
Abstract
The tumor microenvironment is highly complex, and immune escape is currently considered an important hallmark of cancer, largely contributing to tumor progression and metastasis. Named for their capability of killing target cells autonomously, natural killer (NK) cells serve as the main effector cells toward cancer in innate immunity and are highly heterogeneous in the microenvironment. Most current treatment options harnessing the tumor microenvironment focus on T cell-immunity, either by promoting activating signals or suppressing inhibitory ones. The limited success achieved by T cell immunotherapy highlights the importance of developing new-generation immunotherapeutics, for example utilizing previously ignored NK cells. Although tumors also evolve to resist NK cell-induced cytotoxicity, cytokine supplement, blockade of suppressive molecules and genetic engineering of NK cells may overcome such resistance with great promise in both solid and hematological malignancies. In this review, we summarized the fundamental characteristics and recent advances of NK cells within tumor immunometabolic microenvironment, and discussed potential application and limitations of emerging NK cell-based therapeutic strategies in the era of presicion medicine.
Similar content being viewed by others

Background
The diversity of infiltrating stromal cells occurring in human cancers exceeds 30 distinct subgroups, reflecting the huge complexity of the tumor microenvironment (TME), thereby deeply affecting the treatment option for each patient [1]. Attempts have been made to distill this out-of-order situation into a unifying method to better describe actual composition of the TME using both multi-omics and experimental technologies, shedding light on cancer biology. This trend led to a transition in cancer treatment from only targeting tumor cells (like traditional chemotherapy and radiotherapy) to a new generation of approaches emphasizing the modulation of endogenous immune response toward cancer.
The immune system can be generally divided into the innate and adaptive immune systems, both contributing to the recognition and removal of foreign pathogens as well as tumors [2]. Adaptive immunity is mainly composed of cells represented by T and B lymphocytes, which harbor an enormous repertoire of T-cell and B-cell receptors, respectively, that can respond specifically to different antigens in the body. Current immunotherapeutic methods mainly focus on T lymphocytes, especially restoring exhausted CD8+ cytotoxic T cells (CTLs). An example of such approach is immune-checkpoint blockade, with blocking of receptors or ligands that inhibit the activation of CTLs, including programmed cell death protein 1 (PD-1), its main ligand PD-L1, cytotoxic T-lymphocyte antigen 4 (CTLA-4) and lymphocyte-activation gene-3 (LAG-3), by monoclonal neutralizing antibodies [3, 4]. In recent years, the rapid and potent anti-tumor function of innate immunity, which even occurs at a very early stage of tumor progression, has attracted increasing attention. As a subset of whole innate lymphoid cells, natural killer (NK) cells, defined by Herberman in 1976 [5] and often considered a part of type 1 innate-like cells (ILC1s), are currently defined as effector cells similar to CTLs, exerting natural cytotoxicity against primary tumor cells and metastasis by inhibiting proliferation, migration and colonization to distant tissues [6]. Beside their cytotoxic role, NK cells have been reported to produce a large number of cytokines, mainly interferon-γ (IFN-γ), to modulate adaptive immune responses and participate in other related pathways [7, 8]. In addition, as documented in multiple models and experiments, NK cells could distinguish abnormal cells from healthy ones, leading to more specific anti-tumor cytotoxicity and reduced off-target complications [9, 10].
Considering the pivotal role of NK cells in cancer biology, they naturally emerged as a prospective target for cancer therapy, and a growing number of studies and multiple therapeutic agents inhibiting cancer target NK cell-related pathways. In this review, we will review the fundamental characteristics and emerging subpopulations of NK cells. Next, we will mainly use breast cancer (BC) to discuss the plasticity of NK cells in cancer biology and metabolism, as well as current therapeutic regimens, including ongoing clinical trials and FDA-approved therapies targeting NK cells, and future possible approaches for improving cancer treatment.
Development of NK cells
NK cells possess cytotoxic abilities similar to CD8+ T cells functioning in the adaptive immunity but lack CD3 and the T cell receptors (TCRs). Largely circulating in blood and counting for about 5–10% of peripheral blood mononuclear cells (PBMCs), NK cells are found in bone marrow and lymphoid tissues such as the spleen [11, 12]. Similar to other ILCs, NK cells are originated from common lymphoid progenitor (CLP) cells in bone marrow (Fig. 1) with an average renewal cycle of about 2 weeks [12]. During development, a process termed education, which describes the interaction of NK cells expressing immunoreceptor tyrosine-based inhibitory motifs (ITIMs) with major histocompatibility complex-I (MHC-I), helps NK cells become licensed and avoid attacking healthy normal cells [6, 9]. Interestingly, tumor cells always lack or only express low levels of MHC-I to evade CD8+ T cell-mediated cytotoxicity, whereas licensed NK cells are fully activated. However, tumor cells also express molecules that activate NK cells, e.g., MHC class I polypeptide-related sequence A (MICA) and MICB [13, 14], supporting the use of NK cells as anti-cancer agents. In addition, unlicensed NK cells also play important roles in the body, e.g., eliminating murine cytomegalovirus (MCMV) infection and MHC-I+ cells [15].

Development and subgroups of NK cells. In bone marrow, NK cells develop from hematopoietic stem cells (HSCs) through common lymphoid progenitors (CLPs) and NK cell precursors (NKPs), and then migrate to peripheral blood (cNK cells) or tissue (trNK cells). The differentiation of trNK-cells occurs in distinct tissue sites, including the lung, thymus, liver, uterus, skin, subcutaneous adipose tissue, and kidney. In these sites, NK cells have different phenotypic features and functions, which constitute the circulation of NK cells at different stages of maturation. CLA, cutaneous lymphocyte-associated antigen; CCR8, C-C motif chemokine receptor 8; GATA3, GATA binding protein 3; CXCR6, C-X-C motif chemokine receptor 6; KIR, killer cell immunoglobulin-like receptor; CILCP, common innate-like cell precursor
To date, NK cell survival and development is thought to mainly rely on cytokines (especially IL-2 and IL-15) [16,17,18,19] and transcription factors (Nfil3, Id2 and Tox for development, and EOMES and T-bet for maturation) [16, 20]. GRB2-associated binding protein 3 (GAB3) is essential for IL-2 and IL-15-mediated, and its deficiency leads to impaired NK cell expansion [21]. In addition, targeting related signals is a potential option for promoting NK cell-induced cytotoxicity toward cancer. As reported previously, ablation of cytokine-inducible SH2-containg protein (CIS), which negatively regulates IL-15 to restrict NK cell function, could prevent metastasis and also potentiate CTLA-4 and PD-1 blockade therapy in vivo [22].
Identification and molecular features of NK cells
Surface molecules of NK cells
Due to variable expression of surface markers on NK cells, it is hard to use one or two simple molecules or traditional immunohistochemistry to accurately identify this cell type and more importantly, their functional status. However, in humans, in both clinical and research settings, CD3−CD56+ cells are commonly identified as NK cells and can be further divided into the CD56bright and CD56dim subgroups. CD56 is not only a marker but also plays an important role in the terminal differentiation of NK cells since its blockade by monoclonal antibodies obviously inhibits the transition from CD56bright to CD56dim, thus limiting the cytotoxic ability [23]. Consistently, CD3−NK1.1+ and CD3−CD49b+ cells are defined as NK cells in mice. In recent studies, the notion that natural cytotoxicity receptor 46 (NKp46), belonging to natural cytotoxicity receptors (NCRs), should also be included in this panel has been proposed based on the consensus of adding more functional proteins rather than surface molecules into the classification system of NK cells [24, 25].
Activating and inhibitory signals in NK cells
As the main effector cell type in innate immunity, NK cells are capable of killing tumor cells and virus-infected cells at a very early stage. Due to the lack of abundant production of receptors for distinguishing incalculable antigens in the body specifically, they rely on the “missing self” and “induced self” modes to identify target cells by maintaining a precise balance between activating co-stimulatory and inhibitory signals (mainly by functional receptors). Those interacting signals finally decide the activation and functional status of NK cells.
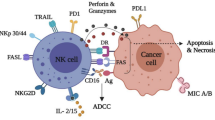
Activating signals include cytokine-binding receptors, integrins, killing-receptors (CD16, NKp40, NKp30 and NKp44), receptors recognizing non-self-antigens (Ly49H) and other receptors (e.g., NKp80, SLAMs, CD18, CD2 and TLR3/9) [26, 27]. In total, the activating receptors of NK cells can be divided into at least three types according to the respective ligands, including MHC-I specific, MHC-I related and MHC-I non-related receptors (Table 1) [13, 28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43]. To emphasize, NCRs, which belong to the third group, include three molecules (NKp30, NKp44 and NKp46), and NKp30 was shown to be capable of recognizing B7-H6 expressed on tumor cells, and could be used as a novel treatment option in the future [35].
Inhibitory signals mainly comprise receptors recognizing MHC-I, such as Ly49s, NKG2A and LLT1, as well as some MHC-I non-related receptors (Table 1) [20, 44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60]. Moreover, MHC-I specific inhibitory receptors can be generally divided into three types according to structure and function: killer cell immunoglobulin-like receptors (KIRs), killer lectin-like receptors (KLRs) and leukocyte immunoglobulin-like receptors (LILRs).
NK cell subpopulations according to site of maturation
Conventional NK (cNK) cells are mainly found in peripheral blood and migrate to a specific location to exert their effects. NK cells also include tissue-resident NK (trNK) cells. The complex process of NK-cell differentiation occurs in several distinct tissues, including bone marrow, liver, thymus, spleen and lymph nodes, and may involve cell circulation at different stages of maturation among these tissues [61]. In bone marrow, blood, spleen and lungs, NK cells are fully differentiated, while that in lymph nodes and intestines are immature and precursory [62]. Single-cell transcriptome ananlysis of bone marrow and blood NK cells helps to illustrate the changes of their characteristics during development. For example, high expreesion of TIM-3, CX3CR1 and ZEB2 represents a more mature status [63]. Many hypotheses have been proposed to describe the motivation of their migration and different biological behaviors of identically originated NK cells in different tissues. The first question could be partly explained by the multi-direction differentiation induced by heterogeneous microenvironments in different tissues, or more straightforward, different phenotypes originated from similar chemokine-recruited peripheral cNK cells.
To conclude, NK cells in various tissues have diverse features, possessing different functions and forming a close relationship with other stromal cells (Fig. 1). In the lung, trNK cells show a different phenotype from that of circulating NK cells (mainly CD56dim) and are considered to express different levels of CD16, CD49a and CD69, with CD56dimCD16+ cells representing the majority of the whole NK family there [64, 65]. Of note, CD69+ cells are the main type of CD56brightCD16− NK cells. However, in the thymus, most NK cells are CD56highCD16−CD127+, highly relying on GATA3 compared with the CD56+CD16+ subgroup [66]. Besides, they produce more effector molecules, including TNF-α and IFN-γ [66, 67]. Similar to the phenotypic features in humans, skin NK cells in the mouse can be generally divided into two types: CD49a+DX5− and CD49a−DX5+ [68, 69]. Similarly, hepatic trNK cells can be classified into two groups, including CD56brightCD16+/− and CD56dimCD16+, both lacking CD3 and CD19 [8]. In addition, CD49a+CD56+CD3−CD19− NK cells have been identified in liver biopsies [70]. Besides, hepatic NK cells can develop memory for structurally diverse antigens, dependent on the surface molecule CXCR6 [71]. In the uterus, most NK cells are CD56brightCD16−, expressing high levels of KIRs [72]. Decidual NK cells are also CD49a+. For skin NK cells, it is intriguing that only few CD56+CD16+ can be detected, which are common in peripheral blood [73]. Interestingly, trNK cells are distinct between subcutaneous (CD56dim) and visceral (CD56bright) adipose tissues, and can be generally divided into three groups according to CD49b and Eomes, showing different expression levels of CD49a (CD49b+Eomes− subgroup) and CD69 (CD49b−Eomes+ subgroup) [74, 75].
Besides different tissue types, NK cells are also highly heterogeneous even in the same organ and the same tissue [61]. Through high-dimensional single-cell RNA-seq, Crinier et al. revealed the heterogeneity of human and mouse NK cells in spleen and blood and identified several subpopulations of NK cells, respectively [76]. As mentioned above, NK cells are considered a subgroup of ILC1s [6]. Although ILC1s are not detectable in many tissues, intra-epithelial ILC1 (ieILC1)-like cells, which highly express IFN-γ, integrins and other cytotoxic molecules similar to the ieILC1s previously described by Fuchs except for different NKp44 expression, could represent the majority of NK cells in the mucosal tissue [61, 77]. Due to their unique features, this cell type represents a subgroup of NK cells other than conventional ILCs. Unlike other trNK cells, DX5−CD11chi liver-resident NK cells participate in autoimmune cholangitis, negatively regulating immune responses, especially by inhibiting the proliferation and function of CD4+ T cells in vivo, which was validated by severer biliary disease in NK-depleted mice resulting from Nfil3 knockdown or treatment with neutralizing antibodies [78].
NK cell subpopulations according to functional molecules
According to surface CD56 expression, NK cells can be divided into CD56bright and CD56dim. CD56dim NK cells are mainly found in peripheral blood, and are always also CD16-positive, expressing high levels of KIR and LFA-1 and showing cell killing ability. CD16 is a key receptor mediating antibody-dependent cell cytotoxicity (ADCC), inducing the phosphorylation of immunoreceptor tyrosine-based activation motif (ITAM) [40, 79, 80]. According to a time-resolved single-cell assay, the cytotoxicity of NK cells is repressed through both necrosis and apoptosis. As a result, FasL/FasR interaction, perforin/granzyme release and Ca2+ influx are all important for NK cell function [81]. However, CD56bright NK cells are similar to helper cells, which mainly secrete cytokines such as IFN-γ, TNF-β and GM-CSF [23]. Researchers even further subgroup these cells into the NK1 and NK2 categories, consistent with Th1 and Th2, mainly secreting IFN-γ and IL-5, respectively [82].
Besides established cytotoxic cNK cells, it has been demonstrated that NK cells could differentiate into antigen-presenting NK (AP-NK) cells [83], helper NK (NKh) cells [84] and regulatory NK (NKreg) cells, each defined by surface molecules and individual functions. A new CD8αα+MHC-II+ phenotype with professional APC capacity was considered to represent unusual AP-NK cells, recognizing and eliminating autoreactive T cells and finally killing them like cNK cells [85]. Human plasmacytoid dendritic cells (DCs) activated by the preventive vaccine FSME upregulate CD56 expression on their surface [86]; in mice, B220+CD11cintNK1.1+ cells have antigen-presenting capacity like DC, hence their name interferon-producing killer DC [87, 88].
Invariant natural killer T cells (iNKT) constitute a subgroup of T cells expressing NK cell-markers. Activated by CD1d-presenting antigens, NKT could secrete not only Th1-type but also Th2-type cytokines to participate in immunity [89, 90]. Th1-polarized iNKT cells exhibit a tumor-depletion phenotype, and Th2-polarized iNKT cells contribute to tumor progression, similar to polarized T cells [91, 92]. Recent studies also highlighted new functional subtypes of iNKT cells. However, in recent years, due to their close relationship with innate immunity, iNKT cells are potentially defined as a special subgroup of ILCs.
NK cells in the tumor microenvironment
Conventional roles of NK cells in immunity
Detection of aberrant cells by NK cells is determined by the intergradation of complex signals such as IL-12, IL-15 and IL-18 [93, 94], and the balance between activating and inhibitory signals interacting with MHC-I on the surface of target cells (Fig. 2). During infection and inflammation, NK cells are recruited and activated in a short period of time, proliferate quickly and contribute largely to both innate and adaptive immune responses [8, 95]. Except for their newly proven regulatory effects, NK cells were first found to directly target infected cells or foreign pathogens; therefore, deficiency in NK cells in both mice and humans results in susceptibility to many viral infections and adverse clinical outcomes, validated by clinicians and researchers.

The complex interaction between NK cells and the extracellular matrix. Exposure of NK cells to the adjacent cells, molecules and metabolites in the extracellular matrix affects their development, maturation, activation and functions. CXCR3, C-X-C motif chemokine receptor 3; NKG2D, nature-killer group 2, member D; IFN-γ, interferon γ; TNF-α, tumor necrosis factor α; IDO, indoleamine 2,3-dioxygenase; MICA, MHC class I polypeptide-related sequence A; PGE2, prostaglandin E2; HCC, hepatocellular carcinoma; CIS, cytokine-inducible SH2-containg protein; TGF-β, transforming growth factor-β; HMGB1, high-mobility group box 1; HIF-1α, hypoxia inducible factor-1α; 27HC, 27-hydroxycholesterol; iNKT, invariant natural killer T; GM2, β-N-acetylhexosaminidase; TCR, T cell receptor
Similar to other innate immune cells that are unable to accurately recognize target cells, NK cells rely on other stromal cells, including DC, which trans-presents IL-15 for NK cell activation [96], and MICA-expressing monocytes, which bind to Fc receptor to enhance antitumor function [97], to fully differentiate and induce effector responses, but surprisingly possess the ability to form immunological memory, termed “trained immunity”. Once considered as a hallmark of adaptive immunity, in recent years, the phenomenon of immunological memory has also been found in innate immune cells, especially in the myeloid lineage, e.g., monocytes and macrophages. In addition, mounting evidence indicates that in humans, NK cells can remember previous exposure to inflammatory microenvironment, and occurrence of similar cytokines could induce trans-differentiation from normal NK cells to memory NK (NKm) cells [98,99,100,101]. This was evidently observed in response to viral infection in humans, prompting the development of NK cell-based vaccines to generate potent effects toward diseases [102, 103]. A large number of NKm cells are observed in the tumor microenvironment, producing high levels of IFN-γ, perforin and granzyme family molecules after re-stimulation [104]. However, concerning to tumors, dysfunction of NK and NKm cells is emerging as an indispensable and undeniable event, leading to not only proliferation of tumor cells but also the formation of distant metastases [101]. It was observed that repeated exposure of NK cells to NK receptor ligand-expressing tumor cells (e.g. NKG2D) finally results in NK cell dysfunction, and effector responses cannot be stimulated in vivo [105, 106]. These results indicate that the formation of NKm cells may not just depend on target cell recognition through surface receptors, and certain cytokines (including IL-12, IL-15 and IL-18) could be key to this process.
Though the half-life of normal NK cells is only for 1–2 weeks, NKm cells can live for 3–4 weeks [107]. This long-term effect truly helps researchers better modulate the function of NK cells in protecting against tumors, and emerging results suggest that NK cells not only rely on MHC-I recognition but also depend on many other signals, shedding light on the use of NK cells and related signaling pathways as future treatment options.
Infiltration of NK cells with cancer genotypes and phenotypes
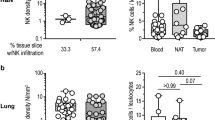
In 2000, an 11-year follow-up study of the Japanese general population, with rigorous use of various related biochemical and immunological markers, indicated that elevated cytotoxic activity of peripheral NK cells is positively associated with reduced cancer risk and vice versa, suggesting the certain importance of natural immune response toward tumors [108]. However, the specific role of NK cells remains controversial and largely depends on distinct cancer types [109]. Even in the same type of cancer, NK cells are highly heterogeneous, characterized by the abundance of surface receptors and the complexity of tumor intrinsic signaling pathways [95, 110]. In the CIBERSORT analysis, NK cells were thoroughly divided into resting and activated subtypes, each contributing to the formation of the tumor microenvironment [111]. In preclinical studies, NK cells were shown to indicate survival and thus therapeutic response in different types of cancer, as detected by immunohistochemistry, immunofluorescence or flow cytometry using different surface and functional markers (Table 2) [46, 56, 112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129]. Although CD3−CD16+CD56+ cells reflect different clinical outcomes in different cancers, functional molecule-positive NK cells, including NKp30+ and NKp46+, indicate favorable survival, pointing out the fact that full activation but not only infiltration density finally determines NK cell-associated immune response.
In BC, besides the ability of total NK cells to reflect favorable survival, peritumoral abundance of NK cells also correlates with elevated pCR rate of neoadjuvant chemotherapy in large and locally advanced breast cancer [130], and vice versa. To address this, it is currently well accepted that tumors are highly heterogeneous, and even one histologic type can be separated into several molecular subtypes. BC can be thoroughly clustered into luminal, HER2-enriched and triple-negative types according to the expression of surface molecules, and both infiltration and activation status of NK cells vary by cluster, e.g., obviously elevated NKG2D in luminal tumors [131]. However, due to the complex and variable functional status of NK cells, their actual role in the TME still awaits further elucidation.
Underlying mechanisms of immune escape and metastasis of cancer
As noted above, NK cells rely on the balance between activating and inhibitory receptors to exert their killing effects, and perforin and the granzyme family of proteins are the main effector molecules. Consistent with cNK cells, as a newly-found subgroup of NK cells, activated NKT cells directly detect and kill CD1d+ tumor cells in several types of cancer [132, 133]. In addition, by expressing high levels of CD40, NKT cells induce DC maturation, thus activating CTL and cNK cells to enhance their anti-tumor effects [134, 135].
Apart from primary tumor proliferation, metastasis remains lethal, accounting for the majority of cancer-associated deaths, and invasion-metastasis cascade is largely attributable to the immune escape [136]. With such rapid and effective ability to target tumor cells directly and indirectly, NK cells are suppressed by tumor-derived molecules, tumor-educated stromal cells (Fig. 2) and tumor cells (Fig. 3), eventually contributing to the progression and multi-step metastatic process of cancer. For example, single-cell analyses found that in lung adenocarcinoma, CD16+ NK cells are hardly infiltrated and present lower granzyme B and CD57 expression compared with normal lung tissue, appearing to form NK cell-excluded TME [137].

Interplay between cancer cells and NK cells during tumorigenesis. The interaction between tumor cells and NK cells changes continuously with NK cell development, tumor progression and metastasis. During the stage of tumorigenesis (a), NK cells recognize tumor cells through various surface molecules and switch to the active status. In the immune control stage (b), NK cells exert killing effects by ADCC, secreting cytokines and generating memory NK cells. Meanwhile, changes in the surface molecules of tumor cells also promote anti-tumor metabolic responses. However, long-term exposure of NK cells to tumor cells, tumor- derived molecules and tumor-educated stromal cells, including fibroblast, monocyte and macrophage, causes NK cells to be in an immunosuppressive state, thereby promoting tumor immune escape and metastasis (c). MHC- I, major histocompatibility complex-I; MICA, MHC class I polypeptide-related sequence A; MICB, MHC class I polypeptide-related sequence B; NCR, natural cytotoxicity receptor; Nfil3, nuclear factor interleukin-3-regulated protein; Id2, inhibitor of DNA binding 2; Tox, thymocyte selection associated high mobility group box; EOMES, eomesodermin; T-bet, T-box transcription factor 21; ADCC, antibody-dependent cell-mediated cytotoxicity; GM-CSF, granulocyte-macrophage colony stimulating factor; PRF1, perforin 1; GZMB, granzyme B; PD-L1, programmed cell death ligand 1; PGE-2, prostaglandin E2; HCC, Hepatocellular Carcinoma; IFN, interfron; TNFα, Tumor Necrosis Factor α;PI3K, Class IA phosphatidylinositol 3 kinase
Class IA phosphatidylinositol 3 kinases (PI3Ks) are involved in growth and survival of normal cells, and mutation of the PI3KCA isoform is commonly found in the genomic landscape of many cancers. Inhibiting abnormally activating signals of PI3KCB with a tested inhibitor called P110β in hematologic malignancies obviously enhances susceptibility of tumors to NK cell activity in vitro, probably through the regulation of MHC-I [138]. Tumor-derived prostaglandin E2 [139], extracellular adenosine [140], fragmented mitochondria in the cytoplasm of tumor-infiltrating NK cells [141] and actin cytoskeleton remodeling [142] also lead to immunosuppression and help potentially metastatic cancer cells to avoid NK cell elimination. Interestingly, in a breast cancer cell line, CDC42 or WASP knockdown does not change the activation status of NK cells but obviously increases the expression of effective granzyme B and overcomes resistance to NK cell-mediated attack [142]. Further investigation in this field might help identify a new signaling pathway or a new marker of NK cell-activation and provide new insights into NK cell-therapy. Similar to the inhibitory role of CD73 in T cell-immunity, tumors can train normal NK cells into CD73+ NK cells which express high levels of checkpoint molecules, including LAG-3, PD-1, and PD-L1, finally resulting in immune escape [57]. As mentioned above, Th2-polarized iNKT cells in the TME contribute to tumor progression through immunosuppressive effects [91], and continuous exposure to ligands expressed on the surface of tumor cells induces the dysfunction of NKm cells that are engaged in long-term anti-tumor immunity [106]. Moreover, absence of NKG2D is a common feature of functionally suppressed NK cells, which is achieved through many different pathways [105, 143], thus could be used as a marker to guide NK cell-related therapies [144].
In addition, NK cell-associated cytotoxicity can also be impaired by stromal cells, for example cancer-associated fibroblasts, monocytes, macrophages and other immune cells. Fibroblasts in TME suppress function of NK cells through downregulating ligands of NK cell activating receptors [145], enhancing tumor-associated macrophages enrichment [146], and producing extracelluar matrix components such as IDO and PGE2 [147]. In human gastric cancer, tumor infiltrating monoctyes/macrophages can reduce IFNγ, TNFα, and Ki-67 expression of NK cells via TGFβ1, thus impairing NK cell function [148]. Meanwhile, the interaction of PD-1+ NK cells and PD-L1+ monoctyes/macrophages in Hodgkin lymphoma results in immune evasion, which can be reversed by PD-1 blockade [149]. The monocytes from hepatocellular carcinoma express CD48, which could block 2B4 on NK cells and induce NK cell dysfunction [150].
Consistent with CD8+ T cells, activated PD-1+ NK cells are inhibited by elevated PD-L1 expression in the TME [58]. Dysfunction of NK cell following surgery has been regarded as a risk factor of metastasis and can be partly explained by disturbing the balance between activating and inhibitory signals. NK cell-mediated metastasis control was found dependent on dectin-1-mediated activation of macrophages, especially the plasma membrane tetraspan molecule MS4A4A [151]. In colorectal cancer, lipid accumulation is common for postoperative patients and facilitates the formation of metastasis by impairing NK cell function, by elevating CD36 expression [152]. Hence, NK cells can be rarely seen in metastatic melanoma and are mainly TIGIT−CD226− which are deprived of cytotoxicity toward MHC-I-defient malignant cells [153]. Interestingly, proved by convincing genetically engineered models, it is the absence of NK cells but not CD8+ T cells that evidently leads to the metastatic dissemination of small cell lung cancer, pushing us to better define the pivotal role of NK cells in both initial progression and later metastasis of cancer [154].
Relationship between NK cell-based and CD8+ T cell-based immunity
NK cells, though belonging to innate immunity, have characteristics similar to cytotoxic CD8+ T cells [95]. Originally proven important in the two-signal activation model of T cells, CD28 is also necessary for optimal cytokine secretion and proliferation of NK cells both in vitro and in vivo [155]. It was shown that IL-21 is important for the maturation and activation of NK cells [156,157,158]. However, data also uncovered the double-sided function of IL-21 in the development of NK cells and surprisingly, its positive role in T-cell-based immune response [159, 160], for example inducing KLRG1+CD8+ T cells during acute intracellular infection [161]. Besides IL-21, NK cells produce multiple other cytokines during their proliferation, maturation and function similar to T cells, including IL-2 [162], IL-7 [163] and IL-15 [164], validating the close relationship between these two types of effector cells in the body’s immune system.
Although sharing many similarities, compared with effector T cells, NK cells are more cytotoxic to tumors and possess lower immunogenicity [10]. As mentioned above, NK cells respond to target cells more quickly and do not need extra ligation of activating receptors [95]. On the contrary, NK cells have the ability of suppressing the function of CD8+ T cells via NKG2D in severe aplastic anemia [165]. It has been underlined that during infection with chronic lymphocytic choriomeningitis virus, NK cell-intrinsic FcRγ signaling could inhibit the expansion of CD8+ T cells [166]. Interestingly, tumor cells that develop checkpoint blockade resistance to CTLs, especially through suppressed MHC-I expression, are more vulnerable to NK cell-based immunity; thus, combination immunotherapy utilizing both NK cells and CD8+ T cells can constitute a future strategy in terms of tumor immune escape [167]. Besides, in tumors that lack MHC-I-related molecules, elevated amounts of HLA-E and HLA-G were observed, indicating the possibility for NK cells to harness this unclassical pathway [168]. More basic researches are urgently needed for better understanding of the complex relationship between these two major effector cells, as NK cells-based treatment is currently highly underestimated.
Crosstalk of NK cells and metabolic signaling in cancer
Immunometabolic disorder as a hallmark of cancer
Similar to high blood pressure and diabetes, cancer is currently regarded as not only a process of pathogenesis but also a social issue. Lipid accumulation in the liver, abnormal glucose metabolism and irregular lifestyle all contribute to tumorigenesis and cancer progression, which eventually prompt research about the underlying mechanisms of this phenomenon. It is admitted that the metabolic competition between tumor and stromal cells largely affects the process of tumorigenesis and cancer progression. In the TME, NK cell function is impaired not only by suppressive cytokines but is also attributable to inappropriate metabolic conditions, including hypoxia, lack of nutrition and abnormal concentrations of tumor-derived products such as lactate, which induces unfavorable acidic condition, hindering the proliferation and cytokine production of CTLs as well (Fig. 2) [169]. As metabolic disorder is currently considered a hallmark of cancer, which shares a close relationship with the microenvironment, the idea of harnessing immunometabolism attracts increasing attention to improve the efficacy of NK cell-dependent anti-tumor therapy.
The normal breast tissue is surrounded by adipose tissue, and obesity is considered a potential risk factor for breast cancer, which is supported by population-based studies, together with high mental pressure, evidently affecting lipid and glucose metabolic pathways. Obesity-induced inflammation in adipose tissue could result in the recruitment of M1-polarized macrophages, neutrophils, NK cells and CD8+ T cells, and higher expression levels of pro-inflammatory cytokines, as well as obvious exclusion of Treg and invariant NKT (iNKT) cells [170]. In 2011, aware of the importance of tumor-promoting inflammation in the TME, Weinberg et al. included this phenomenon into the hallmarks of cancer and particularly highlighted inflammation induced by the innate immune response [1]. Thus, improved understanding of the mechanism by which metabolic activity affects the function of tumor-infiltrating stromal cells, finally resulting in cancer progression and immune escape, would provide clues for developing novel therapeutics for immunometabolic targets.
Metabolic disorder of conventional NK cells in the TME
NK cell function can be altered by different components in the TME. Breast cancer metabolomics data overtly show that lipid and glucose metabolic pathways are highly activated, especially the fatty acid synthase glycolysis pathway, compared with paired peritumoral tissue.
Similar to other lymphocytes, NK cells require energy to survive, and glucose consumption is evidently increased after full activation, while the competition between NK cells and tumor cells could disturb such need. It has been shown that surface transporters, especially glucotransporter 1 (GLUT1), help NK cells utilize glucose to generate ATP and pyruvate, contributing to glycolysis and oxidative phosphorylation [171, 172]. Several studies have pointed out the importance of sufficient glucose supply for NK cell activities, including proliferative capacity, activation status, cytokine production and direct cytotoxicity [173, 174]. Glycolysis and oxidative phosphorylation contribute to maintain the cytotoxic ability of NK cells, as their inhibition highly decreases the expression levels of IFN-γ and Fas ligand [175]. NKG2D is essential for the activation of NK cells, which relies on glycolysis. Researchers have identified several pathways pertaining to the interlinked metabolic activity and NK cell function. Obesity-related inflammation is dependent on the IL-6/Stat-dependent pathway, thereby resulting in a distinct functional status of NK cells [176]. In addition, Assmann et al. highlighted that sterol regulatory element-binding proteins (SREBP) transcription factor-controlled glucose metabolism is essential for metabolic reprogramming in activated NK cells, providing new insights into this process [177]. Accordingly, SREBP inhibitors such as 27-hydroxycholesterol (27HC) are accumulated in the TME, partly affecting SREBP-related glycolysis in ER-positive BC [178]. However, as most studies only focus on GLUT1, other transporters and unclassical pathways should also be paid attention to, paving the way for deeper understanding of the complex relationship between glucose metabolism and NK cell function.
Hypercholesterolemia remains a risk factor for ER-positive BC. In 2013, 27HC, which negatively regulates SREBP, was also found by Nelson et al. to be a bridge linking hypercholesterolemia and BC [179]. It was also found that treating mice submitted to high-cholesterol feed with an inhibitor of CYP27A1, an enzyme important in 27HC biosynthesis, obviously decreases the number of metastases in mice, which reverses immune suppressive environment [180]. Therefore, using drugs designed to decrease blood cholesterol or directly inhibiting the formation of 27HC could be a potential strategy for patients with ER-positive BC. Surprisingly, a recent study suggested that high serum cholesterol and cholesterol accumulation in NK cells increase their anti-tumor ability by facilitating the formation of lipid rafts in the liver-tumor-bearing murine model [181], highlighting the heterogeneous functions of lipid metabolism in cancer.
Hypoxia is also a common feature of cancer, and often mentioned concurrently with low pH in the TME. Previous findings indicated that besides promoting tumorigenesis and cancer progression, hypoxia also stimulates NK cell formation via HIF-1α, initiating a conflict between suppressing and activating this signal [182]. Studies have also shown that low O2 in TME harms the function of NK cells by downregulating activating signals such as NKG2D, NKp30 and CD16, thereby limiting cytokine production and cytotoxicity and resulting in metastasis [183, 184]. In addition to regulating intracellular signals directly, the hypoxic microenvironment could degrade NK cell-secreted functional molecules such as granzyme B [185], together with CTL-based immunity, which is partly rescued by IL-2 [186, 187]. Considering the pivotal roles of interleukin family members in the maintenance of NK cells, we paid more attention to these cytokine-primed metabolic pathways and found more clues under hypoxic conditions.
Besides, tumor cells can directly alter the metabolic status of NK cells. This can be positive as CD25 expression on NK cells is overexpressed after interaction with tumor cells, inducing long-term anti-tumor metabolic responses by promoting glycolysis and NK cell survival, supported by mTORC1/cMYC signaling activation. However, worsening occurs later as the harmful effects overcome the positive impact. For instance, glutamine addiction and high consumption of nutrients remain common in BC. In vitro studies showed arginine deficiency inhibits IFN-γ production by primary human NK cells [188]. Additionally, mTOR signaling within NK cells can be largely suppressed under low-arginine or glutamine conditions, which also affect IL-2-related stimulation process via cMYC [189]. Upon direct contact with tumor cells to form an immune synapse in response to local energy consumption, mitochondria of NK cells are depolarized and lose metabolic energy [190]. Inhibiting PPARα/δ or blocking the transport of lipids into mitochondria reverses NK cell metabolic incapability and restores cytotoxicity [191].
Indeed, previous studies have reported that anti-PD-L1 therapy could reshape metabolic pathways in the tumor microenvironment and re-stimulate exhausted CD8+ T cells for cytotoxicity [192]. Interestingly, NK cells also express the ligands of these checkpoints. Glycoengineering of NK cells enhances their killing ability toward CD22+ lymphoma in a CD22-dependent manner [193]. Blockade of monocarboxylate transporter 1, which regulates cell metabolism, using AZD3965 also potentiates NK cell activity [194]. Studies that focus on translating mature theories into the practical use of NK cells are promising.
Aberrant metabolic features of iNKT cells
As mentioned above, iNKT cells can be polarized into different properties, each possessing distinct functions. The normal breast tissue is surrounded by adipose tissue. Different from conventional T cells, iNKT cells comprise large amounts of stromal cells in adipose tissue, whose infiltration decreases apparently in high-BMI individuals [195, 196].
With invariant TCR on the surface, iNKT cells are termed innate-like T lymphocytes and act on the front line of the immunity battle against cancer [197, 198]. iNKT cells recognize glycolipid signals but not peptides via semi-invariant TCR, and are restricted to glycolipid antigens presented via CD1d-related molecules, which are MHC-like and highly enriched, especially in adipocytes and hepatocytes, linking innate and adaptive immune responses. This process can be altered by metabolic activity. GM2 is a glycosphingolipid that binds the CD1d molecule. Pereira et al. pointed out that GM2 inhibits the activation of iNKT cells in a dose-dependent manner, which might result from its competition with α-GalCer for binding CD1d [199]. Compared with T lymphocytes, iNKT cells show much higher capacity of glycolysis but reduced mitochondrial respiratory activity, resulting in particular molecular features. Under hypoxia, widespread RNA editing can be induced by mitochondrial respiratory inhibition via APOBEC3G, an endogenous RNA editing enzyme [200]. Fu et al. demonstrated that aerobic glycolysis in iNKT cells is highly increased after TCR engagement, which is essential for the production of IFN-γ [90]. This process can also be inhibited by the lack of glucose. Hence, reduced expression of IFN-γ in iNKT cells compared with the normal tissue was confirmed in several tumor types, predicting patient response to the therapy of PD-1 blockade [201,202,203]. In humanized mouse models undergoing PD-1 blockade and CAR-T (with different costimulatory molecules) combination therapy, only those with Δ-CD28 CAR control tumor growth, and in vitro analysis showed that these cells exhibit elevated glycolysis, fatty acid oxidation and oxidative phosphorylation [204]. Overcoming the impaired metabolic function combined with immune checkpoint blockade would be a potential strategy in future researches and clinical practice.
However, what is currently known about iNKT cells is just the tip of the iceberg. Compared with T lymphocytes, it remains unclear how intracellular metabolic signals influence the survival and function of iNKT cells, which deserves further investigation, as this may be the next potential target of cancer immunometabolic therapy after CD8+T cells and NK cells.
NK cells in cancer therapy
As an important effector of innate immunity, though suffering a resistance evolved by tumor cells, NK cells show their potential to be used in clinical practice [205,206,207]. In the past few years, researches about NK cell-related immunotherapy have flourished and the latest development mainly focused on cytokine supplement, monoclonal antibody, modification of internal signal pathway, adoptive transfer and genetic engineering of NK cells. Besides, NK cell-based therapy has achieved favorable results used either alone or in combination with other therapies, which suggests a wide and effective use in malignancies.
Cytokine supplement
IL-15 promotes the development and cytotoxic ability of NK cells, and several clinical trials have illustrated the safety profile of recombinant human IL-15 (rhIL-15) in multiple tumors [208, 209] as well as its agonist, ALT-803, in metastatic lung cancer and post-transplantation patients (Fig. 4) [210, 211]. In an open-label, phase Ib trial, ALT-803 showed a fantastic potential when combined with anti-PD-1 monoclonal antibody (nivolumab) without increasing the incidence of very severe grade 4 or 5 adverse events [211], which evidently could be the future way to enhance rhIL-15 treatment. In addition to soluble IL-15 in the microenvironment, it has been revealed that in the mouse, direct contact with membrane-bound IL-15 on adjacent stromal cells could induce stronger cytotoxic effects in NK cells [212]. Heterodimeric IL-15 can also increase intratumoral NK cell and CD8+ T cell infiltration, elevating the effective rate of current immunotherapy [213].

Possible targets harnessing NK cells in cancer therapy. In order to obtain better clinical efficacy and reduced severe adverse events, the development of NK cell-based therapies that support NK cell maintenance (a), enhance NK cell function (b) and harness abnormal immunometabolic and intracellular microenvironment (c) is essential. rhIL-12/15/18, recombinant human interleukin-12/15/18; CAR-iPS, chimeric antigen receptor-induced pluripotent stem cell; MIC: MHC I chain related molecule; MICA, MHC class I polypeptide-related sequence A; MICB, MHC class I polypeptide-related sequence B; PD-L1, programmed cell death-ligand 1;scFv, single-chain variable fragment; TSA, tumor specific antigen; BiKE, bispecific killer cell engager; TriKE, trispecific killer engager; CAR-NK, chimeric antigen receptor-nature kill; PD-1, programmed cell death protein 1; MerTK, MER proto-oncogene, tyrosine kinase; 27HC, 27-hydroxycholesterol; GLUT1, glucotransporter 1; MCT1, monocarboxylate transporter 1
Apart from IL-15, other interleukins are also synergetic to this process (Table 3). IL-21 enhances tumor rejection in mice via NKG2D-dependent NK cell activity, suggesting IL-21 to be a possible target for immune escape induced by NKG2D elicitation [214]. However, IL-15-dependent expansion of resting NK cells can be suppressed by IL-21, while on the other hand adaptive immune response is enhanced [159], providing substantial insights into this complex network in clinical use. Besides, blocking CIS could promote IL-15-type cytotoxicity and thus results in increased production of IFN-γ [22]. NK cells pre-exposed to IL-12, IL-15 and IL-18 accumulate in the tumor tissue and retain their anti-tumor function both in vitro and in vivo. However, IL-15 alone does not exert such effects [104]. In addition to the inner pathway, treatment with IL-2 and IL-15 obviously enhances glycolysis and oxidative phosphorylation of NK cells, thus promoting the killing ability. In a first-in-human phase I multicenter study, NKTR-214, a novel IL-2 pathway agonist, was found promoting proliferation and activation of NK cells without expansion of Treg cells [215]. For metastatic melanoma refractory to CD8+ T cell cytotoxicity due to the lack of MHC-I, combination of IL-15 and TIGIT blockade shall be effective by stimulating NK cell-mediated immunity [153].
As mentioned above, cytokines (especially IL-12, IL-15 and IL-18) are critical to the formation of NKm cells. Memory-like NK cells supplemented with IL-12, IL-15 and IL-18 also show enhanced responses against acute myeloid leukemia both in vitro and in vivo [107], and are currently assessed in a first-in-human clinical trial. However, studies also pointed out that IL-12 could increase NKG2A expression and inhibit the activation of NK cells [216].
Monoclonal antibodies
As shown previously, in parallel with CD8+ T cells, NK cells can also be suppressed by immune checkpoint molecules. After cetuximab treatment, PD-1+ NK cells are more enriched in the TME and are correlated with favorable clinical outcome in head and neck cancer patients, which was further demonstrated by in vivo experiments and a stage III/IVA clinical trial assessing neoadjuvant cetuximab (NCT01218048) [58]. With PD-1 blockade (nivolumab), cetuximab-induced NK cell activation and function are remarkably enhanced in PD-L1high tumors.
In addition to already-well-defined PD-1 and PD-L1, NK cells with reduced amounts of T-cell immunoglobulins and ITIM domain (TIGIT) show higher levels of cytokine secretion, degranulation activity and cytotoxicity [217], and blockade of TIGIT could prevent exhaustion in NK cells [20]. A recent study also highlighted that TIGIT-depleted NK cells are highly sensitized [218] and develop resistance to MDSC-mediated immunosuppression [219]. Meanwhile, CD96, which shares the same ligand CD155 with CD226 and TIGIT, negatively controls the immune response by NK cells [220] and predicts adverse survival in human hepatocellular carcinoma [56]. Single use of CD96 antibody promotes NK-cell-induced anti-metastatic ability [221], and such effect is largely increased when combined with anti-CTLA-4, anti-PD-1 or doxorubicin chemotherapy [222].
Though prospective, unexpected biological events have been observed in a single-arm phase II study showing that intravenous infusion of 1 mg/kg IPH2101 (a human monoclonal antibody against KIRs) results in severe contraction and obvious inhibition of NK cells in myeloma patients [223]. Lirilumab, a 2nd generation antibody targeting KIR, had encouraging results in a phase I trial, which demonstrated its safety [224]; however, the subsequent phase II trial in AML patients showed no clinical effects. Combination of CIS inhibition with CTLA-4 and PD-1 blockade exerts even greater effects in reducing melanoma metastasis compared with either of these treatments administered alone; thus, CIS inhibition could offer an alternative therapeutic option for patients not responding to other immune checkpoint inhibitors [22].
As an essential receptor for the activation of NK cells, NKG2D is blocked by many ligands (e.g., MICA, MICB, and ULBP1–6) upregulated in tumor cells as a result of abnormal cellular stress in the TME. A recent study demonstrated that antibodies targeting MICA and MICB can prevent NK cell recognition and tumor cell binding, inhibiting tumor growth in fully immunocompetent mouse models as well as humanized mouse models [129]. Furthermore, combination treatment targeting soluble MIC, e.g., MICA and MICB, and PD-L1 shows better effect than monotherapy in vivo [225]. Moreover, soluble MULT1, a high affinity mouse NKG2D ligand stimulates NKG2D in distant NK cells and enhances NK cell tumor immunity [106]. Hence, a clinically used antibody, monalizumab, has been developed targeting NKG2A, an inhibitory checkpoint of NK cells, which not only promotes NK cell function in various preclinical models, as previously characrerized, but also potentiates anti-PD-1 [226] and anti-EGFR (cetuximab) therapy [227]. In addition to antibody, NKG2Anull NK cells, constructed through retroviral transduction of NKG2A blocker which inhibits de novo NKG2A expression, present increased anti-tumor activity in pre-clinical model [228].
In summary, besides the targets close to T cells, including FDA-approved anti-CTLA-4 (ipilimumab) and anti-PD-1 (nivolumab, pembrolizumab) antibodies, others designed specifically for NK cells are also under clinical trials, e.g., anti-KIR (IPH2101, lirilumab) and anti-NKG2A (monalizumab) (Table 3) (Fig. 4).
Cell adoptive therapy and newly arising genetic modification of NK cells
As an applicable option for enhancing autologous immunity, adoptive transfer of NK cells has been implemented to treat certain types of cancer in the past few years [229]. Previous studies of NK adoptive transfer in acute myeloid leukemia patients have presented slightly beneficial effects in controlling disease [230, 231], and a phase II clinical trial in patients with recurrent ovarian or breast cancer showed that adoptive transfer of haploidentical NK cells after lymphodepleting chemotherapy leads to a temporary benefit but its clinical value remains controversial, and is partly limited by recipient reconstitution of regulatory T cells [232]. Apart from adults, a phase I clinical trial applied autologous ex-vivo-expanded NK cells to children with recurrent medulloblastoma and ependymoma and obtained good safety and therapitic efficacy [233]. Interestingly, transfer of NK cells along with CD34+ hematopoietic stem cells shows no added adverse effects but potential therapy response in older patients with acute myeloid leukemia [234]. Combinational application of NK cell infusion with monoclonal antibody provides a new direction of combinational immunotherapy. In a phase I trial, activated autologous NK cell were infused into patients with HER2-positive solid tumor undergoing trastuzmab and showed prelimitary anti-tumor phenotype [235].
In 2009, Fujisaki et al. found that overexpression of telomerase reverse transcriptase could lead to over 100 additional doubling cycles in NK cells, revealing a potential way to overcome the limitation of NK cell amplification in vitro [236]. However, though the adoption of NK cells seems promising in preclinical and clinical researches, many questions exist, e.g., the undeniable fact that NK cells gain self-renewal ability following infusion. Originally designed and considered a one-time-use therapeutic process, a small part of NK cells surprisingly remain alive and proliferate in the human body for months, mediating continuous surveillance against tumor [237]. However, in addition to beating tumor cells, the risk of long-lived NK cells should not be ignored where their “brake” is lost and they might also kill normal cells, even worse, finally increasing the possibilities of NK lymphoma [95, 238]. Hence, the ability of transferred NK cells may be limited by inappropriate persistence or expansion in vitro, and biology-driven methods are commonly used to overcome this issue before adoptive transfer. A recent study indicated that NK cells pre-activated by IL-12, IL-15 and IL-18 suppress graft-versus-host disease but obviously inhibit the generation and function of CD8+ T cells, which could result from mutual competition of IL-2 [239], limiting its value in the field of cancer treatment. The use of mouse models to unveil the detailed features of adoptive NK cells following in vitro proliferation would actually provide deeper insights into this treatment strategy, shedding light on the future implementation of NK cell-based therapy toward cancer.
Genetic modification of immune cells by chimeric antigen receptors (CARs) to target tumor cells directly is a promising therapeutic option in cancer therapy. Kymriah (CTL019, a CAR-T product) by Novartis was approved by the FDA for treating recurrent and refractory acute lymphoblastic leukemia in 2017, and remains under investigation for other indications in several clinical studies [240,241,242]. Two months later, Yescarta by Kite Pharma was approved for diffuse large B cell lymphoma [243]. Due to serious adverse effects induced by CARs, especially cytokine releasing storm (CRS) and neurotoxicity, Actemra (tocilizumab, anti-IL-6 monoclonal antibody) was then approved for CRS, with a further study also showing potential application of Anakinra (an antagonist IL-1 receptor) in such case [244, 245]. In NK cells, antibody engineering approaches optimize NK cell-mediated ADCC to tumor cells through the bispecific killer cell engager (BiKEs) or trispecific killer engager (TriKEs) antibodies (Fig. 4). BiKE connects a single-chain variable fragment (scFv) to the anti-CD16 recognition site with the scFv of a tumor specific antigen, such as CD19/CD20 for non-Hodgkin lymphomas, CD33/CD123 for acute myelogenous leukemia/AML and CD30 for Hodgkin lymphoma, to enhance NK cell recognition of tumor cells [246]. TriKE consists of a BiKE and cytokine IL-15, which boost NK cell function and survival. It was shown that CD19-CD16 BiKE engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies [247]. Meanwhile, CD16-IL15-CD3 TriKE can activate suppressed NK cells and induce NK cell-mediated control of MDS and AML [248]. The advantages of CAR-NK therapy are obvious, including higher possibility of recognizing tumors (including cytokines and apoptosis) and lower incidence of CRS compared with CAR-T (Table 3) [249, 250]. In 2018, Enli Liu et al. transduced cord blood-derived NK cells with a retroviral vector incorporating the genes for CAR-CD19, inducible caspase-9-based suicide gene (iC9) and IL-15, and demonstrated the efficacy and safety in cell lines and the murine model. In the engineered NK cells, CAR-CD19 redirected the specificity of NK cells against leukemia, IL-15 promoted NK cell proliferation, and iC9 allowed NK cells to initiate suicide after killing the target cells [251]. Since CAR-NK cells exhibit striking efficacy and limited toxicity, clinical trials assessing these CAR-NK cells have been launched. In recent phase I and II trials, iC9/CAR-CD19/IL-15 NK cells were prepared ex vivo and infused into patients with relapsed or refractory CD19-positive cancers after lymphodepleting chemotherapy. Among the 11 treated patients, 8 had an objective response, including 7 with complete remission, without major toxic effects [250]. Apart from engaging NK cell with CAR, geneic modification involves deleting surface molecules, e.g., CD38, on NK cells, which evidently elimates fratricide and enhances cytotoxic ability [252].
However, limitations should be mentioned. Due to the complex process of producing CAR-NK cells, the current procedure is too expensive and the effects on solid tumors are far from being satisfactory. CAR-iPS might be a future direction, which can grow in vitro and differentiate into CAR-NK cells in vivo to directly enhance anti-tumor immunotherapy [253]. Recently, Zhu H et al. reprogramed NK cell metabolism by depleting CISH in human iPSC-derived NK cells and obtained satisfactory persistence and anti-tumor activity in vivo, which could be a novel method to generate CAR-NK from CAR-iPS [254].
Refinement of the established therapies
As an important immunosuppressive cytokine that promotes tumor progression, TGF-β and its pathway represent potential opportunities for anti-tumor drug development. The clinical modulation of TGF-β, which is achieved through small-molecule inhibitors and antibodies, is being investigated in a number of clinical trials [255]. As the safety and efficacy of TGF-β blockade therapy have been demonstrated, two studies independently showed that combination treatments of TGF-β blockade with anti-PD-1/PD-L1 therapies have synergistic effects on murine EMT6 breast mammary carcinoma and colorectal cancer [256]. It is admitted that TGF-β inhibits NK cell metabolism, proliferation, cytokine production, cytotoxicity and anti-metastatic functions through mTOR signaling in multiple cancers [257]. Activin-A, a member of the TGF-β superfamily, increases ILC1-like tissue residency features, reduces cytokine production, and suppresses proliferative and metabolic functions in both human and murine NK cells through an alternative SMAD2/3-related pathway with effects similar to those of conventional TGF-β pathway [258]. Therefore, targeting TGF-β, TGF-β superfamily, and their downstream pathways in NK cells may be promising treatment options that enhance the efficacy of current immunotherapies (Fig. 4). Apart from blockage of immunosuppressive cytokine, immunostimulatory agonist have been tested in clinical trials. In a recent phase I/IIa study, BMS-986156, a human glucocorticoid-induced TNF receptor-related protein agonist, appears to increase NK cell proliferation no matter applied with or without nivolumab, and has an adorable safety and efficacy profile [259].
Rapamycin, an inhibitor of the mTOR pathway, and its derivative everolimus are effective for treating breast cancer in clinical trials, including BOLERO-6 and PrE0102 studies, either alone or in combination with endocrine and chemotherapy [260, 261]. However, NK cells highly rely on PI3K-mTOR signaling pathway-dependent metabolic reprogramming to exert their anti-tumor effects. Therefore, in patients with both PI3K-mTOR pathway activation and NK cell-based microenvironment, such inhibitors should be cautiously employed.
In HER2-enriched breast cancer, besides HER2-targeted therapies, anti-GD2 also appears to be promising in preclinical studies. However, GD2-related pathways are essential for ADCC [262], also reducing the effectiveness of NK cells, which provides insights into the complex relationships among such networks to minimize off-target effects exerted by established therapies on immunotherapy.
Radiotherapy was found to kill cancer cells via inducing DNA damage, but recent studies found its ability to cause immunogenic cell death, named immunogenic radiotherapy [263]. There is a growing interest to combine immunogenic radiotherapy and immunotherapy, plus chemotherapy or target therapy. A triple-combination therapy which inhibits PD-1 and MER proto-oncogene tyrosine kinase plus radiotherapy, increases NK cells infiltration in abscopal TME [264]. Adding indoximod, an inhibitor of IDO pathway, to radiotherapy and PD-1 blockade also enhances NK cell activity and shows great clinical response [265]. Another representative drug of triple-combination is selenium-containing nanoparticles,which delivers doxorubicin to the tumor site and releases high energy rays. The rays not only kill tumor cells, promote doxorubicin release, but also produce seleninic acid which enhances NK cell function [266].
Conclusions and perspectives
In this review, we draw a picture about the development and function of NK cells and emphasize their variant roles in cancer biology. NK cells performed anti-tumor immunity through their interplay with cancer cells, stromal cells and extracellular matrix, especially the metabolites. As tumor metabolism and tumor imunnity are both recent attractive research areas, we summarized relationship between NK cell and metabolism, which may provide ideas for cross study of these areas. NK cells often suffer resistance in TME and possible mechanisms have been illustrated in many researches, thus developing NK cell-based therapeutic strategies. In addition, clinical trials taking advantage of NK cells, either used alone or in combination with other therapies, have achieved promising results, paving the way for the future basic and clinical researches of the previously ignored but now prosperous NK cell-based cancer therapy and lighting up hope for patients resistant to current T cell-based immunotherapy.
Due to the rapid progress in understanding the TME, new concepts of immunotherapy keep emerging, which obviously helps promote the utilization of immune response for the treatment of cancer, especially the long forgotten innate component. Although previously considered to be characterized clearly, recent evidence shows that the accurate processes of differentiation, activation and generation of memory NK cells remain controversial. Most clinical trials related to NK cell immunotherapy are still in phases I and II, and mainly treat hematological malignancies. Although the clinically-proven safety of these drugs is helpful for the clinical transformation of NK treatment methods for solid tumors, which have broader prospect and wider application in the future, difficulties to overcome still exist. A future challenge for the implementation of NK cell-based therapy is to better define specific NK cell populations and to identify respective markers, as well as functional and regulatory pathways in each subgroup, thereby using different therapeutic strategies for the treatment of tumors infiltrated with different NK cells. Besides, to avoid off-target effects exerted by established anti-tumor drugs on the microenvironment to inhibit treatment effectiveness, more carefully selected combination therapies should be implemented in future clinical trials.
As most studies now take advantage of NK cells originating from blood, trNK cells are worth further investigating for implication in adoptive cell therapy especially for solid tumors. In the era of precision medicine, defining these questions could prompt new approaches that would permit selective regulation of anti-tumor versus pro-tumor response of NK cells. It is an exciting moment in which attention should be paid to this long-ignored cell population, and future possible targets for improved treatment options may harness tumor intrinsic pathways involving both extrinsic innate and adaptive immunity-related microenvironment.
Availability of data and materials
Not applicable.
Abbreviations
- 27HC:
-
27-hydroxycholesterol
- ADCC:
-
Antibody-dependent cell cytotoxicity
- AML:
-
Acute myeloid leukemia
- AP-NK:
-
Antigen-presenting natural killer
- BC:
-
Breast cancer
- BiKE:
-
Bispecific killer cell engager
- CAR:
-
Chimeric antigen receptor
- CD:
-
Cluster of differentiation
- CIS:
-
Cytokine-inducible SH2-containg protein
- cNK cell:
-
Conventional natural killer cell
- CTLA-4:
-
Cytotoxic T-lymphocyte antigen 4
- CTLs:
-
Cytotoxic T cells
- CRS:
-
Cytokine releasing storm
- DC:
-
Dendritic cell
- EOMES:
-
Eomesodermin
- ER:
-
Estrogen receptor
- FDA:
-
Food and drug administration
- FSME:
-
Influenza, pneumococcal, herpes zoster, early summer meningoencephalitis
- GAB3:
-
GRB2-associated binding protein 3
- GLUT1:
-
Glucotransporter 1
- GM2:
-
β-N-acetylhexosaminidase
- GM-CFS:
-
Granulocyte-macrophage colony stimulating factor
- HER2:
-
Human epidermal growth factor receptor 2
- HIF-1α:
-
Hypoxia inducible factor-1α
- HLA:
-
Human leukocyte antigen
- Id2:
-
Inhibitor of DNA binding 2
- IDO:
-
indoleamine 2,3-dioxygenase
- ieILC1:
-
Intraepithelial type 1 innate-like cell
- IFN-γ:
-
Interferon γ
- IL:
-
Interleukin
- ILC1s:
-
Type 1 innate-like cells
- iNKT:
-
Invariant natural killer T cell
- iPSC:
-
induced pluripotent stem cells
- ITAM:
-
Immunoreceptor tyrosine-based activation motif
- ITIMs:
-
Immunoreceptor tyrosine-based inhibitory motifs
- KIRs:
-
Killer cell immunoglobulin-like receptors
- KLRs:
-
Killer lectin-like receptors
- LAG3:
-
Lymphocyte-activation gene-3
- LFA-1:
-
Lymphocyte function-associated antigen-1
- LILRs:
-
Leukocyte immunoglobulin-like receptors
- MCMV:
-
Murine cytomegalovirus
- MDSC:
-
Myeloid-derived suppressor cell
- MHC-I:
-
Major histocompatibility complex-I
- MIC:
-
MHC I chain related molecule
- MICA:
-
MHC class I polypeptide-related sequence A
- MICB:
-
MHC class I polypeptide-related sequence B
- NCRs:
-
Natural cytotoxicity receptors
- Nfil3:
-
Nuclear factor interleukin-3-regulated protein
- NK cell:
-
Natural killer cell
- NKG2:
-
Nature-killer group 2
- NKh:
-
Helper natural killer
- NKm:
-
Memory natural killer
- NKp30/44/46:
-
Natural cytotoxicity receptor 30/44/46
- NKreg:
-
Regulatory natural killer
- PBMCs:
-
Peripheral blood mononuclear cells
- pCR:
-
Pathologic complete response
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death-ligand 1
- PI3Ks:
-
Class IA phosphatidylinositol 3 kinases
- rhIL-15:
-
Recombinant human interleukin − 15
- scFv:
-
Single-chain variable fragment
- SREBP:
-
Sterol regulatory element-binding proteins
- T-bet:
-
T-box transcription factor 21
- TCRs:
-
T cell receptors
- TGF-β:
-
Transforming growth factor-β
- TIGIT:
-
T-cell immunoglobulins and ITIM domain
- TLR:
-
Toll-like receptor
- TME:
-
Tumor microenvironment
- TNF-α:
-
Tumor necrosis factor α
- Tox:
-
Thymocyte selection associated high mobility group box
- TriKEs:
-
Trispecific killer engager
- trNK cell:
-
Tissue-resident natural killer cell
References
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
Netea MG, Joosten LA, Latz E, Mills KH, Natoli G, Stunnenberg HG, O'Neill LA, Xavier RJ. Trained immunity: A program of innate immune memory in health and disease. Science. 2016;352:aaf1098.
Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, Ivanova Y, Hundal J, Arthur CD, Krebber WJ, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–81.
Pitt JM, Vetizou M, Daillere R, Roberti MP, Yamazaki T, Routy B, Lepage P, Boneca IG, Chamaillard M, Kroemer G, Zitvogel L. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity. 2016;44:1255–69.
Herberman RB, Holden HT, Ting CC, Lavrin DL, Kirchner H. Cell-mediated immunity to leukemia virus- and tumor-associated antigens in mice. Cancer Res. 1976;36:615–21.
Chiossone L, Dumas PY, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol. 2018;18:671–88.
Luetke-Eversloh M, Cicek BB, Siracusa F, Thom JT, Hamann A, Frischbutter S, Baumgrass R, Chang HD, Thiel A, Dong J, Romagnani C. NK cells gain higher IFN-gamma competence during terminal differentiation. Eur J Immunol. 2014;44:2074–84.
Vivier E, Raulet DH, Moretta A, Caligiuri MA, Zitvogel L, Lanier LL, Yokoyama WM, Ugolini S. Innate or adaptive immunity? The example of natural killer cells. Science. 2011;331:44–9.
Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol. 2016;17:1025–36.
Vivier E, Ugolini S, Blaise D, Chabannon C, Brossay L. Targeting natural killer cells and natural killer T cells in cancer. Nat Rev Immunol. 2012;12:239–52.
Cherrier DE, Serafini N, Di Santo JP. Innate Lymphoid Cell Development: A T Cell Perspective. Immunity. 2018;48:1091–103.
Zhang Y, Wallace DL, de Lara CM, Ghattas H, Asquith B, Worth A, Griffin GE, Taylor GP, Tough DF, Beverley PC, Macallan DC. In vivo kinetics of human natural killer cells: the effects of ageing and acute and chronic viral infection. Immunology. 2007;121:258–65.
Bauer S, Groh V, Wu J, Steinle A, Phillips JH, Lanier LL, Spies T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science. 1999;285:727–9.
Amin PJ, Shankar BS. Sulforaphane induces ROS mediated induction of NKG2D ligands in human cancer cell lines and enhances susceptibility to NK cell mediated lysis. Life Sci. 2015;126:19–27.
Sungur CM, Tang-Feldman YJ, Ames E, Alvarez M, Chen M, Longo DL, Pomeroy C, Murphy WJ. Murine natural killer cell licensing and regulation by T regulatory cells in viral responses. Proc Natl Acad Sci U S A. 2013;110:7401–6.
Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis ESC. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell. 2018;172:1022–37 e1014.
Huntington ND. The unconventional expression of IL-15 and its role in NK cell homeostasis. Immunol Cell Biol. 2014;92:210–3.
Huntington ND, Puthalakath H, Gunn P, Naik E, Michalak EM, Smyth MJ, Tabarias H, Degli-Esposti MA, Dewson G, Willis SN, et al. Interleukin 15-mediated survival of natural killer cells is determined by interactions among Bim, Noxa and Mcl-1. Nat Immunol. 2007;8:856–63.
Sathe P, Delconte RB, Souza-Fonseca-Guimaraes F, Seillet C, Chopin M, Vandenberg CJ, Rankin LC, Mielke LA, Vikstrom I, Kolesnik TB, et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat Commun. 2014;5:4539.
Zhang Q, Bi J, Zheng X, Chen Y, Wang H, Wu W, Wang Z, Wu Q, Peng H, Wei H, et al. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat Immunol. 2018;19:723–32.
Sliz A, Locker KCS, Lampe K, Godarova A, Plas DR, Janssen EM, Jones H, Herr AB, Hoebe K. Gab3 is required for IL-2- and IL-15-induced NK cell expansion and limits trophoblast invasion during pregnancy. Sci Immunol. 2019;4(38):eaav3866. https://doi.org/10.1126/sciimmunol.aav3866.
Delconte RB, Kolesnik TB, Dagley LF, Rautela J, Shi W, Putz EM, Stannard K, Zhang JG, Teh C, Firth M, et al. CIS is a potent checkpoint in NK cell-mediated tumor immunity. Nat Immunol. 2016;17:816–24.
Wagner JA, Rosario M, Romee R, Berrien-Elliott MM, Schneider SE, Leong JW, Sullivan RP, Jewell BA, Becker-Hapak M, Schappe T, et al. CD56bright NK cells exhibit potent antitumor responses following IL-15 priming. J Clin Invest. 2017;127:4042–58.
Crome SQ, Nguyen LT, Lopez-Verges S, Yang SY, Martin B, Yam JY, Johnson DJ, Nie J, Pniak M, Yen PH, et al. A distinct innate lymphoid cell population regulates tumor-associated T cells. Nat Med. 2017;23:368–75.
Freud AG, Zhao S, Wei S, Gitana GM, Molina-Kirsch HF, Atwater SK, Natkunam Y. Expression of the activating receptor, NKp46 (CD335), in human natural killer and T-cell neoplasia. Am J Clin Pathol. 2013;140:853–66.
Barrow AD, Martin CJ, Colonna M. The Natural Cytotoxicity Receptors in Health and Disease. Front Immunol. 2019;10:909.
Kruse PH, Matta J, Ugolini S, Vivier E. Natural cytotoxicity receptors and their ligands. Immunol Cell Biol. 2014;92:221–9.
Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, Gallagher MM, Malkki M, Petersdorf E, Dupont B, Hsu KC. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med. 2012;367:805–16.
Zhang X, Feng J, Chen S, Yang H, Dong Z. Synergized regulation of NK cell education by NKG2A and specific Ly49 family members. Nat Commun. 2019;10:5010.
Freud AG, Keller KA, Scoville SD, Mundy-Bosse BL, Cheng S, Youssef Y, Hughes T, Zhang X, Mo X, Porcu P, et al. NKp80 Defines a Critical Step during Human Natural Killer Cell Development. Cell Rep. 2016;16:379–91.
Vance RE, Jamieson AM, Raulet DH. Recognition of the class Ib molecule Qa-1(b) by putative activating receptors CD94/NKG2C and CD94/NKG2E on mouse natural killer cells. J Exp Med. 1999;190:1801–12.
Chan CJ, Andrews DM, McLaughlin NM, Yagita H, Gilfillan S, Colonna M, Smyth MJ. DNAM-1/CD155 interactions promote cytokine and NK cell-mediated suppression of poorly immunogenic melanoma metastases. J Immunol. 2010;184:902–11.
Narni-Mancinelli E, Gauthier L, Baratin M, Guia S, Fenis A, Deghmane AE, Rossi B, Fourquet P, Escalière B, Kerdiles YM, et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci Immunol. 2017;2(10):eaam9628. https://doi.org/10.1126/sciimmunol.aam9628.
Siewiera J, Gouilly J, Hocine HR, Cartron G, Levy C, Al-Daccak R, Jabrane-Ferrat N. Natural cytotoxicity receptor splice variants orchestrate the distinct functions of human natural killer cell subtypes. Nat Commun. 2015;6:10183.
Textor S, Bossler F, Henrich KO, Gartlgruber M, Pollmann J, Fiegler N, Arnold A, Westermann F, Waldburger N, Breuhahn K, et al. The proto-oncogene Myc drives expression of the NK cell-activating NKp30 ligand B7-H6 in tumor cells. Oncoimmunology. 2016;5:e1116674.
Spreu J, Kuttruff S, Stejfova V, Dennehy K, Schittek B, Steinle A. Interaction of C-type lectin-like receptors NKp65 and KACL facilitates dedicated immune recognition of human keratinocytes. Proc National Acad Sci USA. 2010;107:5100–5.
Anikeeva N, Steblyanko M, Fayngerts S, Kopylova N, Marshall DJ, Powers GD, Sato T, Campbell KS, Sykulev Y. Integrin receptors on tumor cells facilitate NK cell-mediated antibody-dependent cytotoxicity. Eur J Immunol. 2014;44:2331–9.
Crozat K, Eidenschenk C, Jaeger BN, Krebs P, Guia S, Beutler B, Vivier E, Ugolini S. Impact of β2 integrin deficiency on mouse natural killer cell development and function. Blood. 2011;117:2874–82.
Sciumè G, De Angelis G, Benigni G, Ponzetta A, Morrone S, Santoni A, Bernardini G. CX3CR1 expression defines 2 KLRG1+ mouse NK-cell subsets with distinct functional properties and positioning in the bone marrow. Blood. 2011;117:4467–75.
Amand M, Iserentant G, Poli A, Sleiman M, Fievez V, Sanchez IP, Sauvageot N, Michel T, Aouali N, Janji B, et al. Human CD56(dim)CD16(dim) Cells As an Individualized Natural Killer Cell Subset. Front Immunol. 2017;8:699.
Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, Pandey R, Campbell KS, Orange JS. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Investigation. 2012;122:3769–80..
Matsumoto M, Tatematsu M, Nishikawa F, Azuma M, Ishii N, Morii-Sakai A, Shime H, Seya T. Defined TLR3-specific adjuvant that induces NK and CTL activation without significant cytokine production in vivo. Nature Communications. 2015;6:6280..
Brennan TV, Lin L, Brandstadter JD, Rendell VR, Dredge K, Huang X, Yang Y. Heparan sulfate mimetic PG545-mediated antilymphoma effects require TLR9-dependent NK cell activation. J Clin Invest. 2016;126:207–19.
Jeanette E Boudreau, Fabio Giglio, Ted A Gooley, Philip A Stevenson, Jean-Benoît Le Luduec, Brian C Shaffer, Raja Rajalingam, Lihua Hou, Carolyn Katovich Hurley, Harriet Noreen, et al: KIR3DL1/HLA-B Subtypes Govern Acute Myelogenous Leukemia Relapse After Hematopoietic Cell Transplantation. Journal of clinical oncology : official journal of the American Soc Clin Oncol 2017, 35:2268–78.
Fauriat C, Ivarsson MA, Ljunggren H-G, Malmberg K-J, Michaëlsson J. Education of human natural killer cells by activating killer cell immunoglobulin-like receptors. Blood. 2010;115:1166–74.
Sun C, Xu J, Huang Q, Huang M, Wen H, Zhang C, Wang J, Song J, Zheng M, Sun H, et al. High NKG2A expression contributes to NK cell exhaustion and predicts a poor prognosis of patients with liver cancer. Oncoimmunol. 2017;6:e1264562.
Kamishikiryo J, Fukuhara H, Okabe Y, Kuroki K, Maenaka K. Molecular basis for LLT1 protein recognition by human CD161 protein (NKRP1A/KLRB1). J Biol Chem. 2011;286:23823–30.
Nowak I, Wilczyńska K, Wilczyński JR, Malinowski A, Radwan P, Radwan M, Kuśnierczyk P. KIR, LILRB and their Ligands' Genes as Potential Biomarkers in Recurrent Implantation Failure. Arch Immunol Ther Exp (Warsz). 2017;65:391–9.
Ito M, Maruyama T, Saito N, Koganei S, Yamamoto K, Matsumoto N. Killer cell lectin-like receptor G1 binds three members of the classical cadherin family to inhibit NK cell cytotoxicity. J Exp Med. 2006;203:289–95.
Yamaji T, Teranishi T, Alphey MS, Crocker PR, Hashimoto Y. A Small Region of the Natural Killer Cell Receptor, Siglec-7, Is Responsible for Its Preferred Binding to ?2,8-Disialyl and Branched ?2,6-Sialyl Residues: A COMPARISON WITH Siglec-9. J Biol Chem. 2002;277:6324–32.
Hernández-Caselles T, Miguel RC, Ruiz-Alcaraz AJ, García-Peñarrubia P. CD33 (Siglec-3) Inhibitory Function: Role in the NKG2D/DAP10 Activating Pathway. J Immunol Res. 2019;2019:6032141.
Markel G, Lieberman N, Katz G, Arnon TI, Lotem M, Drize O, Blumberg RS, Bar-Haim E, Mader R, Eisenbach L, Mandelboim O. CD66a interactions between human melanoma and NK cells: a novel class I MHC-independent inhibitory mechanism of cytotoxicity. J Immunol. 2002;168:2803–10.
McNerney ME, Lee KM, Kumar V. 2B4 (CD244) is a non-MHC binding receptor with multiple functions on natural killer cells and CD8+ T cells. Mol Immunol. 2005;42:489–94.
Cantoni C, Bottino C, Augugliaro R, Morelli L, Marcenaro E, Castriconi R, Vitale M, Pende D, Sivori S, Millo R, et al. Molecular and functional characterization of IRp60, a member of the immunoglobulin superfamily that functions as an inhibitory receptor in human NK cells. Eur J Immunol. 1999;29:3148–59.
Meyaard L, Vries A, Ruiter T, Lanier L, Phillips J, Clevers H. The Epithelial Cellular Adhesion Molecule (EP-Cam) Is a Ligand for the Leukocyte-Associated Immunoglobulin-like Receptor (Lair). J Experimental Med. 2001;194:107–12.
Sun H, Huang Q, Huang M, Wen H, Lin R, Zheng M, Qu K, Li K, Wei H, Xiao W, et al. Human CD96 Correlates to Natural Killer Cell Exhaustion and Predicts the Prognosis of Human Hepatocellular Carcinoma. Hepatology. 2019;70:168–83.
Neo SY, Yang Y, Record J, Ma R, Chen X, Chen Z, Tobin NP, Blake E, Seitz C, Thomas R, et al. CD73 immune checkpoint defines regulatory NK cells within the tumor microenvironment. J Clin Invest. 2020;130:1185–98.
Concha-Benavente F, Kansy B, Moskovitz J, Moy J, Chandran U, Ferris RL. PD-L1 Mediates Dysfunction in Activated PD-1(+) NK Cells in Head and Neck Cancer Patients. Cancer Immunol Res. 2018;6:1548–60.
Balaji GR, Aguilar OA, Tanaka M, Shingu-Vazquez MA, Fu Z, Gully BS, Lanier LL, Carlyle JR, Rossjohn J, Berry R. Recognition of host Clr-b by the inhibitory NKR-P1B receptor provides a basis for missing-self recognition. Nat Commun. 2018;9:4623.
Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin Immunol. 2019;42:101305.
Simoni Y, Fehlings M, Kloverpris HN, McGovern N, Koo SL, Loh CY, Lim S, Kurioka A, Fergusson JR, Tang CL, et al. Human Innate Lymphoid Cell Subsets Possess Tissue-Type Based Heterogeneity in Phenotype and Frequency. Immunity. 2017;46:148–61.
Dogra P, Rancan C, Ma W, Toth M, Senda T, Carpenter DJ, Kubota M, Matsumoto R, Thapa P, Szabo PA, et al. Tissue Determinants of Human NK Cell Development, Function, and Residence. Cell. 2020;180:749–63 e713.
Yang C, Siebert JR, Burns R, Gerbec ZJ, Bonacci B, Rymaszewski A, Rau M, Riese MJ, Rao S, Carlson K-S, et al. Heterogeneity of human bone marrow and blood natural killer cells defined by single-cell transcriptome. Nature Communications. 2019;10:3931.
Hervier B, Russick J, Cremer I, Vieillard V. NK Cells in the Human Lungs. Front Immunol. 2019;10:1263.
Marquardt N, Kekäläinen E, Chen P, Kvedaraite E, Wilson JN, Ivarsson MA, Mjösberg J, Berglin L, Säfholm J, Manson ML, et al. Human lung natural killer cells are predominantly comprised of highly differentiated hypofunctional CD69(−)CD56(dim) cells. J Allergy Clin Immunol. 2017;139:1321–30 e1324.
Vosshenrich CA, García-Ojeda ME, Samson-Villéger SI, Pasqualetto V, Enault L, Richard-Le Goff O, Corcuff E, Guy-Grand D, Rocha B, Cumano A, et al. A thymic pathway of mouse natural killer cell development characterized by expression of GATA-3 and CD127. Nat Immunol. 2006;7:1217–24.
Di Santo JP, Vosshenrich CA. Bone marrow versus thymic pathways of natural killer cell development. Immunol Rev. 2006;214:35–46.
Luci C, Reynders A. Ivanov, II, Cognet C, Chiche L, Chasson L, Hardwigsen J, Anguiano E, Banchereau J, Chaussabel D, et al: Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat Immunol. 2009;10:75–82.
Erick TK, Brossay L. Phenotype and functions of conventional and non-conventional NK cells. Curr Opin Immunol. 2016;38:67–74.
Marquardt N, Beziat V, Nystrom S, Hengst J, Ivarsson MA, Kekalainen E, Johansson H, Mjosberg J, Westgren M, Lankisch TO, et al. Cutting edge: identification and characterization of human intrahepatic CD49a+ NK cells. J Immunol. 2015;194:2467–71.
Paust S, Gill HS, Wang BZ, Flynn MP, Moseman EA, Senman B, Szczepanik M, Telenti A, Askenase PW, Compans RW, von Andrian UH. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat Immunol. 2010;11:1127–35.
Gaynor LM, Colucci F. Uterine Natural Killer Cells: Functional Distinctions and Influence on Pregnancy in Humans and Mice. Front Immunol. 2017;8:467.
Ebert LM, Meuter S, Moser B. Homing and function of human skin gammadelta T cells and NK cells: relevance for tumor surveillance. J Immunol. 2006;176:4331–6.
O'Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, Sun JC. Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity. 2016;45:428–41.
Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O'Connor D, Stutte S, Tavakkoli A, Winters D, et al. Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity. 2017;46:273–86.
Crinier A, Milpied P, Escalière B, Piperoglou C, Galluso J, Balsamo A, Spinelli L, Cervera-Marzal I, Ebbo M, Girard-Madoux M, et al. High-Dimensional Single-Cell Analysis Identifies Organ-Specific Signatures and Conserved NK Cell Subsets in Humans and Mice. Immunity. 2018;49:971–86 e975.
Fuchs A, Vermi W, Lee JS, Lonardi S, Gilfillan S, Newberry RD, Cella M, Colonna M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity. 2013;38:769–81.
Zhao ZB, Lu FT, Ma HD, Wang YH, Yang W, Long J, Miao Q, Zhang W, Tian Z, Ridgway WM, et al. Liver-resident NK cells suppress autoimmune cholangitis and limit the proliferation of CD4(+) T cells. Cell Mol Immunol. 2020;17:178–89.
Ferris RL, Jaffee EM, Ferrone S. Tumor antigen-targeted, monoclonal antibody-based immunotherapy: clinical response, cellular immunity, and immunoescape. J Clin Oncol. 2010;28:4390–9.
Bournazos S, Wang TT, Dahan R, Maamary J, Ravetch JV. Signaling by Antibodies: Recent Progress. Annu Rev Immunol. 2017;35:285–311.
Backes CS, Friedmann KS, Mang S, Knörck A, Hoth M, Kummerow C. Natural killer cells induce distinct modes of cancer cell death: Discrimination, quantification, and modulation of apoptosis, necrosis, and mixed forms. J Biol Chem. 2018;293:16348–63.
Deniz G, Akdis M, Aktas E, Blaser K, Akdis CA. Human NK1 and NK2 subsets determined by purification of IFN-gamma-secreting and IFN-gamma-nonsecreting NK cells. Eur J Immunol. 2002;32:879–84.
Franzoni G, Dei Giudici S, Oggiano A. Infection, modulation and responses of antigen-presenting cells to African swine fever viruses. Virus research. 2018;258:73–80.
Hanna J, Mandelboim O. When killers become helpers. Trends Immunol. 2007;28:201–6.
Wu J, Carlock C, Ross A, Shim J, Lou Y. CD8alphaalpha+MHC Class II+ Cell with the Capacity To Terminate Autoimmune Inflammation Is a Novel Antigen-Presenting NK-like Cell in Rats. J Immunol. 2016;197:4274–82.
Tel J, Smits EL, Anguille S, Joshi RN, Figdor CG, de Vries IJM. Human plasmacytoid dendritic cells are equipped with antigen-presenting and tumoricidal capacities. Blood. 2012;120:3936–44.
GeurtsvanKessel CH, Bergen IM, Muskens F, Boon L, Hoogsteden HC, Osterhaus ADME, Rimmelzwaan GF, Lambrecht BN. Both conventional and interferon killer dendritic cells have antigen-presenting capacity during influenza virus infection. PloS one. 2009;4:e7187.
Bonmort M, Dalod M, Mignot G, Ullrich E, Chaput N, Zitvogel L. Killer dendritic cells: IKDC and the others. Curr Opinion Immunol. 2008;20:558–65.
Almeida CF, Sundararaj S, Le Nours J, Praveena T, Cao B, Burugupalli S, Smith DGM, Patel O, Brigl M, Pellicci DG, et al. Distinct CD1d docking strategies exhibited by diverse Type II NKT cell receptors. Nature Communications. 2019;10:5242.
Fu S, Zhu S, Tian C, Bai S, Zhang J, Zhan C, Xie D, Wang L, Li Z, Li J, et al. Immunometabolism regulates TCR recycling and iNKT cell functions. Sci Signal. 2019;12(570):eaau1788. https://doi.org/10.1126/scisignal.aau1788.
Crosby CM, Kronenberg M. Tissue-specific functions of invariant natural killer T cells. Nat Rev Immunol. 2018;18:559–74.
Hoya M, Nagamatsu T, Fujii T, Schust DJ, Oda H, Akiba N, Iriyama T, Kawana K, Osuga Y, Fujii T. Impact of Th1/Th2 cytokine polarity induced by invariant NKT cells on the incidence of pregnancy loss in mice. Am J Reprod Immunol. 2018;79(3):e12813. https://doi.org/10.1111/aji.12813.
Brigl M, Tatituri RV, Watts GF, Bhowruth V, Leadbetter EA, Barton N, Cohen NR, Hsu FF, Besra GS, Brenner MB. Innate and cytokine-driven signals, rather than microbial antigens, dominate in natural killer T cell activation during microbial infection. J Exp Med. 2011;208:1163–77.
Huntington ND, Vosshenrich CA, Di Santo JP. Developmental pathways that generate natural-killer-cell diversity in mice and humans. Nat Rev Immunol. 2007;7:703–14.
Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–61.
Lucas M, Schachterle W, Oberle K, Aichele P, Diefenbach A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity. 2007;26:503–17.
Campbell AR, Duggan MC, Suarez-Kelly LP, Bhave N, Opheim KS, McMichael EL, Trikha P, Parihar R, Luedke E, Lewis A, et al. MICA-Expressing Monocytes Enhance Natural Killer Cell Fc Receptor-Mediated Antitumor Functions. Cancer Immunol Res. 2017;5:778–89..
Pal M, Schwab L, Yermakova A, Mace EM, Claus R, Krahl AC, Woiterski J, Hartwig UF, Orange JS, Handgretinger R, Andre MC. Tumor-priming converts NK cells to memory-like NK cells. Oncoimmunology. 2017;6:e1317411.
Romee R, Schneider SE, Leong JW, Chase JM, Keppel CR, Sullivan RP, Cooper MA, Fehniger TA. Cytokine activation induces human memory-like NK cells. Blood. 2012;120:4751–60.
Leong JW, Chase JM, Romee R, Schneider SE, Sullivan RP, Cooper MA, Fehniger TA. Preactivation with IL-12, IL-15, and IL-18 induces CD25 and a functional high-affinity IL-2 receptor on human cytokine-induced memory-like natural killer cells. Biol Blood Marrow Transplant. 2014;20:463–73.
Cerwenka A, Lanier LL. Natural killer cell memory in infection, inflammation and cancer. Nat Rev Immunol. 2016;16:112–23.
Hammer Q, Rückert T, Romagnani C. Natural killer cell specificity for viral infections. Nat Immunol. 2018;19:800–8.
Nikzad R, Angelo LS, Aviles-Padilla K, Le DT, Singh VK, Bimler L, Vukmanovic-Stejic M, Vendrame E, Ranganath T, Simpson L, et al. Human natural killer cells mediate adaptive immunity to viral antigens. Sci Immunol. 2019;4(35):eaat8116. https://doi.org/10.1126/sciimmunol.aat8116.
Ni J, Miller M, Stojanovic A, Garbi N, Cerwenka A. Sustained effector function of IL-12/15/18-preactivated NK cells against established tumors. J Exp Med. 2012;209:2351–65.
Coudert JD, Scarpellino L, Gros F, Vivier E, Held W. Sustained NKG2D engagement induces cross-tolerance of multiple distinct NK cell activation pathways. Blood. 2008;111:3571–8.
Deng W, Gowen BG, Zhang L, Wang L, Lau S, Iannello A, Xu J, Rovis TL, Xiong N, Raulet DH. Antitumor immunity. A shed NKG2D ligand that promotes natural killer cell activation and tumor rejection. Science. 2015;348:136–9.
Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, Leong JW, Abdel-Latif S, Schneider SE, Willey S, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8:357ra123.
Imai K, Matsuyama S, Miyake S, Suga K, Nakachi K. Natural cytotoxic activity of peripheral-blood lymphocytes and cancer incidence: an 11-year follow-up study of a general population. Lancet. 2000;356:1795–9.
Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79:4557–66.
Dorner BG, Smith HR, French AR, Kim S, Poursine-Laurent J, Beckman DL, Pingel JT, Kroczek RA, Yokoyama WM. Coordinate expression of cytokines and chemokines by NK cells during murine cytomegalovirus infection. J Immunol. 2004;172:3119–31.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Yang C, Cheng H, Zhang Y, Fan K, Luo G, Fan Z, Huang Q, Lu Y, Jin K, Wang Z, et al. Anergic natural killer cells educated by tumor cells are associated with a poor prognosis in patients with advanced pancreatic ductal adenocarcinoma. Cancer Immunol Immunother. 2018;67:1815–23.
Coca S, Perez-Piqueras J, Martinez D, Colmenarejo A, Saez MA, Vallejo C, Martos JA, Moreno M. The prognostic significance of intratumoral natural killer cells in patients with colorectal carcinoma. Cancer. 1997;79:2320–8.
Okada K, Sadahiro S, Chan LF, Ogimi T, Miyakita H, Saito G, Tanaka A, Suzuki T. The Number of Natural Killer Cells in the Largest Diameter Lymph Nodes Is Associated with the Number of Retrieved Lymph Nodes and Lymph Node Size, and Is an Independent Prognostic Factor in Patients with Stage II Colon Cancer. Oncology. 2018;95:288–96.
Tang YP, Xie MZ, Li KZ, Li JL, Cai ZM, Hu BL. Prognostic value of peripheral blood natural killer cells in colorectal cancer. BMC Gastroenterol. 2020;20:31.
Rea D, Henry G, Khaznadar Z, Etienne G, Guilhot F, Nicolini F, Guilhot J, Rousselot P, Huguet F, Legros L, et al. Natural killer-cell counts are associated with molecular relapse-free survival after imatinib discontinuation in chronic myeloid leukemia: the IMMUNOSTIM study. Haematologica. 2017;102:1368–77.
Wang WT, Zhu HY, Wu YJ, Xia Y, Wu JZ, Wu W, Liang JH, Wang L, Fan L, Li JY, Xu W. Elevated absolute NK cell counts in peripheral blood predict good prognosis in chronic lymphocytic leukemia. J Cancer Res Clin Oncol. 2018;144:449–57.
Shafer D, Smith MR, Borghaei H, Millenson MM, Li T, Litwin S, Anad R, Al-Saleem T. Low NK cell counts in peripheral blood are associated with inferior overall survival in patients with follicular lymphoma. Leuk Res. 2013;37:1213–5.
Zhou XH, Zhang XY, Liang JH, Zhu HY, Wang L, Xia Y, Cao L, Wu W, Fan L, Li JY, Xu W. Low absolute NK cell counts in peripheral blood are associated with inferior survival in patients with mantle cell lymphoma. Cancer Biomark. 2019;24:439–47.
Liu Y, Cheng Y, Xu Y, Wang Z, Du X, Li C, Peng J, Gao L, Liang X, Ma C. Increased expression of programmed cell death protein 1 on NK cells inhibits NK-cell-mediated anti-tumor function and indicates poor prognosis in digestive cancers. Oncogene. 2017;36:6143–53.
Sun H, Liu L, Huang Q, Liu H, Huang M, Wang J, Wen H, Lin R, Qu K, Li K, et al. Accumulation of Tumor-Infiltrating CD49a(+) NK Cells Correlates with Poor Prognosis for Human Hepatocellular Carcinoma. Cancer Immunol Res. 2019;7:1535–46.
Pasero C, Gravis G, Granjeaud S, Guerin M, Thomassin-Piana J, Rocchi P, Salem N, Walz J, Moretta A, Olive D. Highly effective NK cells are associated with good prognosis in patients with metastatic prostate cancer. Oncotarget. 2015;6:14360–73.
Picard E, Godet Y, Laheurte C, Dosset M, Galaine J, Beziaud L, Loyon R, Boullerot L. Lauret Marie Joseph E, Spehner L, et al: Circulating NKp46(+) Natural Killer cells have a potential regulatory property and predict distinct survival in Non-Small Cell Lung Cancer. Oncoimmunology. 2019;8:e1527498.
Fend L, Rusakiewicz S, Adam J, Bastien B, Caignard A, Messaoudene M, Iribarren C, Cremer I, Marabelle A, Borg C, et al. Prognostic impact of the expression of NCR1 and NCR3 NK cell receptors and PD-L1 on advanced non-small cell lung cancer. Oncoimmunology. 2017;6:e1163456.
Jin S, Deng Y, Hao J-W, Li Y, Liu B, Yu Y, Shi F-D, Zhou Q-H. NK cell phenotypic modulation in lung cancer environment. PloS one. 2014;9:e109976.
Muntasell A, Rojo F, Servitja S, Rubio-Perez C, Cabo M, Tamborero D, Costa-Garcia M, Martinez-Garcia M, Menendez S, Vazquez I, et al. NK Cell Infiltrates and HLA Class I Expression in Primary HER2(+) Breast Cancer Predict and Uncouple Pathological Response and Disease-free Survival. Clin Cancer Res. 2019;25:1535–45.
Rathore AS, Goel MM, Makker A, Kumar S, Srivastava AN. Is the tumor infiltrating natural killer cell (NK-TILs) count in infiltrating ductal carcinoma of breast prognostically significant? Asian Pac J Cancer Prev. 2014;15:3757–61.
Kamei R, Yoshimura K, Yoshino S, Inoue M, Asao T, Fuse M, Wada S, Kuramasu A, Furuya-Kondo T, Oga A, et al. Expression levels of UL16 binding protein 1 and natural killer group 2 member D affect overall survival in patients with gastric cancer following gastrectomy. Oncol Lett. 2018;15:747–54.
Mukherjee N, Ji N, Hurez V, Curiel TJ, Montgomery MO, Braun AJ, Nicolas M, Aguilera M, Kaushik D, Liu Q, et al. Intratumoral CD56(bright) natural killer cells are associated with improved survival in bladder cancer. Oncotarget. 2018;9:36492–502.
Verma C, Kaewkangsadan V, Eremin JM, Cowley GP, Ilyas M, El-Sheemy MA, Eremin O. Natural killer (NK) cell profiles in blood and tumour in women with large and locally advanced breast cancer (LLABC) and their contribution to a pathological complete response (PCR) in the tumour following neoadjuvant chemotherapy (NAC): differential restoration of blood profiles by NAC and surgery. J Transl Med. 2015;13:180.
Mamessier E, Sylvain A, Bertucci F, Castellano R, Finetti P, Houvenaeghel G, Charaffe-Jaufret E, Birnbaum D, Moretta A, Olive D. Human breast tumor cells induce self-tolerance mechanisms to avoid NKG2D-mediated and DNAM-mediated NK cell recognition. Cancer Res. 2011;71:6621–32.
Bachy E, Urb M, Chandra S, Robinot R, Bricard G, de Bernard S, Traverse-Glehen A, Gazzo S, Blond O, Khurana A, et al. CD1d-restricted peripheral T cell lymphoma in mice and humans. J Exp Med. 2016;213:841–57.
Haraguchi K, Takahashi T, Nakahara F, Matsumoto A, Kurokawa M, Ogawa S, Oda H, Hirai H, Chiba S. CD1d expression level in tumor cells is an important determinant for anti-tumor immunity by natural killer T cells. Leuk Lymphoma. 2006;47:2218–23.
Joyee AG, Uzonna J, Yang X. Invariant NKT cells preferentially modulate the function of CD8 alpha+ dendritic cell subset in inducing type 1 immunity against infection. J Immunol. 2010;184:2095–106.
Fujii S, Liu K, Smith C, Bonito AJ, Steinman RM. The linkage of innate to adaptive immunity via maturing dendritic cells in vivo requires CD40 ligation in addition to antigen presentation and CD80/86 costimulation. J Exp Med. 2004;199:1607–18.
Lambert AW, Pattabiraman DR, Weinberg RA. Emerging Biological Principles of Metastasis. Cell. 2017;168:670–91.
Lavin Y, Kobayashi S, Leader A, Amir E-AD, Elefant N, Bigenwald C, Remark R, Sweeney R, Becker CD, Levine JH, et al. Innate Immune Landscape in Early Lung Adenocarcinoma by Paired Single-Cell Analyses. Cell. 2017;169:750–65 e717..
Bommarito D, Martin A, Forcade E, Nastke MD, Ritz J, Bellucci R. Enhancement of tumor cell susceptibility to natural killer cell activity through inhibition of the PI3K signaling pathway. Cancer Immunol Immunother. 2016;65:355–66.
Park A, Lee Y, Kim MS, Kang YJ, Park YJ, Jung H, Kim TD, Lee HG, Choi I, Yoon SR. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front Immunol. 2018;9:1859.
Young A, Ngiow SF, Gao Y, Patch A-M, Barkauskas DS, Messaoudene M, Lin G, Coudert JD, Stannard KA, Zitvogel L, et al. A2AR Adenosine Signaling Suppresses Natural Killer Cell Maturation in the Tumor Microenvironment. Cancer Research. 2018;78:1003–16.
Zheng X, Qian Y, Fu B, Jiao D, Jiang Y, Chen P, Shen Y, Zhang H, Sun R, Tian Z, Wei H. Mitochondrial fragmentation limits NK cell-based tumor immunosurveillance. Nat Immunol. 2019;20:1656–67.
Al Absi A, Wurzer H, Guerin C, Hoffmann C, Moreau F, Mao X, Brown-Clay J, Petrolli R, Casellas CP, Dieterle M, et al. Actin Cytoskeleton Remodeling Drives Breast Cancer Cell Escape from Natural Killer-Mediated Cytotoxicity. Cancer Res. 2018;78:5631–43.
Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T, Falcone M, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572:254–9.
Duan S, Guo W, Xu Z, He Y, Liang C, Mo Y, Wang Y, Xiong F, Guo C, Li Y, et al. Natural killer group 2D receptor and its ligands in cancer immune escape. Molecular Cancer. 2019;18:29.
Inoue T, Adachi K, Kawana K, Taguchi A, Nagamatsu T, Fujimoto A, Tomio K, Yamashita A, Eguchi S, Nishida H, et al. Cancer-associated fibroblast suppresses killing activity of natural killer cells through downregulation of poliovirus receptor (PVR/CD155), a ligand of activating NK receptor. Int J Oncol. 2016;49:1297–304.
Zhang R, Qi F, Zhao F, Li G, Shao S, Zhang X, Yuan L, Feng Y. Cancer-associated fibroblasts enhance tumor-associated macrophages enrichment and suppress NK cells function in colorectal cancer. Cell Death Dis. 2019;10:273.
Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, Tai Y, Zhang Q, Chen G. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318:154–61.
Wu Y, Kuang DM, Pan WD, Wan YL, Lao XM, Wang D, Li XF, Zheng L. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57:1107–16.
Vari F, Arpon D, Keane C, Hertzberg MS, Talaulikar D, Jain S, Cui Q, Han E, Tobin J, Bird R, et al. Immune evasion via PD-1/PD-L1 on NK cells and monocyte/macrophages is more prominent in Hodgkin lymphoma than DLBCL. Blood. 2018;131:1809–19.
Peng LS, Zhang JY, Teng YS, Zhao YL, Wang TT, Mao FY, Lv YP, Cheng P, Li WH, Chen N, et al. Tumor-Associated Monocytes/Macrophages Impair NK-Cell Function via TGFbeta1 in Human Gastric Cancer. Cancer Immunol Res. 2017;5:248–56.
Mattiola I, Tomay F, De Pizzol M, Silva-Gomes R, Savino B, Gulic T, Doni A, Lonardi S, Astrid Boutet M, Nerviani A, et al. The macrophage tetraspan MS4A4A enhances dectin-1-dependent NK cell-mediated resistance to metastasis. Nat Immunol. 2019;20:1012–22.
Niavarani SR, Lawson C, Bakos O, Boudaud M, Batenchuk C, Rouleau S, Tai LH. Lipid accumulation impairs natural killer cell cytotoxicity and tumor control in the postoperative period. BMC Cancer. 2019;19:823.
Chauvin JM, Ka M, Pagliano O, Menna C, Ding Q, DeBlasio R, Sander C, Hou J, Li XY, Ferrone S, et al. IL-15 stimulation with TIGIT blockade reverses CD155-mediated NK cell dysfunction in melanoma. Clin Cancer Res. 2020. https://doi.org/10.1158/1078-0432.CCR-20-0575.
Best SA, Hess J, Souza-Fonseca-Guimaraes F, Cursons J, Kersbergen A, Dong X, Rautela J, Hyslop SR, Ritchie ME, Davis MJ, et al. Harnessing natural killer immunity in metastatic small cell lung cancer. J Thorac Oncol. 2020;S1556-0864(20)30394-4). https://doi.org/10.1016/j.jtho.2020.05.008.
Zhuang X, Long EO. CD28 Homolog Is a Strong Activator of Natural Killer Cells for Lysis of B7H7(+) Tumor Cells. Cancer Immunol Res. 2019;7:939–51..
Brady J, Hayakawa Y, Smyth MJ, Nutt SL. IL-21 induces the functional maturation of murine NK cells. J Immunol. 2004;172:2048–58.
Granzin M, Stojanovic A, Miller M, Childs R, Huppert V, Cerwenka A. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. Oncoimmunology. 2016;5:e1219007.
Seo H, Jeon I, Kim BS, Park M, Bae EA, Song B, Koh CH, Shin KS, Kim IK, Choi K, et al. IL-21-mediated reversal of NK cell exhaustion facilitates anti-tumour immunity in MHC class I-deficient tumours. Nat Commun. 2017;8:15776.
Kasaian MT, Whitters MJ, Carter LL, Lowe LD, Jussif JM, Deng B, Johnson KA, Witek JS, Senices M, Konz RF, et al. IL-21 limits NK cell responses and promotes antigen-specific T cell activation: a mediator of the transition from innate to adaptive immunity. Immunity. 2002;16:559–69.
Barjon C, Michaud HA, Fages A, Dejou C, Zampieri A, They L, Gennetier A, Sanchez F, Gros L, Eliaou JF, et al. IL-21 promotes the development of a CD73-positive Vgamma9Vdelta2 T cell regulatory population. Oncoimmunology. 2017;7:e1379642.
Moretto MM, Khan IA. IL-21 Is Important for Induction of KLRG1+ Effector CD8 T Cells during Acute Intracellular Infection. J Immunol. 2016;196:375–84.
Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–87.
Cui G, Staron MM, Gray SM, Ho PC, Amezquita RA, Wu J, Kaech SM. IL-7-Induced Glycerol Transport and TAG Synthesis Promotes Memory CD8+ T Cell Longevity. Cell. 2015;161:750–61.
Clenet ML, Gagnon F, Moratalla AC, Viel EC, Arbour N. Peripheral human CD4(+)CD8(+) T lymphocytes exhibit a memory phenotype and enhanced responses to IL-2, IL-7 and IL-15. Sci Rep. 2017;7:11612.
Chen T, Zhang T, Liu C, Wang C, Ding S, Shao Z, Fu R. NK cells suppress CD8(+) T cell immunity via NKG2D in severe aplastic anemia. Cell Immunol. 2019;335:6–14.
Duhan V, Hamdan TA, Xu HC, Shinde P, Bhat H, Li F, Al-Matary Y, Haussinger D, Bezgovsek J, Friedrich SK, et al. NK cell-intrinsic FcepsilonRIgamma limits CD8+ T-cell expansion and thereby turns an acute into a chronic viral infection. PLoS Pathog. 2019;15:e1007797.
Freeman AJ, Vervoort SJ, Ramsbottom KM, Kelly MJ, Michie J, Pijpers L, Johnstone RW, Kearney CJ, Oliaro J. Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Rep. 2019;28:2784–94 e2785.
de Kruijf EM, Sajet A, van Nes JG, Natanov R, Putter H, Smit VT, Liefers GJ, van den Elsen PJ, van de Velde CJ, Kuppen PJ. HLA-E and HLA-G expression in classical HLA class I-negative tumors is of prognostic value for clinical outcome of early breast cancer patients. J Immunol. 2010;185:7452–9.
Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, Gottfried E, Schwarz S, Rothe G, Hoves S, et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood. 2007;109:3812–9.
Lee YS, Wollam J, Olefsky JM. An Integrated View of Immunometabolism. Cell. 2018;172:22–40.
Piątkiewicz P, Bernat-Karpińska M, Miłek T, Rabijewski M, Rosiak E. NK cell count and glucotransporter 4 (GLUT4) expression in subjects with type 2 diabetes and colon cancer. Diabetol Metab Syndr. 2016;8:38.
Salzberger W, Martrus G, Bachmann K, Goebels H, Heß L, Koch M, Langeneckert A, Lunemann S, Oldhafer KJ, Pfeifer C, et al. Tissue-resident NK cells differ in their expression profile of the nutrient transporters Glut1, CD98 and CD71. PLoS One. 2018;13:e0201170.
Keppel MP, Saucier N, Mah AY, Vogel TP, Cooper MA. Activation-specific metabolic requirements for NK Cell IFN-γ production. J Immunol. 2015;194:1954–62.
Mah AY, Rashidi A, Keppel MP, Saucier N, Moore EK, Alinger JB, Tripathy SK, Agarwal SK, Jeng EK, Wong HC, et al. Glycolytic requirement for NK cell cytotoxicity and cytomegalovirus control. JCI Insight. 2017;2(23):e95128. https://doi.org/10.1172/jci.insight.95128.
Wang Z, Guan D, Wang S, Chai LYA, Xu S, Lam KP. Glycolysis and Oxidative Phosphorylation Play Critical Roles in Natural Killer Cell Receptor-Mediated Natural Killer Cell Functions. Front Immunol. 2020;11:202.
Theurich S, Tsaousidou E, Hanssen R, Lempradl AM, Mauer J, Timper K, Schilbach K, Folz-Donahue K, Heilinger C, Sexl V, et al. IL-6/Stat3-Dependent Induction of a Distinct, Obesity-Associated NK Cell Subpopulation Deteriorates Energy and Glucose Homeostasis. Cell Metab. 2017;26:171–84 e176.
Assmann N, O'Brien KL, Donnelly RP, Dyck L, Zaiatz-Bittencourt V, Loftus RM, Heinrich P, Oefner PJ, Lynch L, Gardiner CM, et al. Srebp-controlled glucose metabolism is essential for NK cell functional responses. Nat Immunol. 2017;18:1197–206.
Wu Q, Ishikawa T, Sirianni R, Tang H, McDonald JG, Yuhanna IS, Thompson B, Girard L, Mineo C, Brekken RA, et al. 27-Hydroxycholesterol promotes cell-autonomous, ER-positive breast cancer growth. Cell Rep. 2013;5:637–45.
Nelson ER, Wardell SE, Jasper JS, Park S, Suchindran S, Howe MK, Carver NJ, Pillai RV, Sullivan PM, Sondhi V, et al. 27-Hydroxycholesterol links hypercholesterolemia and breast cancer pathophysiology. Science. 2013;342:1094–8.
Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, Pillai RV, McDowell HB, Thompson JW, Dubois LG, et al. The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun. 2017;8:864.
Qin WH, Yang ZS, Li M, Chen Y, Zhao XF, Qin YY, Song JQ, Wang BB, Yuan B, Cui XL, et al: High Serum Levels of Cholesterol Increase Antitumor Functions of Nature Killer Cells and Reduce Growth of Liver Tumors in Mice. Gastroenterology. 2020;158:1713–27.
Chabi S, Uzan B, Naguibneva I, Rucci J, Fahy L, Calvo J, Arcangeli ML, Mazurier F, Pflumio F, Haddad R. Hypoxia Regulates Lymphoid Development of Human Hematopoietic Progenitors. Cell Rep. 2019;29:2307–20 e2306.
Balsamo M, Manzini C, Pietra G, Raggi F, Blengio F, Mingari MC, Varesio L, Moretta L, Bosco MC, Vitale M. Hypoxia downregulates the expression of activating receptors involved in NK-cell-mediated target cell killing without affecting ADCC. Eur J Immunol. 2013;43:2756–64.
Engel C, Brügmann G, Lambing S, Mühlenbeck LH, Marx S, Hagen C, Horváth D, Goldeck M, Ludwig J, Herzner AM, et al. RIG-I Resists Hypoxia-Induced Immunosuppression and Dedifferentiation. Cancer Immunol Res. 2017;5:455–67.
Baginska J, Viry E, Berchem G, Poli A, Noman MZ, van Moer K, Medves S, Zimmer J, Oudin A, Niclou SP, et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc Natl Acad Sci U S A. 2013;110:17450–5.
Sarkar S, Germeraad WT, Rouschop KM, Steeghs EM, van Gelder M, Bos GM, Wieten L. Hypoxia induced impairment of NK cell cytotoxicity against multiple myeloma can be overcome by IL-2 activation of the NK cells. PLoS One. 2013;8:e64835.
Huenecke S, Zimmermann SY, Kloess S, Esser R, Brinkmann A, Tramsen L, Koenig M, Erben S, Seidl C, Tonn T, et al. IL-2-driven regulation of NK cell receptors with regard to the distribution of CD16+ and CD16- subpopulations and in vivo influence after haploidentical NK cell infusion. J Immunother. 2010;33:200–10.
Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, Li W, MacKinnon AL, Makkouk A, Marguier G, et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5:101.
Loftus RM, Assmann N, Kedia-Mehta N, O'Brien KL, Garcia A, Gillespie C, Hukelmann JL, Oefner PJ, Lamond AI, Gardiner CM, et al. Amino acid-dependent cMyc expression is essential for NK cell metabolic and functional responses in mice. Nat Commun. 2018;9:2341.
Abarca-Rojano E, Muñiz-Hernández S, Moreno-Altamirano MM, Mondragón-Flores R, Enriquez-Rincón F, Sánchez-García FJ. Re-organization of mitochondria at the NK cell immune synapse. Immunol Lett. 2009;122:18–25.
Michelet X, Dyck L, Hogan A, Loftus RM, Duquette D, Wei K, Beyaz S, Tavakkoli A, Foley C, Donnelly R, et al. Metabolic reprogramming of natural killer cells in obesity limits antitumor responses. Nat Immunol. 2018;19:1330–40.
Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, Stelekati E, McLane LM, Paley MA, Delgoffe GM, Wherry EJ. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity. 2016;45:358–73.
Wang X, Lang S, Tian Y, Zhang J, Yan X, Fang Z, Weng J, Lu N, Wu X, Li T, et al. Glycoengineering of Natural Killer Cells with CD22 Ligands for Enhanced Anticancer Immunotherapy. ACS Cent Sci. 2020;6:382–9.
Beloueche-Babari M, Casals Galobart T, Delgado-Goni T, Wantuch S, Parkes HG, Tandy D, Harker JA, Leach MO. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br J Cancer. 2020;122:895–903.
Exley MA, Hand L, O'Shea D, Lynch L. Interplay between the immune system and adipose tissue in obesity. J Endocrinol. 2014;223:R41–8.
Lynch L. Adipose invariant natural killer T cells. Immunology. 2014;142:337–46.
Bandyopadhyay K, Marrero I, Kumar V. NKT cell subsets as key participants in liver physiology and pathology. Cell Mol Immunol. 2016;13:337–46.
Berzins SP, Smyth MJ, Baxter AG. Presumed guilty: natural killer T cell defects and human disease. Nat Rev Immunol. 2011;11:131–42.
Pereira CS, Ribeiro H, Pérez-Cabezas B, Cardoso MT, Alegrete N, Gaspar A, Leão-Teles E, Macedo MF. The GM2 ganglioside inhibits iNKT cell responses in a CD1d-dependent manner. Mol Genet Metab. 2018;125:161–7.
Sharma S, Wang J, Alqassim E, Portwood S, Cortes Gomez E, Maguire O, Basse PH, Wang ES, Segal BH, Baysal BE. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 2019;20:37.
Tahir SM, Cheng O, Shaulov A, Koezuka Y, Bubley GJ, Wilson SB, Balk SP, Exley MA. Loss of IFN-gamma production by invariant NK T cells in advanced cancer. J Immunol. 2001;167:4046–50.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40.
Williams JB, Li S, Higgs EF, Cabanov A, Wang X, Huang H, Gajewski TF. Tumor heterogeneity and clonal cooperation influence the immune selection of IFN-γ-signaling mutant cancer cells. Nat Commun. 2020;11:602.
Gulati P, Rühl J, Kannan A, Pircher M, Schuberth P, Nytko KJ, Pruschy M, Sulser S, Haefner M, Jensen S, et al. Aberrant Lck Signal via CD28 Costimulation Augments Antigen-Specific Functionality and Tumor Control by Redirected T Cells with PD-1 Blockade in Humanized Mice. Clin Cancer Res. 2018;24:3981–93.
Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nature Reviews Immunol. 2018;18:671–88.
Daher M, Rezvani K. Next generation natural killer cells for cancer immunotherapy: the promise of genetic engineering. Current opinion in immunology. 2018;51:146–53.
Hodgins JJ, Khan ST, Park MM, Auer RC, Ardolino M. Killers 2.0: NK cell therapies at the forefront of cancer control. J Clin Investigation. 2019;129:3499–510.
Conlon KC, Lugli E, Welles HC, Rosenberg SA, Fojo AT, Morris JC, Fleisher TA, Dubois SP, Perera LP, Stewart DM, et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J Clin Oncol. 2015;33:74–82.
Conlon KC, Potter EL, Pittaluga S, Lee CR, Miljkovic MD, Fleisher TA, Dubois S, Bryant BR, Petrus M, Perera LP, et al. IL15 by Continuous Intravenous Infusion to Adult Patients with Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin Cancer Res. 2019;25:4945–54.
Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, DeFor TE, et al. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131:2515–27.
Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, Miller JS, Farhad M, Anderton K, Lindsey K, et al. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19:694–704.
Kobayashi H, Dubois S, Sato N, Sabzevari H, Sakai Y, Waldmann TA, Tagaya Y. Role of trans-cellular IL-15 presentation in the activation of NK cell-mediated killing, which leads to enhanced tumor immunosurveillance. Blood. 2005;105:721–7.
Bergamaschi C, Pandit H, Nagy BA, Stellas D, Jensen SM, Bear J, Cam M, Valentin A, Fox BA, Felber BK, Pavlakis GN. Heterodimeric IL-15 delays tumor growth and promotes intratumoral CTL and dendritic cell accumulation by a cytokine network involving XCL1, IFN-γ, CXCL9 and CXCL10. J Immunother Cancer. 2020;8(1):e000599. https://doi.org/10.1136/jitc-2020-000599.
Takaki R, Hayakawa Y, Nelson A, Sivakumar PV, Hughes S, Smyth MJ, Lanier LL. IL-21 enhances tumor rejection through a NKG2D-dependent mechanism. J Immunol. 2005;175:2167–73.
Bentebibel S-E, Hurwitz ME, Bernatchez C, Haymaker C, Hudgens CW, Kluger HM, Tetzlaff MT, Tagliaferri MA, Zalevsky J, Hoch U, et al. A First-in-Human Study and Biomarker Analysis of NKTR-214, a Novel IL2Rβγ-Biased Cytokine, in Patients with Advanced or Metastatic Solid Tumors. Cancer Discovery. 2019;9:711–21.
Sáez-Borderías A, Romo N, Magri G, Gumá M, Angulo A, López-Botet M. IL-12-dependent inducible expression of the CD94/NKG2A inhibitory receptor regulates CD94/NKG2C+ NK cell function. J Immunol. 2009;182:829–36.
Wang F, Hou H, Wu S, Tang Q, Liu W, Huang M, Yin B, Huang J, Mao L, Lu Y, Sun Z. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur J Immunol. 2015;45:2886–97.
Xu F, Sunderland A, Zhou Y, Schulick RD, Edil BH, Zhu Y. Blockade of CD112R and TIGIT signaling sensitizes human natural killer cell functions. Cancer Immunol Immunother. 2017;66:1367–75.
Sarhan D, Cichocki F, Zhang B, Yingst A, Spellman SR, Cooley S, Verneris MR, Blazar BR, Miller JS. Adaptive NK Cells with Low TIGIT Expression Are Inherently Resistant to Myeloid-Derived Suppressor Cells. Cancer Res. 2016;76:5696–706.
Chan CJ, Martinet L, Gilfillan S, Souza-Fonseca-Guimaraes F, Chow MT, Town L, Ritchie DS, Colonna M, Andrews DM, Smyth MJ. The receptors CD96 and CD226 oppose each other in the regulation of natural killer cell functions. Nat Immunol. 2014;15:431–8.
Roman Aguilera A, Lutzky VP, Mittal D, Li XY, Stannard K, Takeda K, Bernhardt G, Teng MWL, Dougall WC, Smyth MJ. CD96 targeted antibodies need not block CD96-CD155 interactions to promote NK cell anti-metastatic activity. Oncoimmunology. 2018;7:e1424677.
Blake SJ, Stannard K, Liu J, Allen S, Yong MC, Mittal D, Aguilera AR, Miles JJ, Lutzky VP, de Andrade LF, et al. Suppression of Metastases Using a New Lymphocyte Checkpoint Target for Cancer Immunotherapy. Cancer Discov. 2016;6:446–59.
Carlsten M, Korde N, Kotecha R, Reger R, Bor S, Kazandjian D, Landgren O, Childs RW. Checkpoint Inhibition of KIR2D with the Monoclonal Antibody IPH2101 Induces Contraction and Hyporesponsiveness of NK Cells in Patients with Myeloma. Clin Cancer Res. 2016;22:5211–22.
Benson DM, Cohen AD, Jagannath S, Munshi NC, Spitzer G, Hofmeister CC, Efebera YA, Andre P, Zerbib R, Caligiuri MA. A Phase I Trial of the Anti-KIR Antibody IPH2101 and Lenalidomide in Patients with Relapsed/Refractory Multiple Myeloma. Clin Cancer Res. 2015;21:4055–61.
Basher F, Dhar P, Wang X, Wainwright DA, Zhang B, Sosman J, Ji Z, Wu JD. Antibody targeting tumor-derived soluble NKG2D ligand sMIC reprograms NK cell homeostatic survival and function and enhances melanoma response to PDL1 blockade therapy. J Hematol Oncology. 2020;13:74.
Segal N, Naidoo J, Curigliano G, Patel S, Sahebjam S, Papadopoulos K, Gordon M, Wang D, Rueda A, Song X, et al. First-in-human dose escalation of monalizumab plus durvalumab, with expansion in patients with metastatic microsatellite-stable colorectal cancer. Journal of Clinical Oncology. 2018;36:3540.
Cohen R, Bauman J, Salas S, Colevas A, Even C, Cupissol D, Posner M, Lefebvre G, Saada-Bouzid E, Bernadach M, et al. Combination of monalizumab and cetuximab in recurrent or metastatic head and neck cancer patients previously treated with platinum-based chemotherapy and PD-(L)1 inhibitors. Journal of Clinical Oncology. 2020;38:6516.
Kamiya T, Seow SV, Wong D, Robinson M, Campana D. Blocking expression of inhibitory receptor NKG2A overcomes tumor resistance to NK cells. J Clin Invest. 2019;129:2094–106.
Gill S, Olson JA, Negrin RS. Natural killer cells in allogeneic transplantation: effect on engraftment, graft- versus-tumor, and graft-versus-host responses. Biol Blood Marrow Transplant. 2009;15:765–76.
Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–7.
Curti A, Ruggeri L, D'Addio A, Bontadini A, Dan E, Motta MR, Trabanelli S, Giudice V, Urbani E, Martinelli G, et al. Successful transfer of alloreactive haploidentical KIR ligand-mismatched natural killer cells after infusion in elderly high risk acute myeloid leukemia patients. Blood. 2011;118:3273–9.
Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, Jonson AL, Panoskaltsis-Mortari A, Curtsinger J, McKenna D, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy. 2011;13:98–107.
Khatua S, Cooper LJN, Sandberg DI, Ketonen L, Johnson JM, Rytting ME, Liu DD, Meador H, Trikha P, Nakkula RJ, et al. Phase I study of intraventricular infusions of autologous ex-vivo-expanded NK cells in children with recurrent medulloblastoma and ependymoma. Neuro Oncol. 2020. https://doi.org/10.1093/neuonc/noaa047.
Dolstra H, Roeven MWH, Spanholtz J, Hangalapura BN, Tordoir M, Maas F, Leenders M, Bohme F, Kok N, Trilsbeek C, et al. Successful Transfer of Umbilical Cord Blood CD34(+) Hematopoietic Stem and Progenitor-derived NK Cells in Older Acute Myeloid Leukemia Patients. Clin Cancer Res. 2017;23:4107–18.
Lee SC, Shimasaki N, Lim JSJ, Wong AL, Yadav K, Yong WP, Tan LK, Koh LP, Poon MLM, Tan SH, et al. 1Phase I trial of expanded, activated autologous NK cell infusions with trastuzumab in patients with HER2-positive cancers. Clin Cancer Res. 2020. https://doi.org/10.1158/1078-0432.CCR-20-0768.
Fujisaki H, Kakuda H, Imai C, Mullighan CG, Campana D. Replicative potential of human natural killer cells. Br J Haematol. 2009;145:606–13.
Sun JC, Beilke JN, Bezman NA, Lanier LL. Homeostatic proliferation generates long-lived natural killer cells that respond against viral infection. J Exp Med. 2011;208:357–68.
Min-Oo G, Lanier LL. Cytomegalovirus generates long-lived antigen-specific NK cells with diminished bystander activation to heterologous infection. J Exp Med. 2014;211:2669–80.
Huber CM, Doisne JM, Colucci F. IL-12/15/18-preactivated NK cells suppress GvHD in a mouse model of mismatched hematopoietic cell transplantation. Eur J Immunol. 2015;45:1727–35.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–17.
Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, Waldron E, Chakraborty A, Awasthi R, Levine BL, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130:2317–25.
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, Bader P, Verneris MR, Stefanski HE, Myers GD, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med. 2018;378:439–48.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, Braunschweig I, Oluwole OO, Siddiqi T, Lin Y, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med. 2017;377:2531–44.
Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016;127:3321–30.
Norelli M, Camisa B, Barbiera G, Falcone L, Purevdorj A, Genua M, Sanvito F, Ponzoni M, Doglioni C, Cristofori P, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med. 2018;24:739–48.
Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. 2017;31:64–75.
Romanski A, Uherek C, Bug G, Seifried E, Klingemann H, Wels WS, Ottmann OG, Tonn T. CD19-CAR engineered NK-92 cells are sufficient to overcome NK cell resistance in B-cell malignancies. J Cell Mol Med. 2016;20:1287–94.
Felices M, Sarhan D, Brandt L, Guldevall K, McElmurry R, Lenvik A, Chu S, Tolar J, Taras E, Spellman SR, et al. CD16-IL15-CD33 Trispecific Killer Engager (TriKE) Overcomes Cancer-Induced Immune Suppression and Induces Natural Killer Cell-Mediated Control of MDS and AML Via Enhanced Killing Kinetics. Blood. 2016;128:4291.
Herrera L, Santos S, Vesga MA, Anguita J, Martin-Ruiz I, Carrascosa T, Juan M, Eguizabal C. Adult peripheral blood and umbilical cord blood NK cells are good sources for effective CAR therapy against CD19 positive leukemic cells. Sci Rep. 2019;9:18729.
Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, Nassif Kerbauy L, Overman B, Thall P, Kaplan M, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020;382:545–53.
Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, Orange J, Wan X, Lu X, Reynolds A, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–31.
Naeimi Kararoudi M, Nagai Y, Elmas E, Pereira MSF, Ali SA, Imus PH, Wethington D, Borrello I, Lee DA, Ghiaur G. CD38 deletion of human primary NK cells eliminates daratumumab-induced fratricide and boosts their effector activity. Blood. 2020. https://doi.org/10.1182/blood.2020006200.
Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell. 2018;23:181–92 e185.
Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, Meng Z, Wu C, Guan KL, Malmberg KJ, Kaufman DS. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell. 2020;S1934-5909(20)30206-X. https://doi.org/10.1016/j.stem.2020.05.008.
Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, Sevillano M, Ibiza S, Cañellas A, Hernando-Momblona X, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. 2018;554:538–43.
Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel Iii EE, Koeppen H, Astarita JL, Cubas R, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544–8.
Viel S, Marçais A, Guimaraes FS, Loftus R, Rabilloud J, Grau M, Degouve S, Djebali S, Sanlaville A, Charrier E, et al. TGF-β inhibits the activation and functions of NK cells by repressing the mTOR pathway. Sci Signal. 2016;9:ra19.
Robson NC, Wei H, McAlpine T, Kirkpatrick N, Cebon J, Maraskovsky E. Activin-A attenuates several human natural killer cell functions. Blood. 2009;113:3218–25.
Heinhuis KM, Carlino M, Joerger M, Di Nicola M, Meniawy T, Rottey S, Moreno V, Gazzah A, Delord JP, Paz-Ares L, et al. Safety, Tolerability, and Potential Clinical Activity of a Glucocorticoid-Induced TNF Receptor-Related Protein Agonist Alone or in Combination With Nivolumab for Patients With Advanced Solid Tumors: A Phase 1/2a Dose-Escalation and Cohort-Expansion Clinical Trial. JAMA Oncol. 2019;6:1–8.
Kornblum N, Zhao F, Manola J, Klein P, Ramaswamy B, Brufsky A, Stella PJ, Burnette B, Telli M, Makower DF, et al. Randomized Phase II Trial of Fulvestrant Plus Everolimus or Placebo in Postmenopausal Women With Hormone Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Metastatic Breast Cancer Resistant to Aromatase Inhibitor Therapy: Results of PrE0102. J Clin Oncol. 2018;36:1556–63.
Jerusalem G, de Boer RH, Hurvitz S, Yardley DA, Kovalenko E, Ejlertsen B, Blau S, Özgüroglu M, Landherr L, Ewertz M, et al. Everolimus Plus Exemestane vs Everolimus or Capecitabine Monotherapy for Estrogen Receptor-Positive, HER2-Negative Advanced Breast Cancer: The BOLERO-6 Randomized Clinical Trial. JAMA Oncol. 2018;4:1367–74.
Keating SE, Zaiatz-Bittencourt V, Loftus RM, Keane C, Brennan K, Finlay DK, Gardiner CM. Metabolic Reprogramming Supports IFN-gamma Production by CD56bright NK Cells. J Immunol. 2016;196:2552–60.
Golden EB, Apetoh L. Radiotherapy and immunogenic cell death. Semin Radiat Oncol. 2015;25:11–7.
Caetano MS, Younes AI, Barsoumian HB, Quigley M, Menon H, Gao C, Spires T, Reilly TP, Cadena AP, Cushman TR, et al. Triple Therapy with MerTK and PD1 Inhibition Plus Radiotherapy Promotes Abscopal Antitumor Immune Responses. Clin Cancer Res. 2019;25:7576–84.
Watanabe T, Gaedicke S, Guffart E, Firat E, Niedermann G. Adding Indoximod to Hypofractionated Radiotherapy with Anti-PD-1 Checkpoint Blockade Enhances Early NK and CD8(+) T-Cell-Dependent Tumor Activity. Clin Cancer Res. 2020;26:945–56.
Gao S, Li T, Guo Y, Sun C, Xianyu B, Xu H. Selenium-Containing Nanoparticles Combine the NK Cells Mediated Immunotherapy with Radiotherapy and Chemotherapy. Adv Mater. 2020;32:e1907568.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (81922048, 81874112, 81874113, 81572583 and 81502278), the Fok Ying-Tong Education Foundation for College Young Teachers (171034), the Training Plan of Excellent Talents in Shanghai Municipality Health System (2017YQ038), the “Chen Guang” project supported by Shanghai Municipal Education Commission and Shanghai Education Development Foundation (17CG01), Shanghai Pujiang Program (18PJD007), and the Training Plan of Excellent Talents of Fudan University Shanghai Cancer Center (YJYQ201602). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Z.-M.S., Y.-Z.J. and S.W. designed and finalized the study; S.W. wrote the paper; T.F. participated in revising the review. All the authors approved the final version submitted.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
The content of this manuscript has not been previously published and is not under consideration for publication elsewhere. All of the authors are aware of and agree to the content of the paper and their being listed as a co-author of the paper.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, SY., Fu, T., Jiang, YZ. et al. Natural killer cells in cancer biology and therapy. Mol Cancer 19, 120 (2020). https://doi.org/10.1186/s12943-020-01238-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-020-01238-x