°
A total of 998 preantral follicles were examined (mean ± SD for each group: 199.6 ± 24.3). The distribution of preantral follicles at each developmental stage was similar among storage groups. Most of the preantral follicles were at primordial stage (77.2 ± 1.2%), while primary (15.7 ± 1.1%) and secondary follicles (6.8 ± 1.2%) were represented in lower percentages.
The percentage of morphologically normal follicles at different storage times is shown in Fig. 1. Storage for 24 h maintained the proportion of follicles with normal morphology (86.3 ± 2.8%) similar to control values (87.7 ± 2.5%). A progressive decrease (P < 0.05) in the percentage of normal follicles was observed following 48 h (65.0 ± 6.1%), 72 h (39.7 ± 2.8%) and 96 h (20.0 ± 3.2%) of storage compared to 0 and 24 h groups.

a Percentage (mean ± S.E.M) of morphologically normal preantral follicles after different time of ovary storage at 4 °C. b Representative images of sections of ovarian tissue (I) and different classes of preantral follicles (II-IV) stained by hematoxylin and eosin. Different superscripts (a, b, c, d) among storage times indicate P < 0.05. n = number of preantral follicles analyzed
Effect of ovary storage at 4 °C on the organization of the microtubular cytoskeleton
A storage time-dependent change of the tubulin organization was evident in cellular components of ovarian tissue (Fig. 2). In control sections, large amounts of tubulin were visible in the granulosa cell layers and a well detectable microtubular network was present throughout the cytoplasm of follicle-enclosed oocytes (Fig. 2 a, b). The tubulin distribution was progressively disrupted when increasing the storage time. Ovary storage at 4 °C for 24 h (Fig. 2 c,d) and 48 h (Fig. 2 e, f) caused a decrease in tubulin staining, which became irregularly distributed in granulosa and stromal cells. Storage for more extended periods (72 and 96 h) progressively induced tubulin alterations (Fig. 2 g,h,i, l). Similarly, the network of microtubules within the oocyte became irregular and disorganized following storage; spots of tubulin staining (see arrow in Fig. 2 f, h, and l) were observed inside the cytoplasm of the primary/secondary follicle-enclosed oocytes. The alterations in the tubulin network and the presence of tubulin spots in the oocyte cytoplasm were more evident when the storage time was prolonged.

Representative images of tubulin organization of preantral follicles after different times of ovary storage at 4 °C. Tubulin staining of granulosa and stromal cells is progressively reduced with increasing storage time (a-i) and bright spots of tubulin appear in the cytoplasm of follicle enclosed oocyte (d-l, white arrow). Nuclei were stained with Hoechst 33,342 (blue). Scale bar = 20 μm (a, c, e, g, i); 10 μm (b, d, f, h, l)
Effect of ovary storage at 4 °C on the DNA integrity of preantral follicles
The presence of characteristic bright points (foci) within oocyte nuclei after immunostaining for γH2AX was considered to represent the induction of DNA DSBs. The percentage of follicles expressing distinct γH2AX foci and the number of foci they presented were assessed in preantral follicles after different times of ovary storage at 4 °C (Fig. 3). Since the control samples had very few oocytes with more than three foci (Fig. 3a, A, C), detection of three or more foci\nucleus was considered an indicator of DNA damage induced by ovary storage (Fig. 3a, B, D).

a. Representative images of fluorescent immunodetection of γH2AX phosphorylation: follicle-enclosed oocytes with nuclei without bright foci (A, C) or expressing green fluorescent foci (B, D, white arrows). b Percentage (mean ± S.E.M) of preantral follicles with 3 or more foci/oocyte after different time of ovary storage at 4 °C. Nuclei were stained with propidium iodide (red). Scale bar =10 μm. Different superscripts (a, b) among storage times indicate P < 0.05. n = number of preantral follicles analyzed per storage time
Low percentages of γH2AX-positive preantral follicles with > 3 foci/oocyte were observed in the 24 h stored group (6.2 ± 2.3) and did not differ from the control group (0.3 ± 0.3%). In contrast, γH2AX-positive foci were significantly (P < 0.05) more abundant in preantral follicles from ovaries stored for 48 h (16.6 ± 2.6%), 72 h (21.8 ± 2.1%) and 96 h (23.7 ± 3.1%) compared to control and 24 h stored groups (Fig. 3b).
No staining was observed in individual granulosa cells or in any other ovarian cell types.
Experiment 2
Effect of ovary storage at 4 °C on the meiotic competence of oocytes
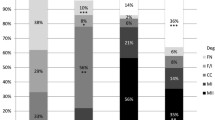
The in vitro meiotic progression of oocytes retrieved from antral follicles of ovaries stored at 4 °C for 0, 24, 48, 72 and 96 h is summarized in Table 1. A significantly higher (P < 0.05) number of oocytes in the 96 h group stayed at the GV stage compared to the other groups. The percentage of oocytes that resumed meiosis (GVBD/ MI stage) did not differ among the five groups. The percentages of oocytes reaching MII stage were similar among 0, 24 and 48 h stored groups. A significantly lower (P < 0.05) MII rate was detected following storage for 72 and 96 h. The degeneration rate significantly increased with storage time (P < 0.05).
Effect of ovary storage at 4 °C on intracellular levels of GSH, ROS, and lipid peroxidation of in vitro matured oocytes
The intracellular level of GSH was not statistically different among groups (Fig. 4a, a’).

Effect of ovary storage time at 4 °C on intracellular GSH (a) and ROS levels (b) of in vitro matured oocytes. Epifluorescence photomicrographs of MII oocytes that were stained with CellTracker Blue to determine the level of GSH (a’) and with 2′7′-dichlorodihydrofluorescein diacetate (H2DCFDA) to detect ROS (b’). Different superscripts (a, b, c, d) among storage times indicate P < 0.05. Scale bar =50 μm
A significantly higher level (P < 0.05) of ROS was recorded in in vitro matured oocytes recovered from ovaries stored for 48, 72 and 96 h compared to oocytes obtained from ovaries stored for 0 and 24 h (Fig. 4b, b’).
The levels of lipid peroxidation of in in vitro matured oocytes retrieved from ovaries stored for 72 and 96 h were higher than those in oocytes from 0, 24 and 48 h groups (Fig. 5a, b).

a Levels of lipid peroxidation as measured by the ratio of red: green fluorescence of in vitro matured oocytes recovered from ovaries stored at 4 °C for different times. b Epifluorescence photomicrographs of matured oocytes that were stained with BODIPY. Scale bar =100 μm. Different superscripts (a, b) among storage times indicate P < 0.05.
Effect of ovary storage at 4 °C on oocyte developmental competence
The proportion of oocytes that progressed to the first cleavage stage after in vitro fertilization was similar among 0, 24 and 48 h stored groups. A significantly (P < 0.05) decrease of cleavage rate was observed following 72 and 96 h of ovary storage (Table 2). The percentages of blastocysts relative to the number of cleaved embryos (developmental competence) and to the total number of oocytes (blastocyst yield) were lower in 48, 72 and 96 h stored groups compared to those in 0 and 24 h groups (Table 2, Fig. 6). Cell numbers in blastocysts from 0 and 24 h groups were similar. Only one blastocyst with 98 cells developed after 48 h of ovary storage.

In vitro produced blastocysts obtained after in vitro maturation and fertilization of oocytes recovered from ovaries stored for 0, 24 and 48 h at 4 °C under phase contrast microscopy (a, b, c) and under epifluorescent microscopy after staining with Hoechst 33,342 (a’, b′,c′). Scale bar =100 μm
Discussion
This study is the first to examine the effect of cat ovary storage at 4 °C for an extended period of time (up to 96 h) on the morphology, microtubular cytoskeleton and DNA damage in preantral follicles. We also investigated the in vitro meiotic and developmental competence of oocytes recovered from antral follicles and parameters of oxidative stress as indicators of the quality of matured oocytes. The main findings of the research were: a) storage up to 24 h did not affect the morphology and DNA integrity of preantral follicles, whereas the organization of the microtubular cytoskeleton was moderately altered; b) extended storage times (48, 72, 96 h) caused progressive structural changes of preantral follicles as showed by morphological abnormality, disassembling of microtubules and DNA damage; c) ovary storage up to 48 h did not influence the in vitro meiotic maturation ability of the oocytes retrieved from antral follicles but only the oocytes stored within the ovary for 24 h developed to blastocyst stage in percentage similar to control oocytes; d) the duration of storage did not affect the GSH levels, whereas increased levels of ROS were detected starting from 48 h and lipid peroxidation at 72 and 96 h of storage.
Hypothermic storage (0–7 °C) of organs is normally used to minimize tissue metabolism and prevent ischemic injury, and the maximum storage time depends on the specific organs. Several studies described transportation at cold temperature of mammalian, including human, ovarian tissue for fertility preservation. Indeed, the transportation of human ovarian tissue at low temperature and within a period of 4 to 26 h didn’t affect human ovarian tissue morphology, viability, and follicle development in vitro and in vivo [53,54,55,56,57]. On the other hand, a study by Klocke et al. [58] investigating the effects that storage at 4 °C for 24 or 48 h before vitrification has on the human ovarian cortex highlighted significant negative consequences within 24 h.
Some investigators [55, 56, 59] recommended cooling human ovarian tissue to low positive temperatures before cryopreservation independently of transportation purposes. The Storage of human ovarian cortex for 24 h at 5 °C before cryopreservation increased the viability of the follicles after thawing and in vitro culture in the chorioallantoic membrane system [56]. Moreover, the same group also showed, after in vitro culture, a significant increase of neo-vascularisation in human ovarian tissue pre-cooled at 5 °C for 24 h compared to non-cooled control tissue [55] and a reduced translocation of phosphatidylserine, a membrane component with a key role in the mechanisms of necrosis and apoptosis [59].
Certainly the proof of the efficiency of ovarian tissue storage at cold temperatures during transportation are the successful births reported in European countries where ovarian transportation for long distance is conducted [29, 60,61,62,63].
Evidence from animal studies has shown that chilling ovarian tissue at 4 °C during transportation provides optimal conditions to preserve follicular morphology and viability for longer times than at higher temperatures. In the mouse, storage at 4 °C for up to 24 h has no deleterious effect on histological morphology of ovarian tissue or on mature gametes [27]. Similarly, ovarian follicles from sheep and goat [23, 64] were successfully preserved for up to 24 h and ovarian follicles from zebu cows for up to 18 h [65]. In the canine [24] and equine models [25], the morphology of preantral follicles was best maintained when ovaries were stored at 4 °C for shorter periods (12 and 4 h respectively).
Our study clearly demonstrated that morphological changes in the preantral follicle population of cat ovaries stored at 4 °C was modest for at least 24 h, maintaining about 86.3% of morphologically normal follicles. Longer storage times caused a progressive damage to preantral follicles as shown by the percentages of normal follicles following 48 h (65%), 72 h (39.7%) and 96 h (20%) of storage.
Vitrification is an ideal option for preserving ovarian tissue especially for wildlife species because it doesn’t require specialized equipment and can be performed in the field. To date, there are only few studies on vitrification of feline ovarian tissue [66,67,68,69,70,71] and, despite recent progress, this procedure still requires modification and optimization. The percentage of vitrified-warmed morphologically normal follicles, immediately after warming, ranged from 5.9% [66] to a maximum 58.8% [69] under the optimal conditions. The storage at 4 °C can be a useful method for short-term preservation of ovarian tissue because the normal morphology of preantral follicles remains high even after 48 h storage, allowing xenotrasplantation into recipient animals or in vitro culture of follicles.
Information regarding the influence of cold storage on preantral follicles of cat ovaries is scarce. A study by Cocchia et al. [12] explored cell viability in cat ovaries stored at 4 °C up to 72 h and reported a storage time-dependent increase in the percentage of apoptotic areas in the ovarian tissue. Recently, the impact of holding of cat ovaries at 4 °C for 24 and 72 h followed by vitrification has been investigated by monitoring the survival and morphology of preantral follicles with neutral red staining, histology and ultrastructural analysis by transmission electron microscopy [22]. Results indicated a reduction in the number of intact and viable follicles after 24 and 72 h storage without significant differences between groups and time points. As reported by the Authors, the large variation in the number of follicles evaluated and the high standard deviations probably masked differences between groups and time points. The variability of sample numbers as well as differences in experimental design and methodology make it difficult to compare these data with our results. Indeed, cortical slices of ovaries stored for 24 and 72 h were incubated for 16 h with neutral red before viability and morphology assessment. The small number of follicles utilized for transmission electron microscopy affected the accuracy of the ultrastructural evaluation, although it revealed the preservation of subcellular structures after cooling for 24 h. Authors finally concluded that the ovarian tissue can be maintained for a maximum of 24 h at 4 °C before cryopreservation.
A further goal of our study was to investigate the response of the microtubular cytoskeleton of preantral follicles to chilling at 4 °C. The presence of an intact cytoskeleton and its dynamic organization are crucial during folliculogenesis and oogenesis [72]. The effect of cold temperature is known to induce depolimerization of microtubules [73] in mammalian cells. Chilling injuries on meiotic spindles of fully grown oocytes have been extensively reported in several species [74,75,76]. A previous study on mice [42] investigated the effect of chilling at 0 °C on the cytoskeleton organization of isolated early preantral follicles. The results showed that chilling for only 1 min was sufficient to cause depolymerization of microtubules in the oocytes and the surrounding granulosa cell layer. In the present study, an alteration of microtubular network in preantral follicles of ovaries stored at 4 °C for different times has been detected. Observed changes included tubulin depolimerization and disassembling in the granulosa cell layer and the appearance of tubulin spots in the oocytes of preantral follicles. The extent of microtubule modifications depended on the time of ovary storage: these started from 24 h of storage and became more evident following 48, 72 and 96 h. Further studies are needed to understand the reversibility of the cooling effect on the microtubular cytoskeleton. Preliminary evaluations (data not shown) indicated that the microtubule modification was fully reversible when ovaries stored at 4 °C for 24 and 48 h were re-warmed at 38.5 °C for 2 h.
Beside the morphological and structural analysis of ovarian tissue, the preservation of DNA integrity is fundamental in order to understand the effect of low temperature on preantral follicles. DNA double-strand breaks in oocytes enclosed in preantral follicles have never been investigated after storing of cat ovaries at 4 °C. Cocchia et al. [12] previously evaluated apoptosis by TUNEL assay in the cat ovarian tissue stored at 4 °C up to 72 h and indicated that the addition of the antioxidant enzyme superoxide dismutase (SOD) to transportation medium reduced cellular apoptosis, albeit these differences were not statistically relevant. Double-strand breaks of DNA (DSBs) are a marker of severe DNA damage, which can activate apoptosis [45] if not repaired. We have demonstrated that ovary storage at 4 °C for over 24 h induces DSBs in oocytes enclosed in preantral follicles and that DNA damage increases as storage is prolonged. Additional analysis is needed to assess the DNA repair capability of oocytes to ultimately confirm the exact extent of the DNA damage.
Our explorative study focuses mainly on the evaluation of morphological and structural modifications in preantral follicles in feline ovarian tissue after storage at 4 °C for a prolonged period. The next step should be to validate the quality and survival of stored ovarian tissue by assessing the developmental competence of the preantral follicles following xenotrasplantation of ovarian cortex into immunodeficient recipients or after in vitro culture.
The second part of our study was aimed to determine the adverse effects of cold storage on the oocytes harvested from antral follicles by analyzing their meiotic and developmental competence after in vitro fertilization.
Our findings indicated that when ovaries were stored at 4 °C for 24 and 48 h, oocytes retained the ability to progress to metaphase II at a rate similar to control oocytes, while a significant reduction of nuclear maturation was registered after 72 and 96 h of storage. The storage of ovarian tissue at 4 °C (at least for 48 h) may offer the possibility to preserve viability and meiotic competence of oocytes retrieved from antral follicles otherwise compromised by vitrification of ovarian tissue. Indeeed, Luvoni et al... [66] reported that oocytes recovered from vitrified ovarian tissue showed higher degeneration rate and lower rate of meiosis resumption compared to those collected from fresh tissue.
Our data corroborate the results previously reported by other Authors [4, 6, 9], showing that ovary storage at 4 °C for 24 and 48 h has no effect on the in vitro meiotic competence of cat oocytes. Conversely, Evecen et al. [77] documented that, while storing ovaries for 24 h at 4 °C did not affect the meiotic competence of oocytes in vitro, 48 h of storage decreased it dramatically. Unaffected IVM rates were reported when cat ovaries were stored at 4 °C up to 72 h; however, only oocytes stored within the ovary for 24 h could produce blastocysts after in vitro fertilization [9, 10]. These developed into live kittens after transfer to recipients [10]. Similarly, our results showed that cleavage frequency, in vitro development to blastocyst stage and blastocyst cell number did not were not different after recovery of oocytes from fresh and 24 h stored ovaries. Moreover, we observed that the developmental competence of oocytes declined markedly after 24 h of storage. Recently, Cocchia et al [12] showed that SOD supplementation in the transportation medium of domestic cat ovaries enhances embryo production after 48 and 72 h of storage.
Our rates of blastocyst formation in the 24 h stored group (22.9%) are higher than those obtained after in vitro maturation of cat oocytes in presence of relaxin [11] or with the addition of SOD during ovary storage [12].
One of the main factors contributing to the poor quality of in vitro matured oocytes may be oxidative stress (OS). Under physiological conditions the antioxidant defense mechanism of oocytes and ovaries, consisting of enzymatic antioxidants such as superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPx) and non-enzymatic antioxidants (GSH), detoxifies excess ROS maintaining the oxidant/antioxidant balance. However, increased levels of ROS beyond the physiological range may lead to OS and cause a wide range of molecular damages, including lipid peroxidation and protein and DNA damage resulting in deterioration of oocyte quality [78, 79]. The ROS scavenging ability of oocytes decreases under pathological conditions, different types environmental stress, and aging or elevated oxygen tension during in vitro manipulation [80, 81]. Glutathione is the major non-protein sulphydryl compound in mammalian cells and protects them from oxidative damage [82]. The level of GSH in oocytes increases as the oocyte resumes meiosis, and higher concentrations are found in mature oocytes than in immature ones [83,84,85].
Several studies underlined that the GSH content in oocytes at the end of IVM appears to reflect the degree of cytoplasmic maturation and quality of oocytes [86,87,88,89].
Our results showed that when ovaries were stored for different times at 4 °C the content of intracellular GSH of in vitro matured oocytes was slightly lower than that in control oocytes, but without statistical difference among groups. We speculate that, given the availability of cysteine in the maturation medium, GSH may be properly synthesized as long as the oocytes retain the ability to complete maturation. A previous study provided evidence that the availability of constitutive amino acids, in particular cysteine, is the limiting factor for GSH synthesis in mammalian oocytes [87].
Beside constant levels of GSH, the analysis of the selected oxidative stress markers in matured oocytes showed a progressive increase in ROS levels from 48 h of ovary storage associated with elevated lipid peroxidation at 72 and 96 h of storage. Lipids are highly susceptible to the attack of ROS [90] and their peroxidation may damage the oocyte membrane, modifying its structure and function.
Long-term storage of ovaries at 4 °C (72 and 96 h) may have altered the activity of other enzymes which are involved in modulating the antioxidant defense system in oocytes during in vitro maturation. Further evaluation is necessary to improve knowledge on the redox state of oocytes recovered from cold stored ovaries.
In light of these results, we suggest that oxidative stress is one of the factors hindering the developmental capacity of in vitro matured oocytes following ovary storage for more than 24 h. Strategies aimed at reducing oxidative stress, including the addition of antioxidants in the oocyte maturation medium or in the transportation medium of the ovaries, may improve oocyte quality.
Collectively, the results provide additional information on the effect of cold storage on preantral follicles and on oocytes from antral follicles and may help in setting up programs for fertility conservation, especially for wild feline species which die in geographic areas located far from ARTs centers.
Conclusions
In summary, the results of our study revealed that morphological and structural changes of preantral follicles are minimal after storage for up to 24 h. These alterations progressively increase from 48 h onwards and become more severe at 72 and 96 h of storage. The extension of the storage period beyond 24 h negatively influences the oxidative status of oocytes after in vitro maturation and their developmental competence.
References
IUCN 2018.The IUCN Red List of Threatened Species.Version 2018-1. http://www.iucnredlist.org
Pope CE, Gomez MC, Dresser BL. In vitro embryo production and embryo transfer in domestic and non-domestic cats. Theriogenology. 2006;66:1518–4.
Amstislavsky S, Lindeberg H, Luvoni G. Reproductive technologies relevant to the genome resource bank in Carnivora. Reprod Domest Anim. 2012;47:164–75.
Luu VV, Namula Z, Do LT, Sato Y, Taniguchi M, Karja NW, Otoi T. Nuclear status and DNA fragmentation of oocytes from porcine, bovine and feline ovaries stored at 4 degrees C for 5 days. Cryo Letters. 2014;35:48–53.
Naoi H, Otoi T, Shimamura T, Karja NW, Agung B, Shimizu R, Taniguchi M, Nagai T. Developmental competence of cat oocytes from ovaries stored at various temperature for 24 h. J Reprod Dev. 2007;53:271–7.
Otoi T, Murakami M, Ooka A, Karja NW, Suzuki T. Effects of size and storage temperature on meiotic competence of domestic cat oocytes. Vet Rec. 2001;148:116–8.
Johnston LA, Donoghue AM, O'Brien SJ, Wildt DE. Rescue and maturation in vitro of follicular oocytes collected from nondomestic felid species. Biol Reprod. 1991;45:898–906.
Wood TC, Montali RJ, Wildt DE. Follicle-oocyte atresia and temporal taphonomy in cold-stored domestic cat ovaries. Mol Reprod Dev. 1997;46:190–200.
Wolfe BA, Wildt DE. Development to blastocysts of domestic cat oocytes matured and fertilized in vitro after prolonged cold storage. J Reprod Fertil. 1996;106:135–41.
Pope CE, Gomez MC, King AL, Harris RF, Dresser BL. Embryos produced in vitro after recovery of oocytes from cat ovaries stored at 4 °C for 24–28 h retain the competence to develop into live kittens after transfer to recipients. Theriogenology. 2003;59:308.
Luu VV, Hanatate K, Tanihara F, Sato Y, Do LT, Taniguchi M, Otoi T. The effect of relaxin supplementation of in vitro maturation medium on the development of cat oocytes obtained from ovaries stored at 4 degrees C. Reprod Biol. 2013;13:122–6.
Cocchia N, Corteggio A, Altamura G, Tafuri S, Rea S, Rosapane I, Sica A, Landolfi F, Ciani F. The effects of superoxide dismutase addition to the transport medium on cumulus-oocyte complex apoptosis and IVF outcome in cats (Felis catus). Reprod Biol. 2015;15:56–64.
Jewgenow K, Paris MC. Preservation of female germ cells from ovaries of cat species. Theriogenology. 2006;66:93–100.
Comizzoli P, Holt WV. Recent advances and prospects in germplasm preservation of rare and endangered species. Adv Exp Med Biol. 2014;753:331–56.
Bosch P, Hernandez-Fonseca HJ, Miller DM, Wininger JD, Massey JB, Lamb SV, Brackett BG. Development of antral follicles in cryopreserved cat ovarian tissue transplanted to immunodeficient mice. Theriogenology. 2004;61:581–94.
Wiedemann C, Hribal R, Ringleb J, Bertelsen MF, Rasmusen K, Andersen CY, Kristensen SG, Jewgenow K. Preservation of primordial follicles from lions by slow freezing and xenotransplantation of ovarian cortex into an immunodeficient mouse. Reprod Domest Anim. 2012;47(Suppl 6):300–4.
Fujihara M, Comizzoli P, Keefer CL, Wildt DE, Songsasen N. Epidermal growth factor (EGF) sustains in vitro primordial follicle viability by enhancing stromal cell proliferation via MAPK and PI3K pathways in the prepubertal, but not adult, cat ovary. Biol Reprod. 2014;90:86.
Alves AE, Padilha-Nakaghi LC, Pires-Butler EA, Apparicio M, Silva N, Motheo TF, Vicente W, Luvoni GC. Viability and growth of feline preantral follicles in vitro cultured with insulin growth factor and epidermal growth factor supplemented medium. Reprod Domest Anim. 2016;52(Suppl 2):93–7.
Green LJ, Shikanov A. In vitro culture methods of preantral follicles. Theriogenology. 2016;86:229–38.
Vilela JM, Leonel EC, D'Oliveira L, Paiva RE, Miranda-Vilela AL, Amorim CA, Pic-Taylor A, Lucci CM. Culture of domestic cat ovarian tissue in vitro and in the chick embryo chorioallantoic membrane. Theriogenology. 2016;86:1774–81.
Eppig JJ, O'Brien MJ. Development in vitro of mouse oocytes from primordial follicles. Biol Reprod. 1996;54:197–207.
Martins JLA, Lopes MD, de Souza FF, Possebon FS, Wibbelt G, Jewgenow K. Cat preantral follicle survival after prolonged cooled storage followed by vitrification. Cryobiology. 2018;81:94–100.
Silva JR, Lucci CM, Carvalho FC, Bao SN, Costa SH, Santos RR, Figueiredo JR. Effect of coconut water and Braun-Collins solutions at different temperatures and incubation times on the morphology of goat preantral follicles preserved in vitro. Theriogenology. 2000;54:809–22.
Lopes CA, dos Santos RR, Celestino JJ, Melo MA, Chaves RN, Campello CC, Silva JR, Bao SN, Jewgenow K, de Figueiredo JR. Short-term preservation of canine preantral follicles: effects of temperature, medium and time. Anim Reprod Sci. 2009;115:201–14.
Gomes RG, Andrade ER, Lisboa LA, Ciquini A, Barreiros TR, Fonseca NA, Seneda MM. Effect of holding medium, temperature and time on structural integrity of equine ovarian follicles during the non-breeding season. Theriogenology. 2012;78:731–6.
Henry L, Fransolet M, Labied S, Blacher S, Masereel MC, Foidart JM, Noel A, Nisolle M, Munaut C. Supplementation of transport and freezing media with anti-apoptotic drugs improves ovarian cortex survival. J Ovarian Res. 2016;9:4.
Kamoshita K, Okamoto N, Nakajima M, Haino T, Sugimoto K, Okamoto A, Sugishita Y, Suzuki N. Investigation of in vitro parameters and fertility of mouse ovary after storage at an optimal temperature and duration for transportation. Hum Reprod. 2016;31:774–81.
Fisch B, Abir R. Female fertility preservation: past, present and future. Reproduction. 2018; 156(1):F11-F27.
Jensen AK, Kristensen SG, Macklon KT, Jeppesen JV, Fedder J, Ernst E, Andersen CY. Outcomes of transplantations of cryopreserved ovarian tissue to 41 women in Denmark. Hum Reprod. 2015;30:2838–45.
Rosendahl M, Schmidt KT, Ernst E, Rasmussen PE, Loft A, Byskov AG, Andersen AN, Andersen CY. Cryopreservation of ovarian tissue for a decade in Denmark: a view of the technique. Reprod BioMed Online. 2011;22:162–71.
Von Wolff M, Montag M, Dittrich R, Denschlag D, Nawroth F, Lawrenz B. Fertility preservation in women-a practical guide to preservation techniques and therapeutic strategies in breast cancer, Hodgkin's lymphoma and borderline ovarian tumours by the fertility preservation network. FertiPROTEKT. Arch Gynecol Obstet. 2011;284(2):427–35.
Bastings L, Liebenthron J, Westphal JR, Beerendonk CCM, Van der Ven H, Meinecke B, Montag M, Braat DDM, Peek R. Efficacy of ovarian tissue cryopreservation in a major European center. J Assist Reprod Genet. 2014;31(8):1003–12.
Woodruff TK. The Oncofertility consortium--addressing fertility in young people with cancer. Nat Rev Clin Oncol. 2010;7:466–675.
Gracia CR, Chang J, Kondapalli L, Prewitt M, Carlson CA, Mattei P, Jeffers S, Ginsberg JP. Ovarian tissue cryopreservation for fertility preservation in cancer patients: successful establishment and feasibility of a multidisciplinary collaboration. J Assist Reprod Genet. 2012;29:495–502.
Duncan FE, Zelinski M, Gunn AH, Pahnke JE, O’Neill CL, Songsasen N, Woodruff DR, Woodruff TK. Ovarian tissue transport to expand access to fertility preservation: from animals to clinical practice. Reproduction. 2016;152(6):201–10.
Armstrong AG, Kimler BF, Smith BM, Woodruff TK, Pavone ME, Duncan FE. Ovarian tissue cryopreservation in young females through the Oncofertility Consortium's National Physicians Cooperative. Future Oncol. 2018;14(4):363–78.
Irving Zeidman MD. Chemical factors in the mutual adhesiveness of epithelial cells. AACR Journals. 1947;7:386–9.
Ueda MJ, Takeichi M. Two mechanisms in cell adhesion revealed by effects of divalent cations. Cell structure and function. 1976;1:377–88.
Bristol-Gould S, Woodruff TK. Folliculogenesis in the domestic cat (Felis catus). Theriogenology. 2006;66:5–13.
Dowing KH, Nogales E. Tubulin and microtubule structure. Cell Biology. 1998;10:16–22.
Comizzoli P, Wildt DE, Pukazhenthi BS. Effect of 1,2-propanediol versus 1,2-ethanediol on subsequent oocytes maturation, spindle integrity, fertilization, and embryo development in vitro in the domestic cat. Biol Reprod. 2004;71:598–604.
Vanhoutte L, Cortvrindt R, Nogueira D, Smitz J. Effects of chilling on structural aspects of early preantral mouse follicles. Biol Reprod. 2004;70:1041–8.
Yin XJ, Lee YH, Jin JY, Kim NH, Kong IK. Nuclear and microtubule remodeling and in vitro development of nuclear transferred cat oocytes with skin fibroblasts of the domestic cat (Felis silvestris catus) and leopard cat (Prionailurus bengalesis). Anim Reprod Sci. 2006;95:307–15.
Jin YX, Cui XS, Yu XF, Han JY, Kong IK, Kim NH. Alterations of spindle and microfilament assembly in aged cat oocytes. Reprod Dom Anim. 2010;45:865–71.
Petrillo SK, Desmeules P, Truong TQ, Devine PJ. Detection of DNA damage in oocytes of small ovarian follicles following phosphoramide mustard exposures of cultured rodent ovaries in vitro. Toxicol Appl Pharmacol. 2011;253:94–102.
Bogliolo L, Leoni G, Ledda S, Zedda MT, Bonelli P, Madau L, Santucciu C, Naitana S, Pau S. M-phase promoting factor (MPF) and mitogen activated protein kinases (MAPK) activities of domestic cat oocytes matured in vitro and in vivo. Cloning Stem Cells. 2004;6(1):15–23.
Sovernigo TC, Adona PR, Monzani PS, Guemra S, Barros F, Lopes FG, Leal C. Effects of supplementation of medium with different antioxidants during in vitro maturation of bovine oocytes on subsequent embryo production. Reprod Domest Anim. 2017;52(4):561–9.
Sobinoff AP, Beckett EL, Jarnicki AG, Sutherland JM, McCluskey A, Hansbro PM, McLaughlin EA. Scrambled and fried: cigarette smoke exposure causes antral follicle destruction and oocyte dysfunction through oxidative stress. Toxicol Appl Pharmacol. 2013;271(2):156–67.
Bogliolo L, Leoni G, Ledda S, Naitana S, Zedda M, Carluccio A. Pau S:intracytoplasmic sperm injection of in vitro matured oocytes of domestic cats with frozen-thawed epididymal spermatozoa. Theriogenology. 2001;56(5):955–67.
Niżanski W, Dejneka GJ, Klimowicz M, Dubiel A. Evaluating some properties of domestic cat epididymal spermatozoa and their cryopreservation. Med Weter. 2005;61:173–8.
Tsutsui T, Wada M, Anzai M, Hori T. Artificial insemination with frozen epididymal sperm in cats. J Vet Med Sci. 2003;3:397–9.
Sananmuang T, Tharasanit T, Nguyen C, Phutikanit N, Techakumphu M. Culture medium and embryo density influence on developmental competence and gene expression of cat embryos. Theriogenology. 2011;75:1708–19.
Schmidt KL, Ernst E, Byskov AG, Andersen AN, Andersen CY. Survival of primordial follicles following prolonged transportation of ovarian tissue prior to cryopreservation. Hum Reprod. 2003;18:2654–9.
Isachenko E, Isachenko V, Nawroth F, Rahimi G, Weiss JM. Effect of long-term exposure at suprazero temperatures on activity and viability of human ovarian cortex. Fertil Steril. 2009;91:1556–9.
Isachenko V, Isachenko E, Mallmann P, Rahimi G. Long-time cooling of human ovarian tissue before cryopreservation as obvious procedure: stimulation of follicular development and neo-vascularisation. Clin Lab. 2012;58:1293–300.
Isachenko V, Isachenko E, Mallmann P, Rahimi G. Increasing follicular and stromal cell proliferation in cryopreserved human ovarian tissue after long-term precooling prior to freezing: in vitro versus chorioallantoic membrane (CAM) xenotransplantation. Cell Transplant. 2013;22:2053–61.
Laronda MM, Duncan FE, Hornick JE, Xu M, Pahnke JE, Whelan KA, Shea LD, Woodruff TK. Alginate encapsulation supports the growth and differentiation of human primordial follicles within ovarian cortical tissue. J Assist Reprod Genet. 2014;31(8):1013–28.
Klocke S, Tappehorn C, Griesinger G. Effects of supra-zero storage on human ovarian cortex prior to vitrification-warming. Reprod BioMed Online. 2014;29(2):251–8.
Isachenko V, Todorov P, Isachenko E, Rahimi G, Tchorbanov A, Mihaylova N, Manoylov I, Mallmann P, Merzenich M. Long-time cooling before cryopreservation decreased translocation of phosphatidylserine (Ptd-L-Ser) in human ovarian tissue. PLoS One. 2015;10(6):e0129108.
Andersen CY, Rosendahl M, Byskov AG, Loft A, Ottosen C, Dueholm M, Schmidt KL, Andersen AN, Ernst E. Two successful pregnancies following autotransplantation of frozen/thawed ovarian tissue. Hum Reprod. 2008;23:2266–72.
Ernst E, Bergholdt S, Jorgensen JS, Andersen CY. The first woman to give birth to two children following transplantation of frozen/thawed ovarian tissue. Hum Reprod. 2010;25:1280–1.
Dittrich R, Lotz L, Keck G, Hoffmann I, Mueller A, Beckmann MW, van der Ven H, Montag M. Live birth after ovarian tissue autotransplantation following overnight transportation before cryopreservation. Fertil Steril. 2012;97:387–90.
Muller A, Keller K, Wacker J, Dittrich R, Keck G, Montag M, Van der Ven H, Wachter D, Beckmann MW, Distler W. Retransplantation of cryopreserved ovarian tissue: the first live birth in Germany. Dtsch Arztebl Int. 2012;109:8–13.
Andrade ER, Rodrigues AP, Amorim CA, Carvalho FC, Dode MA, Figueiredo JR. Short term maintenance of sheep preantral follicles in situ in 0.9% saline and Braun-Collins solution. Small Rumin Res. 2001;41:141–9.
Lucci CM, Kacinskis MA, Rumpf R, Bao SN. Effects of lowered temperatures and media on short-term preservation of zebu (Bos indicus) preantral ovarian follicles. Theriogenology. 2004;61:461–72.
Luvoni GC, Tessaro I, Apparício M, Ruggeri E, Luciano AM, Modina SC. Effect of vitrification of feline ovarian cortex on follicular and oocyte quality and competence. Reprod Domest Anim. 2012;47(3):385–91.
Tanpradit N, Comizzoli P, Srisuwatanasagul S, Chatdarong K. Positive impact of sucrose supplementation during slow freezing of cat ovarian tissues on cellular viability, follicle morphology, and DNA integrity. Theriogenology. 2015;83:1553–61.
Brito DCC, Domingues SFS, Silva JK, Wu X, Santos RR, Pieczarka JC. Detrimental effect of phenol red on the vitrification of cat (Felis catus) ovarian tissue. Biopreserv Biobank. 2016;14:17–22.
Mouttham L, Comizzoli P. The preservation of vital functions in cat ovarian tissues during vitrification depends more on the temperature of the cryoprotectant exposure than on the sucrose supplementation. Cryobiology. 2016;73(2):187–95.
Brito DCC, Domingues SFS, Rodrigues APR, Maside C, Lunardi FO, Wu X, Figueiredo JR, Pieczarka JC, Santos RR. Cryopreservation of domestic cat (Felis catus) ovarian tissue: comparison of two vitrification methods. Theriogenology. 2018;111:69–77.
Demirel MA, Acar DB, Ekim B, Çelikkan FT, Alkan KK, Salar S, Erdemli EA, Özkavukçu S, Yar SS, Kanca H, Baştan A. The evaluation of xenotransplantation of feline ovarian tissue vitrified by needle immersed vitrification technique into male immunodeficient mice. Cell Tissue Bank. 2018;19(1):133–47.
Albertini DF. Cytoplasmic microtubular dynamics and chromatin organization during mammalian oogenesis and oocyte maturation. Mutat Res. 1992;296:57–68.
Brinkley BR, Cartwright J Jr. Cold-labile and cold-stable microtubules in the mitotic spindle of mammalian cells. Ann N Y Acad Sci. 1975;253:428–39.
Pickering SJ, Johnson MH. The influence of cooling on the organization of the meiotic spindle of the mouse oocyte. Hum Reprod. 1987;2:207–16.
Vincent C, Johnson MH. Cooling, cryoprotectants, and the cytoskeleton of the mammalian oocyte. Oxf Rev Reprod Biol. 1992;14:73–100.
Liu RH, Sun QY, Li YH, Jiao LH, Wang WH. Effects of cooling on meiotic spindle structure and chromosome alignment within in vitro matured porcine oocytes. Mol Reprod Dev. 2003;65:212–8.
Evecen M, Cirit U, Demir K, Karaman E, Hamzaoglu AI, Bakirer G. Developmental competence of domestic cat oocytes from ovaries stored at various durations at 4 degrees C temperature. Anim Reprod Sci. 2009;116:169–72.
Devine PJ, Perreault SD, Luderer U. Roles of reactive oxygen species and antioxidants in ovarian toxicity. Biol Reprod. 2012;86:27.
Prasad S, Tiwari M, Pandey AN, Shrivastav TG, Chaube SK. Impact of stress on oocyte quality and reproductive outcome. J Biomed Sci. 2016;23:36.
Combelles CM, Gupta S, Agarwal A. Could oxidative stress influence the in-vitro maturation of oocytes? Reprod BioMed Online. 2009;18:864–80.
Lord T, Aitken RJ. Oxidative stress and ageing of the post-ovulatory oocyte. Reproduction. 2013;146:217–27.
Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta. 2003;333:19–39.
Perreault SD, Barbee RR, Slott VL. Importance of glutathione in the acquisition and maintenance of sperm nuclear decondensing activity in maturing hamster oocytes. Dev Biol. 1988;125:181–6.
Yoshida M, Ishigaki K, Nagai T, Chikyu M, Pursel VG. Glutathione concentration during maturation and after fertilization in pig oocytes: relevance to the ability of oocytes to form male pronucleus. Biol Reprod. 1993;49:89–94.
Miyamura M, Yoshida M, Hamano S, Kuwayama M. Glutathione concentration during maturation and fertilization in bovine oocytes. Theriogenology. 1995;43:282.
Funahashi H, Day BN. Effect of cumulus cells on glutathione content of porcine oocytes during in vitro maturation. J Anim Sci. 1995;73:90.
Furnus CC, de Matos DG, Picco S, Garcia PP, Inda AM, Mattioli G, Errecalde AL. Metabolic requirements associated with GSH synthesis during in vitro maturation of cattle oocytes. Anim Reprod Sci. 2008;109:88–99.
Jiao GZ, Cao XY, Cui W, Lian HY, Miao YL, Wu XF, Han D, Tan JH. Developmental potential of prepubertal mouse oocytes is compromised due mainly to their impaired synthesis of glutathione. PLoS One. 2013;8:e58018.
Abazari-Kia AH, Mohammadi-Sangcheshmeh A, Dehghani-Mohammadabadi M, Jamshidi-Adegani F, Veshkini A, Zhandi M, Cinar MU, Salehi M. Intracellular glutathione content, developmental competence and expression of apoptosis-related genes associated with G6PDH-activity in goat oocyte. J Assist Reprod Genet. 2014;31:313–21.
Repetto M, Semprine J, Boveris A. Lipid peroxidation: chemical mechanism, biological implications and analytical determination. In: InTech, editor. Lipid Peroxidation; 2012. p. 4–29.
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Author information
Authors and Affiliations
Contributions
AP, LB designed the study, analyzed data and wrote the paper. AP, GPB, FA, SG, DB, LF, EA, LS conducted experimental procedures of the study and collaborated in analyzing data and drafting the paper. SP and MT Z performed ovariectomies. LB supervised experiments and performed critical data analysis. All authors read and approved the paper.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No experimental animals have been used in this work. The samples (ovarian tissues) were collected during routine ovariectomies at the Veterinary Teaching Hospital of the University, following the basic criteria of good veterinary surgery practices.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Piras, A.R., Burrai, G.P., Ariu, F. et al. Structure of preantral follicles, oxidative status and developmental competence of in vitro matured oocytes after ovary storage at 4 °C in the domestic cat model. Reprod Biol Endocrinol 16, 76 (2018). https://doi.org/10.1186/s12958-018-0395-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-018-0395-1