
Abstract
Background
Tracking targets of natural products is one of the most challenging issues in fields ranging from pharmacognosy to biomedicine. It is widely recognized that the biocompatible nanoparticle (NP) could function as a “key” that opens the target “lock”.
Results
We report a functionalized poly-lysine NP technique that can monitor the target protein of arctigenin (ATG) in vivo non-invasively. The NPs were synthesized, and their morphologies and surface chemical properties were characterized by transmission electron microscopy (TEM), laser particle size analysis and atomic force microscopy (AFM). In addition, we studied the localization of ATG at the level of the cell and the whole animal (zebrafish and mice). We demonstrated that fluorescent NPs could be ideal carriers in the development of a feasible method for target identification. The distributions of the target proteins were found to be consistent with the pharmacological action of ATG at the cellular and whole-organism levels.
Conclusions
The results indicated that functionalized poly-lysine NPs could be valuable in the multimodal imaging of arctigenin.
Similar content being viewed by others


Background
Natural products have supplied a myriad of compounds with complex structures as well as diverse biological activities. Conventional natural products research, which involves structural determinations and chemical syntheses of complex natural products, was highly successful until the 1980s. Unfortunately, such research became less productive in the 1990s [1, 2]. It has become significantly more difficult to find the targets of natural products or discover new concepts in chemistry and biology from natural product binding. Therefore, natural products research needs to employ novel strategies based on targets that are involved in biological phenomena and processes [3, 4]. However, it is widely recognized that exploring targets of natural products is an immense undertaking, although many successful methods for identifying targets of small molecules have been reported [5], such as FG-beads technology [6], photoaffinity beads technology [7], the fishing-rod strategy [8], the “compact” molecular probe (CMP) approach [9], the yeast 3-hybrid (Y3H) system [10], and the database-dependent (DBD) strategy [11, 12]. These technologies have conventionally been used for identifying targets of small molecules [13]; however, the most serious failing of these approaches is the difficulty in target tracking in situ [14]. Although the use of quantum dots for tags could solve the problem of target labeling, the fact that these probes are so large that their localization is not totally in situ cannot not be ignored. More importantly, the problems of obtaining the localization information of the target protein from the body, tissue or cells, excluding non-specific protein interference, and elucidating the relationship between the corresponding bioactivity and indications remain a challenge.
Recently, it was discovered that some nanoparticles (NPs) with unique physical properties have wide usage in a variety of fields, such as the life sciences, pharmacology, and the environmental sciences [15,16,17,18]. Interestingly, NPs are an important tool, which had drawn significant attention in the fields of molecule imaging and protein capturing [19, 20]. The past 5 years have witnessed several advances that have established luminescent NPs as a highly sensitive technology for real-time optical imaging in small animals [21, 22]. Additionally, recent developments have demonstrated that such NPs could be directed against specific cells through proper surface functionalization with targeting ligands [23, 24]. Despite great effort being devoted to the development of bio-functional self-assembling materials [25], the use of self-assembling NPs in identifying natural product targets is largely unexplored. Indeed, imaging in multiple organisms could prove to be a powerful analytical tool in revealing the targets of natural products.
In this strategy, we used arctigenin (ATG), a phenyl propanoid dibenzyl butyrolactone lignan from Arctium lappa L. (commonly known as burdock) as a model compound. It has been widely used in traditional Chinese medicine (TCM) and in Europe and North America for hundreds of years [26, 27]. The biological activities and pharmacological functions reported for ATG include anti-inflammatory, anti-cancer, anti-diabetic, anti-heat shock, antimicrobial and antiviral activities [27,28,29,30,31]. In particular, the target identification process may lead to the discovery biologically intriguing phenomena that could open up new frontiers in chemical biology.
Therefore, we attempted to investigate the targets of ATG using self-assembling NPs. We report here that the use of ATG conjugated poly-lysine fluorescent NPs would be advantageous in such cases; they enable visualization in contrast to affinity beads, and affinity between the ligand and its target is retained. By using the self-assembling NP strategy, we succeeded in imaging the cytosolic target protein of ATG in vivo. This integrated system solved the issue of a natural product interacting with multiple targets and turning on diverse biological activities. This technique may help discover the targets of natural products and benefit drug research and development.
Methods
Reagents and materials
Propargylic acid was purchased from Sigma-Aldrich (St. Louis, U.S.A.). N-Hydroxysuccinimide (NHS) and 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) were purchased from Alfa Aesar (Massachusetts, U.S.A.). Arctigenin (ATG) was purchased from Phytomarker Co., Ltd. (Tianjin, China). Poly-lysine with an average molecular weight of 20 kDa was purchased from Zhengzhou Bainafo Bioengineering Co., Ltd. (Henan, China). NHS-rhodamine and sulfo-SADP were purchased from Pierce (Rockford, U.S.A.). All other chemicals and solvents used were analytical grade. The water used throughout this work was reagent-grade water produced by the Milli-Q Water Purification System of Nihon Millipore Ltd. (Tokyo, Japan).
The self-assembly and modification of poly-lysine fluorescent NPs
Briefly, peptide solutions were prepared by dissolving the poly-lysine powder (100 mg, 5 μmol) in 10 mL of phosphate-buffered saline (PBS) buffer (pH 7.4), and then NHS-rhodamine (2 mg, 3.8 μmol) was thoroughly dispersed in an ultrasonic bath. In a reactor equipped with a stirrer, an organic mixture of 200# petrol (13 mL), tetrachloromethane (12 mL), and span-80 (1 mL) (1/1/0.08, v/v/v, respectively) were stirred for 30 min at room temperature. The mixture of the organic and peptide solutions was thoroughly dispersed using a homogenizer (FA25, Fluco, England), added to the reactor, and dispersed at 12,000 rpm. Subsequently, petroleum ether was added slowly to the mixture. Immediately after the delamination process, PBS solution was added, and self-assembly occurred during the biphasic equilibrium process to room temperature. The aqueous phase was collected, and the process was repeated 3 times to obtain poly-lysine fluorescent NPs. The crude product was centrifuged for 10 min at 1000 rpm, and the precipitate was discarded. Finally, the supernatant was centrifuged for 10 min at 12,000 rpm. The supernatant fluid containing the poly-lysine fluorescent self-assembling NPs was stored at 4 °C.
Sulfo-SADP (15 mg, 0.03 mmol) was dissolved in 0.2 mL of borate buffer (0.2 mol/L pH 8.0), and 1 mL poly-lysine fluorescent NPs was added. The mixture was stirred at room temperature for 4 h. The azide functionalized poly-lysine fluorescent NPs were collected by ultrafiltration (Millipore, USA) and washed with 20 mL of phosphate-buffered saline (PBS, pH 7.4) several times until sulfo-SADP could no longer be detected in the filtrate. The general strategy for synthesis of blank fluorescent NPs (BF NPs) is shown in Fig. 1A.

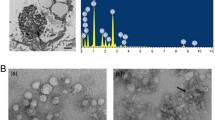
The characterization of NPs. A The general strategy for the synthesis of blank fluorescent NPs (BF NPs) and ATG conjugated poly-lysine fluorescent NPs (AF NPs). B TEM images of AF NPs; scale bar 100 nm. The particle size distribution of AF NPs was determined; the mean diameter of the NPs was 33 nm. AFM images of AF NPs were obtained; the height of the NPs was 32.54 nm
Drug conjugation to poly-lysine fluorescent NPs
The synthesis of alkynyl-ATG is depicted in the electronic Additional file 1 (scheme 1). Alkynyl-ATG (17 mg, 0.04 mmol) was dissolved in 0.2 mL of borate buffer (0.2 mol/L pH 8.0), and 2 mL of azide BF NPs was added, followed by the catalyst (0.2 mmol/L Tris-triazoleamine, 1.0 mmol/L CuSO4, and 2.0 mmol/L sodium ascorbate). The mixture was stirred at room temperature for 1 h. The functionalized NPs were was added to a 50 mL 3kD ultrafiltration tube and washed with 20 mL of PBS several times until small molecules could no longer be detected in the filtrate. The general strategy for synthesis of ATG bearing poly-lysine fluorescent NPs (AF NPs) is shown in Fig. 1A.
Characterization of NPs
A 10-μL portion of the sample solution was placed on newly clipped mica and washed with 1 mL of distilled water and then air-dried. The samples were examined using an atomic force microscope (AFM, Veeco Company, Multimode, NanoIIIa) in tapping mode in air at room temperature. The morphology and structure of the functionalized NPs were studied using a transmission electron microscope (JEOL JEM-2010, USA). The particle hydrodynamic size was measured using a Counter laser size analyzer (Beckman Coulter, USA). The PEI coating of the NPs was checked using a Perkin-Elmer (Norwalk, USA) Spectrum GX Fourier transform infrared (FT-IR) spectrometer (Nicolet NEXUS 670) and KBr pellets. The chemical and physical stability of AF NPs was evaluated during 1 month storage at 4 °C, in terms of mean particle size, polydispersity index(PDI) and zeta potential. Visual inspection was also employed to detect any crystallization process or mold formation.
Zebrafish imaging in vivo
Zebrafish (AB strain) were cultured under a 14 h light/10 h dark cycle at 28.5 °C. Fertilized eggs were collected and cultured in standard system water (KCl 0.05 g/L, NaHCO3 0.025 g/L, NaCl 3.5 g/L, and CaCl2 0.1 g/L, with 1 mg/L methylene blue, pH 7.0) until 4 days post fertilization (dpf). Zebrafish larvae at 4 dpf were exposed to BF NPs and AF NPs for 1, 2 and 3 h, respectively. Zebrafish were anesthetized by immersing in tricaine for bioimaging. The specimens were photographed with a fluorescence stereomicroscope (OLYMPUS SZX 10, Japan) fitted with a digital camera. The parameters of the camera remained unchanged when taking pictures for the untreated control groups, BF NP groups and AF NP groups. The larvae of each group were collected and fixed in 4% paraformaldehyde (pH 7.0) for tissue sectioning.
Cell culture and animals
The BEAS-2B cell line, derived from human bronchial epithelial cells, was obtained from the American Type Culture Collection (Rockville, MD). BEAS-2B cells were cultured using DMEM/F12 medium that was supplemented with 10% FBS, 100 U/mL penicillin and 0.1 mg/mL streptomycin and maintained at 37 °C in 5% CO2. The medium was replenished every 3 days, and the cells were sub-cultured once they reached 80–90% confluence. Male Kunming mice (20–22 g) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd. (Beijing, China). All animals were kept in an animal room with a temperature of 23 ± 2 °C, a humidity of 60 ± 5%, and a 12-h dark to light cycle, with free access to food and water. The animals were housed under the above conditions for a 3-day acclimation period.
Animal model of acute lung injury (ALI) and treatment
Thirty animals were randomly divided into three groups—blank, control and model groups—each containing ten mice. Lipopolysaccharide (LPS) (5 mg/kg) was administered intratracheally to induce ALI. NPs (10 mg/kg) were intravenously injected 1 day after LPS administration [29]. The chosen doses of these drugs were based on our previous studies and preliminary experiments.
Mouse imaging in vivo
The mice were anaesthetized intraperitoneally with chloral hydrate (10 g/kg). Chronic bronchitis was induced by endotracheal instillation of LPS, and this was followed by NP injection (0.2 mL) through the caudal vein 1 day later [32]. After the disease was successfully induced, the animals were randomly divided into two groups. Model groups received AF NPs, intravenous injection (i.v.), and were subjected to in vivo imaging continuously for 1 h. Control groups received BF NPs, and the above procedure was repeated. The excitation wavelength was 550 nm, and the emission wavelength range was 600–700 nm.
The mice (n = 10) were sacrificed at pre-determined time points immediately after the imaging procedure. Blood was dropt by heart puncture. Then a peristaltic pump with saline flushing was used to wash the vessels through the ventricles until the eluate was colorless. The organs (heart, liver, lung, kidney, and spleen) of the control and model groups were harvested and cooled in liquid nitrogen. Then, the organs were ground repeatedly until they were powdered. Anhydrous ethanol was added to the powder, and it was sonicated for 5 min in ice-cold water. Then, the mixture was centrifuged for 10 min at 12,000 rpm. The supernatant fluid containing rhodamine-bound poly-lysine polymers was collected, and these were detected using a luminoscope (Tecan Safire 2 type, Switzerland). The excitation wavelength was 550 nm and the emission wavelength 592 nm. A part of the lung tissue was harvested and quickly fixed in 4% paraformaldehyde. The lung tissue was dehydrated, paraffin embedded, and sliced. After hematoxylin and eosin (H&E) staining of lung tissue slices, pathological changes were observed using a CKX41 microscope (Olympus Co., Japan).
Cellular localization of poly-lysine fluorescent NPs
BEAS-2B cells were washed with pre-cooled PBS and treated with 100 μL of AF NPs or BF NPs. After 3 h of incubation, the cells were washed with PBS, fixed using 4% paraformaldehyde, and permeabilized with 1% Triton X-100 for 30 min. Then, the cells were labeled with DAPI, and fluorescence images were obtained with a confocal microscope (TCS SP5, Leica, Germany). The excitation wavelengths were 365 and 552 nm, and the emission wavelengths were 465 and 590 nm, respectively.
In parallel, cells were incubated at 37 °C for 6 h, and then, 0.25% trypsin was added (Gibco, 25200-056, final concentration 0.05%), and the sample was incubated for 1 min. The cell suspension was washed in DMEM/10% FBS to inactivate trypsin, followed by two washes in PBS. Digested cells were then resuspended in Hank’s balanced salt solution containing 1% bovine serum albumin, 2 mM EDTA (FC buffer) and kept at 4 °C before flow cytometry analysis.
Two aliquots were placed in tubes for flow cytometry analysis. Fluorescence-minus-one controls were generated for the NPs used and compensations were set using BD Plus CompBeads and FACSDiva software. Data collection was performed using a Becton–Dickinson LSR II flow cytometer with ultraviolet (20 mW), violet (25 mW), blue (20 mW) and red (17 mW) lasers with the default filter configuration and utilizing the High Throughput Sampler attachment. At least 10,000 events were collected per well. The results were analyzed using Cell Quest software (Becton–Dickinson Co., USA). FCS 3.0 files were exported to Flow Jo version 9.3. In all cases, dead cells and doublets were excluded prior to analyzing marker expression.
Results
NP molecular design and morphology assay
Self-assembling NPs have been extensively explored as an efficient carrier for fluorescent probes and drug delivery due to their potential ability to label the target. We therefore designed and synthesized azido functionalized poly-lysine fluorescent NPs and alkynyl modified ATG for drug loading. The proposed ATG carrying fluorescent NPs (AF NPs) were formed based on click chemistry. The poly-lysine in the NPs might have two functions. One is to increase the hydrophilicity of the resulting peptides for self-assembly. The other is to endow the self-assembling NPs with active amino-groups to link fluorescence dyes and functional groups. The alkynyl modified ATG could be easily obtained by standard organic synthesis and was purified by HPLC. The purity and identity of the compound was determined by 1H NMR and 13C NMR (see Additional file 1: Figure S1).
After successful synthesis, the morphology of the self-assembling NPs was characterized by TEM. As shown in Fig. 1B, poly-lysine self-assembled into uniform NPs and showed a regular spherical structure. Figure 1B presents the particle size distribution of the NPs; the mean diameter was approximately 33 nm, which was consistent with the results of TEM analysis. The AFM images of the AF NPs are shown in Fig. 1B. The similar morphologies observed in the AFM images are noteworthy. The height of each NP, which as approximately 32.54 nm, was similar to that observed with TEM. These results suggested that functionalization of the poly-lysine NPs had negligible effects on their self-assembling properties and morphologies. The results of stability studies of AF NPs during 1 month storage at 4 °C were stable, maintaining nanometric dimensions of the dispersed particles and PDI and Zeta potential values similar to those of the corresponding freshly prepared systems. It suggested the good physical–chemical stability and highlighting the preservative effect of poly-lysine.
Zebrafish imaging in vivo
To examine the tissue localization of NPs in zebrafish, both BF NPs and AF NPs were added to standard system water for 1–3 h, and they were visualized in vivo by fluorescence detection. It was apparent that in the non-NP-treated control group, development was not disturbed, and no fluorescence was detected, whereas other groups of zebrafish showed specific fluorescence (Fig. 2A). Interestingly, the fluorescence intensity of BF NP-treated zebrafish remained almost unchanged from 1 to 2 h. Notably, the fluorescence intensity was significantly clustered and increased with an increase in AF NP incubation time. Tissue localization analysis showed that the fluorescent AF NPs were present as an intense red on the gills of the zebrafish, especially in the epithelium of the branchial arch, as shown in Fig. 2B. In contrast, with BF NPs, a weak fluorescence was observed 2 h after treatment. These results clearly indicated that AF NPs were ideal carriers in developing a suitable method for target identification, as they were specifically located on the gills of the zebrafish.

BF NP and AF NP bioimaging in 4 dpf zebrafish in vivo. A The untreated control group is represented in a; b and c represent BF NPs treated for 1 and 2 h, respectively; d, e and f represent AF NPs treated for 1, 2 and 3 h, respectively. B Sections from the treated groups of BF NPs (a–c) and the treated groups of AF NPs (d–f) at 2 h post treatment are shown. The scale bar indicates 500 µm
Evaluation of targeting specificity in vivo
The targeting specificity of AF NPs was determined by fluorescent in vivo imaging. BF NPs and AF NPs were administered to control and LPS-induced pneumonia model mice, respectively, via tail vein injection. After 30 min of observation, only AF NPs were found enriched in the model group (Fig. 3B), and especially in the thoracic cavity. There was also a slight enrichment of BF NPs at the same site. No significant differences were found in the organs of the control group (Fig. 3A). To further evaluate their tissue distribution, NPs were isolated from the organs. The fluorescence intensity of rhodamine from the different organs was determined and is presented in Fig. 3C. There were significant differences in fluorescence intensity between the groups. Compared with the control group, LPS stimulation resulted in a prominent increase in the lung (P < 0.01) and a slight increase in the heart sizes (P < 0.05). However, other organs showed no significant changes (P > 0.05). Additionally, the liver, spleen and kidneys showed no significant changes in morphology after the injection of AF NPs. H&E staining of lung slices (Fig. 3D) indicated greater numbers of AF NPs in the bronchial epithelial cells of the lung in the model group (indicated as arrows in Fig. 3D). BF NPs were not found in lung slices in the model group indicating that the specific targets of AF NPs were inflammation-associated epithelial cells in the lung tissue.

BF NP and AF NP bioimaging in mice in vivo. A Representative images from control mice. The bar plot shows the distribution of fluorescence signal intensity. B Representative images from model mice. C Bar plots represent the concentrations of NPs in different tissues. The values shown represent the mean values ± sem. D Immunohistochemical staining was performed using diverse types of lung tissue
Cross-membrane uptake of AF NPs
We investigated the ability of AF NPs to cross the membrane barrier and bind to its targets in vitro (Figs. 4A a, e). For this purpose, DAPI (4′,6-diamidino-2-phenylindole) and then NPs were added to the culture medium of BEAS cells, which were imaged by confocal microscopy (Figs. 4A b, f). Treatment with BF NPs did not result in effective internalization in cultured BEAS-2B cell lines (Fig. 4A g). However, the fluorescence signal could be clearly visualized in AF NP-treated BEAS-2B cells, indicating their internalization by the cells (Fig. 4A c). As shown in Figs. 4A d, h, our data demonstrated the ability of AF NPs to cross the membrane and bind with the target protein in the cytoplasm. The results of flow cytometry analysis suggested that the fluorescence intensity of rhodamine positive cells was markedly increased from 2 × 103 in the BF NP group to 104 in the AF NP group. Interestingly, the side scatter (SSC) of both groups remained almost unchanged (Fig. 4B). These analyses showed that the AF NPs were highly specific and could be a reliable tool for targeted protein capture.

Target localization using BF NPs and AF NPs. A General strategy for fluorescent labeling of the target protein. BEAS-2B cells were treated with AF or BF NPs, followed by the DAPI fluorescent marker. Fluorescence and DIC images are shown. All scale bars 20 μm. B The histogram of flow cytometry analysis is shown in a. The scatter diagram of flow cytometry analysis is shown in b
Discussion
As is generally known, tissue targeting can be achieved by taking advantage of the enhanced permeability and retention time resulting from a chaotic tissue microenvironment [33]. The molecules in circulation cannot reach the tubule until they are filtered by the glomerular filtration barrier. It is estimated that the mean diameter of the glomerular filtration barrier is approximately 40–60 nm. The sizes of the target NPs should not exceed this range [34]. Fortunately, multiple observations confirmed that the mean diameter of our AF NPs was approximately 33 nm, and grafting with poly-lysine preserved its biocompatibility. Furthermore, the concept is based on ATG itself having high affinity for its target and modification of a functional probe to keep its original affinity intact. Structure–activity relationship studies have shown that modifications at the hydroxyl group of ATG had a limited impact on its biological activity [35]. Therefore, the combination of alkynylated ATG and azide-modified NPs with introduction of rhodamine by copper-catalyzed azide-alkyne cycloaddition (CuAAC) [36] was expected to give a good signal-to-noise ratio in target identification experiments. The present approach is different from traditional magnetic protein capture, enrichment or fluorescent NP location tracking [37] and provided optimum results in which not only the specific target was traced but also enriched by the same AF NPs tool. The experiments demonstrated that ATG-bound self-assembling fluorescent NPs are a highly sensitive optical tool to monitor the target in real time and are especially useful for intracellular target protein enrichment [38, 39].
Fluorescence tracking showed that ATG conjugation effectively eliminated non-specific distribution and improved the tissue-targeting efficiency. Interestingly, the AP NPs localized in the epithelium of the branchial arch in zebrafish. This result is somewhat at odds with the observation that the lung is the major site of ATG localization under standard physiological conditions [29].We also noticed a seemingly selective biodistribution in the lung of the LPS-induced pneumonia mouse. This result is in good agreement with a large body of pharmaceutical research on ATG; the fruits of Arctium lappa L. are an often-used herbal drug in TCM for the treatment of respiratory diseases [31]. Lung homing of ATG, as determined by H&E staining and analysis of tissues, is most certainly associated with a rapid stress reaction in the lung capillary bed and is typical of tissue targeting based on enhanced permeability.
An important problem observed in target identification using molecular probes is that the presence of linker and tag moieties may decrease the affinity with the target as well as cause nonspecific adsorption. Reduction of nonspecific binding to proteins is important for the success of target identification approaches.
The mechanism of action of ATG appears to be multifaceted but was not fully understood previously. It has been proposed that the immunomodulatory effects of ATG were partly responsible for its anti-inflammatory activity [40,41,42] as well as its therapeutic value in the treatment of acute lung injury [29, 31]. Identification and validation of targets for bioactive small molecules is important in chemical biology and drug development. Recent technological developments and innovations have contributed significantly to these efforts. Herein, we focused on an approach involving a novel herbal remedy conjugated to poly-lysine fluorescent self-assembling NPs that enables the tracking of bioactive compounds to detect cytosolic targets.
Conclusions
The strategy was shown to be effective for the synchronous tracking of targets. Additionally, our results indicate that the identified targets of ATG are strongly linked with its pharmacologic action. Although the generality of this approach needs to be verified by testing many other molecules, the results suggest the utility of the NP approach in increasing the success rates of the biochemical labeling of protein targets of natural products. It is important to choose other appropriate methods depending on the small molecule; multiple approaches may be necessary to accelerate target identification and understand the mode of action of investigative compounds.
References
Ganesan A. The impact of natural products upon modern drug discovery. Curr Opin Chem Biol. 2008;12:306–17.
Ueda M. Chemical biology of natural products on the basis of identification of target proteins. Chem Lett. 2012;41:658–66.
Zhang SD, Lu WQ, Liu XF, Diao YY, Bai F, Wang LY, Shan L, Huang J, Li HL, Zhang WD. Fast and effective identification of the bioactive compounds and their targets from medicinal plants via computational chemical biology approach. MedChemComm. 2011;2:471–7.
Futamura Y, Muroi M, Osada H. Target identification of small molecules based on chemical biology approaches. Mol BioSyst. 2013;9:897–914.
Takahashi D, Hirono S, Hayashi C, Igarashi M, Nishimura Y, Toshima K. Photodegradation of target oligosaccharides by light-activated small molecules. Angew Chem Int Ed. 2010;122:10294–8.
Sakamoto S, Kabe Y, Hatakeyama M, Yamaguchi Y, Handa H. Development and application of high-performance affinity beads: toward chemical biology and drug discovery. Chem Rec. 2009;9:66–85.
Kanoh N, Honda K, Simizu S, Muroi M, Osada H. Photo-cross-linked small-molecule affinity matrix for facilitating forward and reverse chemical genetics. Angew Chem Int Ed. 2005;117:3625–8.
Sato S, Kwon Y, Kamisuki S, Srivastava N, Mao Q, Kawazoe Y, Uesugi M. Polyproline-rod approach to isolating protein targets of bioactive small molecules: isolation of a new target of indomethacin. J Am Chem Soc. 2007;129:873–80.
Ueda M, Manabe Y, Otsuka Y, Kanzawa N. Cassia obtusifolia MetE as a cytosolic target for potassium isolespedezate, a leaf-opening factor of Cassia plants: target exploration by a compact molecular-probe strategy. Chem Asian J. 2011;6:3286.
Chidley C, Haruki H, Pedersen MG, Muller E, Johnsson K. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat Chem Biol. 2011;7:375–83.
Burgett AW, Poulsen TB, Wangkanont K, Anderson DR, Kikuchi C, Shimada K, Okubo S, Fortner KC, Mimaki Y, Kuroda M. Natural products reveal cancer cell dependence on oxysterol-binding proteins. Nat Chem Biol. 2011;7:639–47.
Nishimura S, Arita Y, Honda M, Iwamoto K, Matsuyama A, Shirai A, Kawasaki H, Kakeya H, Kobayashi T, Matsunaga S. Marine antifungal theonellamides target 3β-hydroxysterol to activate Rho1 signaling. Nat Chem Biol. 2010;6:519–26.
Dixon N, Wong LS, Geerlings TH, Micklefield J. Cellular targets of natural products. Nat Prod Rep. 2007;24:1288–310.
Guiffant D, Tribouillard D, Gug F, Galons H, Meijer L, Blondel M, Bach S. Identification of intracellular targets of small molecular weight chemical compounds using affinity chromatography. Biotechnol J. 2007;2:68–75.
Manna L, Milliron DJ, Meisel A, Scher EC, Alivisatos AP. Controlled growth of tetrapod-branched inorganic nanocrystals. Nat Mater. 2003;2:382–5.
Choi CL, Alivisatos AP. From artificial atoms to nanocrystal molecules: preparation and properties of more complex nanostructures. Annu Rev Phys Chem. 2010;61:369–89.
Talapin DV, Lee JS, Kovalenko MV, Shevchenko EV. Prospects of colloidal nanocrystals for electronic and optoelectronic applications. Chem Rev. 2010;110:389–458.
Asem H, Zhao Y, Ye F, Barrefelt A, Abedi-Valugerdi M, El-Sayed R, El-Serafi I, Abu-Salah KM, Hamm J, Muhammed M, Hassan M. Biodistribution of biodegradable polymeric nano-carriers loaded with busulphan and designed for multimodal imaging. J Nanobiotechnology. 2016;14:82.
Miki K, Oride K, Inoue S, Kuramochi Y, Nayak RR, Matsuoka H, Harada H, Hiraoka M, Ohe K. Ring-opening metathesis polymerization-based synthesis of polymeric nanoparticles for enhanced tumor imaging in vivo: synergistic effect of folate-receptor targeting and PEGylation. Biomaterials. 2010;31:934–42.
Sun C, Sze R, Zhang M. Folic acid-PEG conjugated superparamagnetic nanoparticles for targeted cellular uptake and detection by MRI. J Biomed Mater Res Part A. 2006;78:550–7.
de Chermont QLM, Chaneac C, Seguin J, Pelle F, Maitrejean S, Jolivet JP, Gourier D, Bessodes M, Scherman D. Nanoprobes with near-infrared persistent luminescence for in vivo imaging. Proc Natl Acad Sci USA. 2007;104:9266–71.
Maldiney T, Richard C, Seguin J, Wattier N, Bessodes M, Scherman D. Effect of core diameter, surface coating, and PEG chain length on the biodistribution of persistent luminescence nanoparticles in mice. ACS Nano. 2011;5:854–62.
Wu BY, Wang HF, Chen JT, Yan XP. Fluorescence resonance energy transfer inhibition assay for alpha-fetoprotein excreted during cancer cell growth using functionalized persistent luminescence nanoparticles. J Am Chem Soc. 2011;133:686–8.
Thambi T, Son S, Lee DS, Park JH. Poly(ethylene glycol)-b-poly(lysine) copolymer bearing nitroaromatics for hypoxia-sensitive drug delivery. Acta Biomater. 2016;29:261–70.
Miao X, Cao W, Zheng W, Wang J, Zhang X, Gao J, Yang C, Kong D, Xu H, Wang L, Yang Z. Switchable catalytic activity: selenium-containing peptides with redox-controllable self-assembly properties. Angew Chem Int Ed Engl. 2013;52:7781–5.
Cho MK, Jang YP, Kim YC, Kim SG. Arctigenin, a phenylpropanoid dibenzylbutyrolactone lignan, inhibits MAP kinases and AP-1 activation via potent MKK inhibition: the role in TNF-alpha inhibition. Int Immunopharmacol. 2004;4:1419–29.
Chan YS, Cheng LN, Wu JH, Chan E, Kwan YW, Lee SM, Leung GP, Yu PH, Chan SW. A review of the pharmacological effects of Arctium lappa (burdock). Inflammopharmacology. 2011;19:245–54.
Zhao Z, Yin Y, Wang Z, Fang R, Wu H, Jiang M, Bai G, Luo GA. Arctigenin exhibits relaxation effect on bronchus by affecting transmembrane flow of calcium. Biol Trace Elem Res. 2013;156:181–7.
Shi X, Sun H, Zhou D, Xi H, Shan L. Arctigenin attenuates lipopolysaccharide-induced acute lung injury in rats. Inflammation. 2014:1–9.
Jurado-Ramos A, Ropero Romero F, Cantillo Banos E, Salas Molina J. Minimally invasive endoscopic techniques for treating large, benign processes of the nose, paranasal sinus, and pterygomaxillary and infratemporal fossae: solitary fibrous tumour. J Laryngol Otol. 2009;123:457–61.
Fang R, Cui Q, Sun J, Duan X, Ma X, Wang W, Cheng B, Liu Y, Hou Y, Bai G. PDK1/Akt/PDE4D axis identified as a target for asthma remedy synergistic with beta2 AR agonists by a natural agent arctigenin. Allergy. 2015;70:1622–32.
Huang Y, Gao J, Meng XM, Jiang GL, Yang YR, Li H, Liu J, Li J. Involvement of mitogen-activated protein kinase activation in cyclooxygenase-2 and transforming growth factor-beta production in alveolar macrophage from chronic bronchitis rats. Immunopharmacol Immunotoxicol. 2011;33:645–51.
Danhier F, Feron O, Preat V. To exploit the tumor microenvironment: passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J Control Release. 2010;148:135–46.
Liu Z, Xiong M, Gong J, Zhang Y, Bai N, Luo Y, Li L, Wei Y, Liu Y, Tan X, Xiang R. Legumain protease-activated TAT-liposome cargo for targeting tumours and their microenvironment. Nat Commun. 2014;5:4280.
Shen S, Zhuang J, Chen Y, Lei M, Chen J, Shen X, Hu L. Synthesis and biological evaluation of arctigenin ester and ether derivatives as activators of AMPK. Bioorg Med Chem. 2013;21:3882–93.
Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed Engl. 2001;40:2004–21.
Zhu D, Tang J, Shen Y. Zwitterionic poly(lysine methacrylate) brush as an effective carrier for drug delivery. J Control Release. 2015;213:e27–8.
Wu X, Yang Y, Dou Y, Ye J, Bian D, Wei Z, Tong B, Kong L, Xia Y, Dai Y. Arctigenin but not arctiin acts as the major effective constituent of Arctium lappa L. fruit for attenuating colonic inflammatory response induced by dextran sulfate sodium in mice. Int Immunopharmacol. 2014;23:505–15.
Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Identification of a primary target of thalidomide teratogenicity. Science. 2010;327:1345–50.
Kang HS, Lee JY, Kim CJ. Anti-inflammatory activity of arctigenin from Forsythiae Fructus. J Ethnopharmacol. 2008;116:305–12.
Zhao F, Wang L, Liu K. In vitro anti-inflammatory effects of arctigenin, a lignan from Arctium lappa L., through inhibition on iNOS pathway. J Ethnopharmacol. 2009;122:457–62.
Cho JY, Kim AR, Yoo ES, Baik KU, Park MH. Immunomodulatory effect of arctigenin, a lignan compound, on tumour necrosis factor-alpha and nitric oxide production, and lymphocyte proliferation. J Pharm Pharmacol. 1999;51:1267–73.
Authors’ contributions
BG and FXZ designed the study; CQX and HYY performed experiments, acquired and analyzed data, drafted and edited the manuscript; WYN, LX and CDY performed Zebrafish imaging; MXY and WWY synthesized and characterized the nanoparticles; LY and WZY performed cell imaging and flow cytometry analysis; TJ, WQ and JM studied the localization at the level of mice. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Consent to publication
All authors agree to be published.
Ethic approval
Animal treatment and maintenance were performed in accordance with the Principle of Laboratory Animal Care (NIH Publication no. 85-23, revised 1985), and the Animal Ethics Committee of Nankai University approved the experimental protocol.
Funding
This project was financially supported by the National Natural Science Foundation of China (Nos. 81503214, 81373506, 81430095).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional file
12951_2017_263_MOESM1_ESM.docx
Additional file 1. Supplementary information about alkyne-ATG synthesis, purification, characteristics and FI-IR analysis of functionalized NPs.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Cui, Q., Hou, Y., Wang, Y. et al. Biodistribution of arctigenin-loaded nanoparticles designed for multimodal imaging. J Nanobiotechnol 15, 27 (2017). https://doi.org/10.1186/s12951-017-0263-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12951-017-0263-8