
Abstract
Interleukin (IL)-35 is a relatively newly discovered member of IL-12 cytokine family that is unique in that it is a dimer formed by two subunits. The review documents the structure, secretion and signal transduction of IL-35, the regulation effect of IL-35 on B cells and T cells as well as the adoptive transfer of IL-35+ regulatory B cells (Breg), therapeutic prospects of recombinant IL-35 (rIL-35) and IL-35 regulation role in various diseases. B-cell regulation expands the regulatory range of IL-35 and alters the view that IL-10 is the chief immune mechanism for Breg cells which secrete IL-35. IL-35 induces Breg cells, which then can induce Treg cells. IL-35 also plays an immunomodulatory role in the human body.
Similar content being viewed by others


Background
Interleukin (IL)-35 cytokine is a relatively newly discovered member of IL-12 family which are unique in structure as they are dimer formed by two subunits. Existing family members (IL-12, −23, −27) are similar in structure, receptor binding, and downstream signaling pathways that positively or negatively regulate the immune system. IL-35 also strongly inhibits immune function and this review will state the structure and secretion of IL-35, its effect and regulation in B cells and T cells as well as the therapeutic prospects of recombinant IL-35 (rIL-35) and IL-35 regulation role in various diseases.
Interleukin-35 (IL-35) structure and secretion
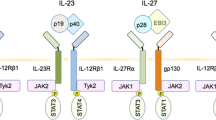
IL-35 was found by Niedbala [1] and Collison [2] almost simultaneously and it is reported to contain IL-12α chain p35 and IL-27β chain Epstein-Barr virus-induced gene 3 (Ebi3) connected by disulfide bond. IL-35, initially named at the 13th International Congress of Immunology, is the new focus of cytokines research. IL-35 is similar to other IL-12 family members which are heterodimeric glycoproteins formed with disulfide-linked α (p19, p28, or p35) and β (p40 or EBi3) chains. The α-chain has 4-α-helical bundles, a typical cytokine structure, and the β-chain is homologous to the soluble cytokine receptor. p35 and p40 combine to form IL-12; p19 and p40 combine to form IL-23; p28 and Ebi3 combine to form IL-27 [3]. IL-35 is composed of p35 and Ebi3, and it differs from the expression and secretion way of other IL-12 members. In response to bacteria, bacterial products, or intracellular parasites, IL-12, IL-23, and IL-27 are secreted by activated antigen-presenting cells, including B cells, monocyte, macrophages and dendritic cells [4,5,6]. IL-35 was initially reported to be produced by Treg cells and was essential for maximizing the inhibitory role of Treg cells [2]. Recently studies suggest that regulatory B cells (Breg) also produce IL-35 and rIL-35 fusion proteins can induce Breg cells to secret IL-10 and IL-35 [7, 8].
IL-35 receptors and signal transduction
Peptide chain sharing is common to the IL-12 family as they bind to receptors to activate signal transducer and activator of transcription (STAT) proteins [9]. IL-35 is uniquely anti-inflammatory cytokine in contrast to other IL-12 pro-inflammatory cytokines [9]. This difference is thought to be associated with the receptors and signaling pathways specific to IL-35 and future studies should confirm these assertions. An obstacle to understanding the molecular mechanism underlying IL-35 is the lack of clarity about the IL-35 receptor (IL-35R) and its signal transduction pathway [9]. Collison’s group reported that mouse IL-35R differed from traditional cytokine receptors. IL-35R is composed of dimers which are IL-12Rβ2 homodimers, gp130 homodimers or IL- IL-12Rβ2/gp130 heterodimer [6]. IL-35 binds to IL-35R and initiates signal transduction and exert biological function. IL-12Rβ2 or gpl30 homodimer activates STAT4 or STAT1, but only IL-35R in the IL-12Rβ2/gp130 heterodimer form can induce STAT1- and STAT4-activated signaling pathways to mediate Treg cell function and positively or negatively feedback regulate IL-35 gene expression [2]. IL-35 is reported to activate STAT1 and STAT4 in T cells [7] where IL-35 binds to gp130 and IL-12Rβ2 receptors [2], both of which depend on JAK-STAT signaling to introduce intracellular responses. However in B cells, IL-35 signaling mediates STAT1 and STAT3 activation through IL-12Rβ2: IL-27Ra heterodimers [8] (Fig. 1).

IL-35 signal transduction in T and B cells. Signaling through gp130 or IL12Rb2 homodimers to STAT1 or STAT4 separately in T cells, while formation of the gp130/IL12Rb2 heterodimer is required for both STAT1 and STAT4 activation in Treg cells (left panel). B cells respond to IL-35 through the IL27Ra/IL12Rb2 heterodimer to STAT1 and STAT3 (right panel)
IL-35 regulation and signal transduction in Breg cells
B cells have been traditionally thought to contribute to immune defense by secreting antibodies and antigen presentation but they also have function in immune regulation as Breg cells [10]. There are multiple B cell subsets which have immune regulation function, such as CD138+ plasma cells [11], B10 cells (CD1dhiCD5hi) [10], CD21hiCD23hiCD24hi transitional 2-marginal zone precursor cells [12], and Tim-1+ B cells [13], but Breg cells do not have a unified determined phenotype. It is generally recognized that Breg cells play a role in immunosuppressive function by secreting IL-10 under the stimulation of toll-like receptor (TLR) agonists, CD40L, and IL-21 [14].
Furthermore immunosuppressive pathways of Breg cells may not solely depend on IL-10 [15] as research suggests that Breg cells can produce IL-35 and that rIL-35 can induce Breg cells to secrete IL-10 and IL-35 [7, 8]. B-cell-derived IL-35 also acts on T cell-induced Foxp3+ Treg cells [7, 15] (Fig. 2). Although rIL-35 inhibits B220hi B cell proliferation, it selectively induces CD19+CD5+B220lo Breg cell proliferation in vivo [7].

IL-35 regulation in Breg cells. The immunosuppressive cytokine IL-35 induces the expansion of Breg cells, which secrete IL-35 to protect from autoimmune disease. Secretion of B cell–derived IL-35 seems to have an autocrine role through activation of the IL-35 receptor (IL-12Rb2 and IL-27Rb) to expand or induce Breg cells. B cell–derived IL-35 also acts on T cells to induce a Foxp3+ Treg cell population
Unlike T cells, inhibition of gp130 (with small interfering RNA [siRNA] or neutralizing antibodies) does not affect IL-35-mediated of B cell proliferation or IL-10 secretion. In contrast, silence of IL-12Rß2 and IL-27Ra in B cells completely blocks inhibition function of IL-35. Thus IL-35 may mediate biological activity in different cell types via different receptors and STAT signaling pathways and future studies are required to confirm those [14].
IL-35 regulation and signal transduction in T cells
IL-35 is a novel inhibitory cytokine that may be specifically produced by Treg cells. It is required for maximal suppressive activity of Treg. In addition, IL-35 can block the proliferation of Th1 and Th17 cells by limiting early T cell rest on the G1 phase of cell division [16]. Although IL-35 can inhibit Th1 proliferation, it is resistant to Treg conversion due to the potent inhibition of Ebi3 and p35 transcription by IFNγ from Th1. Moreover, IL-35 blocks Th2 development by repressing GATA3 and IL-4 expression and limiting Th2 proliferation. IL-35 can also mediate conversion of Th2 cells to Treg, although this can be blocked by IFN-γ [17].
Like TGF-β and IL-10, IL-35 can induce the development of an induced regulatory T cell (iTreg) population, iTr35, which suppressed T-cell proliferation via IL-35 [2]. iTreg do not express Foxp3, IL-10, and TGF-β (Fig. 3). iTr35 were as effective as nTregs at restoring immune homeostasis and preventing autoimmunity disease in Foxp3−/− mice by limiting the proliferation of T cells and so prevented the modeling of EAE and IBD and promoted the proliferation of B16 tumor cells. Adoptive transfer IL-35 treatment increased the proliferation of Foxp3+CD39+ CD4+ T cells which secreted IL-10 for autoimmune protection in a collagen-induced arthritis model [18]. However, whether the presence of IL-35 can mediate Treg amplification under physiological conditions is still uncertain.

iTreg generate. Naïve human or murine T cells were stimulated with IL-35 and converted into a regulatory population – iTr35 cells – which exhibit a highly restricted gene signature
(CD4 + Foxp3 − EBI3 + p35 + IL10 - TGFβ −).
While gp130 is fairly ubiquitously expressed, IL-12Rβ2 is expressed mainly on the surfaces of activated T cells, natural killer cells, B cells, and dendritic cells [19]. IL-12Rβ2 is undetectable on most resting T cells, but can be rapidly up-regulated by exposure to IL-2, IFN-γ, IL-12, IL-27, and TNF-α. Indeed, IL-2 or IL-27 pretreatment increases T cell sensitivity to IL-35 mediated suppression [9].
IL-35 mediates the inhibitory effect on T cells through the signal pathway of STAT1 and STAT4, but also lead to the pro-inflammatory effects by activating the STAT molecule of IFN-γ and IL-12, in which the key difference is that IL-35 induced STAT1-STAT4 heterodimer formation [20, 21].
Recombinant IL-35
Highly purified heterodimeric cytokine IL-35 is difficult to obtain and this is a limitation to immunology research, especially for elucidating the role of this cytokine in autoimmune, infectious diseases and tumor immunity. Transgenic technology has been applied to infect cells to establish a rIL-35 mouse model in which rIL-35 is a heterodimer of p35 and Ebi3 [7]. Similar to IL-27, IL-35 is not secreted as a disulfide-linked heterodimer as Ebi3 associates non-covalently with the IL-12p35 [22]. The native Ebi3/p35 heterodimer would be difficultly to isolate in vivo because only about 4% of the secreted Ebi3 co-precipitated with the IL-12p35 in vitro over expression studies in COS7 cells [22]. Meanwhile, as the absence of IL-12p35 substantial amount of the Ebi3 degraded in the ER (endoplasmic reticulum), thereby reducing the bioavailability of ebi3. These contribute to the low levels of IL-35 in vivo [23]. Others generated recombinant mouse IL-35 (rIL-35) using a bicistronic vector containing IRES (internal ribosomal entry site) that allowed stoichio etric expression of the Ebi3 and IL-12p35 [2]. Another approach that has been used is to construct a heterodimeric protein covalently linking Ebi3 and IL-12p35 [24].
IL-35regulation in various diseases
(1) IL-35 and autoimmune diseases.
Early studies mainly used a model of p35 or ebi3 deficient mice. However, the absence of IL-12α affects the function of IL-12 as well as that of IL-27 and IL-35. Despite these complexities, Ebi3−/− and Il-12a−/− mice model also provide the proof that IL-35 have immune suppressive function in some autoimmune diseases such as experimental autoimmune uveitis (EAU) [7], experimental allergic encephalomyelitis (EAE) [2], collagen-induced arthritis (CIA) [25], encephalitis [26], and other conditions. That IL-35 fusion protein obtained from genetic engineering produced data consistent with the knockout model results and studies indicates that injection of rIL-35 can be used to protect against allergic airway model [27] as well as colitis [20] and autoimmune diabetes [28]. Studies show that IL-35 regulates immune function via T cells in many diseases. In NOD transgenic mice, infusion of IL-35 over expressing islet cells significantly alleviated symptoms of diabetes and local inflammatory responses, reduced the number of effector CD4+ and CD8+ T cells in pancreatic islet tissue, inhibited infiltration of T cells from transitioning from G1 to S phases, and decreased effector T cells proliferation [28]. In a type II CIA model, infusion of rIL-35 reduced local inflammatory responses by inhibiting Th17 cell differentiation. In an Ebi3 subunit knockout mouse model, enhanced Th17 cell infiltration and aggravated inflammatory response was noted [1].
To study a potential regulatory role of IL-35+ Breg, a model of EAU was established and rIL-35 or IL-35+ Breg could control the incidence and development of EAU. IL-35+ Breg cell-mediated protection depended on induction and proliferation of endogenous Breg cells and Foxp3+ Treg cells and inhibition of pathogenic Th1 and Th17 effector cells [7]. Adoptive infusion of rIL-35-mediated Breg cells improved the animal health after EAU eradication as well. This suggested that in vitro study of functional rIL-35+ Breg cells may give insight into potential roles for Breg and IL-35 Breg cells in autoimmune disease and cancer. IL-35-derived Treg cells (i.e., iTR35) can maintain Breg cells, but IL-35+ Breg cells can also induce Treg cells. Therefore, in an inflammation-derived IL-35-dependent regulatory environment, different cell populations generated by IL-35 may interact via positive feed forward mechanisms and induce iTR35 and IL-35 + Breg cells through infectious tolerance [7] (Fig. 4).

a B cell–derived IL-35 can suppress T-cell and monocytic responses. IL-10, IL-35, and TGF-β induce the Treg cell population. b Breg cells activate Treg cells through IL-10 production and B7 costimulatory molecules. Activated Treg cells release IL-10, suppresses the autoreactive Th1response in patients with EAE. IL-10– and TGF-β–producing Breg cells can suppress T-cell responses. c A subset of Breg cells also acquires the ability to produce IL-10. Breg cells from the tumor bed support Treg expansion in vitro and in vivo, suppress T cell proliferation in vitro, suppress IFN-γ production and inhibit CD8+ cytolytic T cell activity. These B regulatory properties inhibit the anti-tumor response and lead to enhanced tumor growth
IL-35 and tumor immunity
Previous work suggests that IL-35 plays a role in tumor immune escape. Ebi3 expression was increased in Hodgkin’s lymphoma [29] and more Treg cells and inhibitory cytokines were noted in peripheral blood and tumor microenvironments of patients with pancreatic or breast carcinomas [30]. Ebi3 expression in lung cancer cells has also been found to be associated with tumor progression and siRNA-mediated down-regulation of the Eib3 gene inhibited proliferation of lung cancer cells [31]. Similarly, inoculation of b16 tumor cells in Ebi3-knockout mice showed enhanced anti-tumor immunity relative to wild-type mice and the metastatic potential of tumor cells were suppressed. In an IL-35-positive tumor cell microenvironment, studies showed that there were significant increase in CD11b + Gr1+ myeloid-derived suppressor cells (MDSC) and vascular endothelial growth factors promoted tumor angiogenesis [32]. These MDSCs were immunosuppressive and inhibited cytotoxic T cells. In this way, tumor cell-generated IL-35 may protect against cytotoxic T-cell-mediated damage. In contrast, applications of neutralizing antibody to IL-35 significantly inhibited tumor cell proliferation [32].
Although many studies have shown that IL-35 contributes to immunosuppressive function in the mouse model, but this effect is limited in humans [33]. No expression of IL-35-derived Treg cells had been observed in early investigations in humans. This suggests that IL-35 may not be constitutively expressed in humans. However, it can be produced in specific tissues and cell types after a certain inflammatory stimulus. Recent studies have shown that, under strong stimulation, Treg can produce IL-35, and CD4+ T cells under the stimulation of IL-35 can express IL-35similar to iTr35 in mice [34]. Similarly, in humans, CD8+ Treg can suppress the immune response against prostate cancer by expressing CTLA-4 and IL-35 [35]. Although more research is required to draw any conclusions, preliminary information suggests that elevated serum IL-35 is associated with tumor malignancy [36,37,38] and clinical stage and decreased IL-35 is associated with autoimmune disease and chronic infection [39,40,41]. (Fig. 4).
IL-35 and infectious diseases
IL-35, as an inhibitory cytokine, plays an important role in infectious diseases. The study suggested that Mycobacterium tuberculosis (M. tuberculosis) could induce T cell proliferation and foster IFN-γ production in p35-deficient mice, P40 deficient mice, and wild-type mice, thus eliminating the pathogen. Mice lacking p40 have been found to be less able to show antigen-specific responses than those lacking the p35 subunit. However, their ability to counteract infection is less pronounced than that of wild-type mice [42]. In addition, the protective responses can be induced in wild-type and p35-deficient mice by inoculating with vaccines, and increased the secretion of IFN-γ and IL-17. However, P40-deficient mice did not produce antigen-specific IFN-γ or IL-17, and increased the infection load of bacteria. In addition, treatment of p35-deficient mice with Candida albicans (C. albicans) reduced the rate of fungal infection and was associated with little to no obvious symptoms of infection relative to P19-deficient mice. In p35-deficient mice and those in which p35 gene was disrupted, the immunosuppressive function of IL-35 was inhibited, which impaired anti-fungal immunity [43]. The CD4+ T cells in the peripheral blood of patients with chronic hepatitis B also showed high levels of expression of p35 and EB13 protein, indicating that IL-35 was related to the immune response of chronic hepatitis B patients [44]. These results showed that, during the acute infection process, IL-35 preferentially activated Th1 cells, stimulated proliferation of Treg cells, and inhibited the differentiation of Th17 cells, thereby preventing excessive tissue damage caused by the clearing of pathogens. In the chronic infection and inflammation, IL-35 selectively inhibited effector cells, including Th17 cells, which slowed down the development of autoimmune diseases [1]. (Fig. 4).
Conclusions
B-cell regulation expands the regulatory range of IL-35 and alters the view that IL-10 is the chief immune mechanism for Breg cells which secrete IL-35. IL-35 signaling mediates STAT1 and STAT3 activation in B cells by binding to IL-12Rβ2 and IL-27Ra receptors and inducing Treg cells which provide mutual stimulation under inflammation and amplification of regulatory cells. IL-35-induced B cells also transform to secretary IL-35+ Breg cells and rIL-35 or IL-35+ Breg cells control the incidence and development of EAU. IL-35 induces Breg cells, which then can induce Treg cells. In the case of inflammation, cells which can generate IL-35 provide mutual stimulation, resulting in amplification of regulatory cells. Recent researchers have found that in some inflammatory stimulus conditions, IL-35 also play an immunomodulatory role in the human body, so we can look forward to further exploring immunotherapy approaches through IL-35.
Future studies may address whether IL-35 shares its receptors with other IL-12 members and rIL-35 may help clarify these biological effects and identify other cell types or subtypes involved in immune regulation by producing IL-35. How IL-35 inhibits cell proliferation, why different cells mediate different signaling pathways, and potential prospects of clinical use of IL-35 (rIL-35 and IL-35 Breg cells) as a chemotherapeutic or to treat autoimmune diseases or organ transplantation also await further study.
Abbreviations
- Breg:
-
Regulatory B cells
- CIA:
-
Collagen-induced arthritis
- EAU:
-
Experimental autoimmune uveitis
- Ebi3:
-
Epstein-Barr virus-induced gene 3
- ER:
-
endoplasmic reticulum
- IL-35:
-
Interleukin-35
- IL-35R:
-
IL-35 receptor
- iTR35:
-
IL-35-derived Treg cells
- MDSC:
-
Myeloid-derived suppressor cells
- rIL-35:
-
recombinant IL-35
- siRNA:
-
small interfering RNA
- STAT:
-
Signal transducer and activator of transcription
- TLR:
-
Toll-like receptor
- Treg:
-
Regulatory T cells
References
Niedbala W, Wei XQ, Cai B, Hueber AJ, Leung BP, IB MI. IL-35 is a novel cytokine with therapeutic effects against collagen-induced arthritis through the expansion of regulatory T cells and suppression of Th17 cells. Eur J Immunol. 2007;37:3021–9.
Collison LW, Workman CJ, Kuo TT, Boyd K, Wang Y, Vignali KM, et al. The inhibitory cytokine IL- 35 contributes to regulatory T- cell function. Nature. 2007;450:566–9.
Tedder TF, Leonard WJ. Autoimmunity:regulatory B cells—IL-35 and IL-21regulate the regulators. Nat Rev Rheumatol. 2014;10:452–3.
Ma X, Trinchieri G. Regulation of interleukin-12 production in antigen-presenting cells. Adv Immunol. 2001;79:55–92.
O’Shea JJ, Paul WE. Regulation of T (H) 1 differentiation—controlling the controllers. Nat Immunol. 2002;3:506–8.
Hunter CA. New IL-12-family members: IL-23 and IL-27, cytokines with divergent functions. Nat Rev Immunol. 2005;5:521–31.
Wang RX, Yu CR, Dambuza IM, Mahdi RM, Dolinska MB, Sergeev YV, et al. Interleukin-35 induces regulatory B cells that suppress autoimmune disease. Nat Med. 2014;20:633–41.
Shen P, Roch T, Lampropoulou V, O'Connor RA, Stervbo U, Hilgenberg E, et al. IL-35-producing B cells are critical regulators of immunity during autoimmune and infectious diseases. Nature. 2014;507:366–70.
Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol. 2012;13:290–9.
Yanaba K, Bouaziz JD, Haas KM, Poe JC, Fujimoto M, Tedder TF, et al. A regulatory B cell subset with a unique CD1dhiCD5+ phenotype controls T cell-dependent inflammatory responses. Immunity. 2008;28:639–50.
Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, et al. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during salmonella typhimurium infection. Immunity. 2010;33:777–90.
Evans JG, Chavez-Rueda KA, Eddaoudi A, Meyer-Bahlburg A, Rawlings DJ, Ehrenstein MR, et al. Novel suppressive function of transitional 2 B cells in experimental arthritis. J Immunol. 2007;178:7868–78.
Ding Q, Yeung M, Camirand G, Zeng Q, Akiba H, Yagita H, et al. Regulatory B cells are identified by expression of TIM-1 and can be induced through TIM-1 ligation to promote tolerance in mice. J Clin Invest. 2011;121:3645–56.
Yoshizaki A, Miyagaki T, DiLillo DJ, Matsushita T, Horikawa M, Kountikov EI, et al. Regulatory B cells control T-cell autoimmunity through IL-21-dependent cognate interactions. Nature. 2012;491:264–8.
Wilson MS, Taylor MD, O'Gorman MT, Balic A, Barr TA, Filbey K, et al. Helminth-induced CD19+CD23hi B cells modulate experimental allergic and autoimmune inflammation. Eur J Immunol. 2010;40:1682–96.
Wirtz S, Billmeier U, Mchedlidze T, Blumberg RS, Neurath MF. Interleukin-35 mediates mucosal immune responses that protect against T-cell-dependent colitis. Gastroenterology. 2011;141:1875–86.
Vignali DA, Kuchroo VK. IL-12 family cytokines: immunological playmakers. Nat Immunol. 2012;13:722–8.
Collison LW, Chaturvedi V, Henderson AL, Giacomin PR, Guy C, Bankoti J, et al. IL-35-mediated induction of a potent regulatory T cell population. Nat Immunol. 2010;11:1093–01.
Presky DH, Yang H, Minetti LJ, Chua AO, Nabavi N, Wu CY, et al. A functional interleukin 12 receptor complex is composed of two beta-type cytokine receptor subunits. Proc Natl Acad Sci U S A. 1996;93:14002–7.
Canda-Sánchez A, Salgado FJ, Pérez-Díaz A, Varela-González C, Arias P, Nogueira M. Differential distribution of both IL-12Rbeta chains in the plasma membrane of human T cells. J Membr Biol. 2009;227:1–12.
Maldonado RA, Soriano MA, Perdomo LC, Sigrist K, Irvine DJ, Decker T, et al. Control of T helper cell differentiation through cytokine receptor inclusion in the immunological synapse. J Exp Med. 2009;206:877–92.
Devergne O, Birkenbach M, Kieff E. Epstein-Barr virus-induced gene 3 and the p35 subunit of interleukin 12 form a novel heterodimeric hematopoietin. Proc Natl Acad Sci U S A. 1997;94:12041–6.
Devergne O, Hummel M, Koeppen H, Le Beau MM, Nathanson EC, Kieff E, et al. A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J Virol. 1996;70:1143–53.
Aparicio-Siegmund S, Moll JM, Lokau J, Grusdat M, Schroder J, Plohn S, et al. Recombinant p35 from bacteria can form interleukin (IL-)12, but not IL-35. PLoS One. 2014;9:e107990.
Kochetkova I, Golden S, Holderness K, Callis G, Pascual DW. IL-35 stimulation of CD39+ regulatory T cells confers protection against collagen II-induced arthritis via the production of IL-10. J Immunol. 2010;184:7144–53.
Tirotta E, Duncker P, Oak J, Klaus S, Tsukamoto MR, Gov L, et al. Epstein-Barr virus-induced gene 3 negatively regulates neuroinflammation and T cell activation following coronavirus-induced encephalomyelitis. J Neuroimmunol. 2013;254:110–6.
Huang CH, Loo EX, Kuo IC, Soh GH, Goh DL, Lee BW, et al. Airway inflammation and IgE production induced by dust mite allergen-specific memory/effector Th2 cell line can be effectively attenuated by IL-35. J Immunol. 2011;187:462–7.
Bettini M, Castellaw AH, Lennon GP, Burton AR, Vignali DA. Prevention of autoimmune diabetes by ectopic pancreatic beta-cell expression of interleukin-35. Diabetes. 2012;61:1519–26.
Niedobitek G, Päzolt D, Teichmann M, Devergne O. Frequent expression of the Epstein-Barr virus (EBV)-induced gene, EBI3, an IL-12 p40-related cytokine, in Hodgkin and reed-Sternberg cells. J Pathol. 2002;198:310–6.
Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–61.
Nishino R, Takano A, Oshita H, Ishikawa N, Akiyama H, Ito H, et al. Identification of Epstein-Barr virus-induced gene 3 as a novel serum and tissue biomarker and a therapeutic target for lung cancer. Clin Cancer Res. 2011;17:6272–86.
Wang Z, Liu JQ, Liu Z, Shen R, Zhang G, Xu J, et al. Tumor-derived IL-35 promotes tumor growth by enhancing myeloid cell accumulation and angiogenesis. J Immunol. 2013;190:2415–23.
Bardel E, Larousserie F, Charlot-Rabiega P, Coulomb-L'Herminé A, Devergne O. Human CD4+ CD25+ Foxp3+ regulatory T cells do not constitutively express IL-35. J Immunol. 2008;181:6898–05.
Seyerl M, Kirchberger S, Majdic O, Seipelt J, Jindra C, Schrauf C, et al. Human rhinoviruses induce IL-35-producing Treg via induction of B7-H1 (CD274) and sialoadhesin (CD169) on DC. Eur J Immunol. 2010;40:321–9.
Olson BM, Jankowska-Gan E, Becker JT, Vignali DA, Burlingham WJ, McNeel DG. Human prostate tumor antigen-specific CD8+ regulatory T cells are inhibited by CTLA-4 or IL-35 blockade. J Immunol. 2012;189:5590–01.
Wu H, Li P, Shao N, Ma J, Ji M, Sun X, et al. Aberrant expression of Treg-associated cytokine IL-35 along with IL-10 and TGF-beta in acute myeloid leukemia. Oncol Lett. 2012;3:1119–23.
Zeng JC, Zhang Z, Li TY, Liang YF, Wang HM, Bao JJ, et al. Assessing the role of IL-35 in colorectal cancer progression and prognosis. Int J Clin Exp Pathol. 2013;6:1806–16.
Jin P, Ren H, Sun W, Xin W, Zhang H, Hao J. CirculatingIL-35 in pancreatic ductal adenocarcinoma patients. Hum Immunol. 2014;75:29–33.
Lin Y, Huang Y, Lu Z, Luo C, Shi Y, Zeng Q, et al. Decreased plasma IL-35 levels are related to the left ventricular ejection fraction in coronary artery diseases. PLoS One. 2012;7:e52490.
Mao H, Gao W, Ma C, Sun J, Liu J, Shao Q, et al. Human placental trophoblasts express the immunosuppressive cytokine IL-35. Hum Immunol. 2013;74:872–7.
Chen C, Deng Y, Chen H, Wu X, Cheng S, Xu Y, et al. Decreased concentration of IL-35 in plasma of patients with asthma and COPD. Asian Pac J Allergy Immunol. 2014;32:211–7.
Cooper AM, Kipnis A, Turner J, Magram J, Ferrante J, Orme IM. Mice lacking bioactive IL-12 can generate protective, antigen-specific cellular responses to mycobacterial infection only if the IL-12 p40 subunit is present. J Immunol. 2002;168:1322–7.
Conti HR, Shen F, Nayyar N, Stocum E, Sun JN, Lindemann MJ, et al. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J Exp Med. 2009;206:299–11.
Shi YY, Dai MJ, Wu GP, Zhou PP, Fang Y, Yan XB. Levels of interleukin-35 and its relationship with regulatory T-cells in chronic hepatitis B patients. Viral Immunol. 2015;28:93–100.
Acknowledgements
None.
Funding
Not applicable.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
AH and LC drafted the initial manuscript, MH, JN and JJW modified the manuscript as submitted. KJ corrected/approved the final manuscript as submitted. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Huang, A., Cheng, L., He, M. et al. Interleukin-35 on B cell and T cell induction and regulation. J Inflamm 14, 16 (2017). https://doi.org/10.1186/s12950-017-0164-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12950-017-0164-5