
Abstract
Background and aims
Although studies on the relation between arrhythmias and the action potential duration (APD) have been carried out, most of them are based only on electrophysiological factors of the heart and lack experiments that consider cardiac mechanical and electromechanical characteristics. Therefore, we conducted this study to clarify the relevance of the shortening of APD of a cell in relation to the mechanical contraction activity of the heart and the associated risk of arrhythmia.
Methods
The human ventricular model used in this study has two dynamic characteristics: electrophysiological conduction and mechanical contraction. The model simulating electrophysiological characteristics was consisted of lumped parameter circuit that can mimic the phenomenon of ion exchange through the cell membrane of myocyte and consisted of 214,319 tetrahedral finite elements. In contrast, the model simulating mechanical contraction characteristics was constructed to mimic cardiac contraction by means of the crossbridge of a myofilament and consisted of 14,720 hermite-based finite elements to represent a natural 3D curve of the cardiac surface. First, we performed a single cell simulation and the electrophysiological simulation according to the change of the APD by changing the electrical conductivity of the I Ks channel. Thus, we confirmed the correlation between APD and intracellular Ca2+ concentration. Then, we compared mechanical response through mechanical simulation using Ca2+ data from electrical simulation.
Results
The APD and the sum of the intracellular Ca2+ concentrations showed a positive correlation. The shortened APD reduced the conduction wavelength of ventricular cells by shortening the plateau and early repolarization in myocardial cells. The decrease in APD reduced ventricular pumping efficiency by more than 60% as compared with the normal group (normal conditions). This change is caused by the decline of ventricular output owing to reduced ATP consumption during the crossbridge of myofilaments and decreased tension.
Conclusion
The shortening of APD owing to increased electrical conductivity of a protein channel on myocardial cells likely decreases the wavelength and the pumping efficiency of the ventricles. Additionally, it may increase tissue sensitivity to ventricular fibrillation, including reentry, and cause symptoms such as dyspnea and dizziness.
Similar content being viewed by others

Background
The heart’s blood pumping activity is caused by the repeated contraction and relaxation of the cardiac muscle. Contractions are caused by electrical conduction/propagation and the mechanical behavior of myocardial cells [1]. The action potential (AP) of ventricular myocardial cells leads to the contraction of myofilaments through the transmembrane passage of various electrical ions (especially Ca2+ ions) [2]. The AP may cause secretion or excretion of these ions, as well as myocardial contractility. Changes in AP may cause abnormal transmembrane currents in myocardial cells, which in turn may cause cardiac arrhythmias and other heart diseases.
There are many potential cause for membrane current abnormalities in myocardial cells. For example, abnormality of the protein channels inside the cell membrane directly affects the movement of ions entering and exiting the cell, thereby changing the action potential duration (APD) [3]. This is because the K+ channel plays an important role in determining the repolarization of the cell’s AP. Therefore, both the gain and loss of the K+ channel function can induce arrhythmia by affecting the APD [4, 5]. If the APD is reduced owing to changes in the K+ channel, the conduction wavelength of the heart is shortened, and the reentry can be easily induced. Conversely, prolonged APD can induce torsades de pointes tachycardia, leading to cardiac death [5, 6].
Clinical studies in 2008 discussed this phenomenon. According to Ravens et al., the higher the membrane conductivity on the K+ channel, the shorter the APD, facilitating the generation of reentrant waves [5]. It has been suggested that changes in APD may play a role in arrhythmogenesis and myocardial disease [7]. Animal experiment in 2017 demonstrated a linear correlation between myocardial contraction and APD, owing to the relationship between APD and ATP conductivity in K+ channels [8]. In addition, a computer simulation study showed that K+ channel abnormalities can affect arrhythmogenesis by predicting changes in APD at the single-cell level according to gain and loss of the K+ channel function [4].
These studies observed the electrophysiological effects of APD on the K+ channel at the single cell level, not in three-dimensional tissue. However, it is critical to observe such electrophysiological phenomena and the mechanical behavior of the heart in three-dimensional tissue, because the electrical stimulus for the ventricles in heterogeneously transferred from cell to cell or tissue to tissue. Furthermore, to thoroughly understand cardiac activity, it is necessary to conduct quantitative analysis of the electromechanical behavior of the heart in three-dimensional space.
We recently developed image-based three-dimensional ventricular electromechanical models, that can quantitatively compare the energy consumed by electrophysiological phenomena during mechanical beating [9,10,11]. In the present study we observed changes in APD according to the electrical conductivity of the K+ channel at the single-cell level, and extended these results to three-dimensional ventricular tissue.
Methods
Model of cellular electrophysiology and crossbridge dynamics
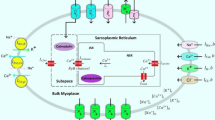
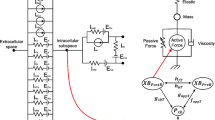
The human ventricular model used in this study has two dynamic characteristics: electrophysiological conduction and mechanical contraction. The part of the model simulating electrophysiological characteristics consisted of a lumped parameter circuit to mimic the phenomenon of ion exchange through the cell membrane of a myocyte. In Fig. 1, Ip, K represents the current due to the K+ pump, Ito denotes the transient outward K+ current, INa, K is the current by the Na+–K+ ion exchange pump, Ip, Ca is the current of the sarcoplasmic Ca2+ pump, and INa, Ca is the current mediated by the Na+–Ca2+ ion exchange pump. EK, ECa, and ENa represent equilibrium potentials of K+, Ca2+, and Na+ ions, respectively, whereas Cm denotes the membrane capacitance due to the phospholipid bilayer in ventricular cells. IKi is the inward rectifier K+ current, IKs is the K+ current due to the slow delayed rectifier, ICa, L represents the L-type inward Ca2+ current, and ICa, b denotes the background Ca2+ current. INa, b represents the background Na+ current, and INa is the fast inward Na+ current. Irel means the Ca2+ current that is released from the junctional sarcoplasmic reticulum (JSR), Ileak denotes the Ca2+ current that leaks from the JSR, and Iup is the Ca2+ uptake current into the network sarcoplasmic reticulum (NSR).

Schematic diagram of electromechanical simulation. The left side of the diagram is a ventricular model of electrophysiological simulation. The electrical components represent the currents, pumps, and ion exchangers from the Ten Tusscher ion model, which emulates the cell membrane for ion transport and the sarcoplasmic reticulum within cardiac cells. “I” represents the ion currents, and “E” is equilibrium potentials of each ion. (For more details, see the text.) The mechanical components on the right side are myofilament models proposed by Rice et al. [14]. N xb and P xb are nonpermissive and permissive confirmation of regulatory proteins, respectively. K np and K pn are transition rates, K np (TCa Tot )7.5 is the forward rate of the nonpermissive-to-permissive transition, and, working in the opposite direction, and K pn (TCa Tot )−7.5 is the backward rate of the permissive-to-nonpermissive transition. g xbT is the ATP-consuming detachment transition rate, h fT and h bT are the forward transition rate and the backward transition rate, respectively; f aapT is the crossbridge attachment rate of transition to the first strongly bound state XB PreR , and g aapT is the reverse rate. XB PreR represents prerotated states of the myosin head in relation to binding. XB PostR denotes a strongly bound myosin head
The part of the model simulating mechanical contraction characteristics was constructed to mimic cardiac contraction by means of the crossbridge of a myofilament. In Fig. 1, XB PreR represents prerotated states of the myosin head in relation to binding and contributes to stiffness but does not generate force in the absence of net motion. XB PostR denotes a strongly bound myosin head and represents the isomerization to induce strain in the extensible neck region. The force due to the crossbridge can be subdivided into an active force and a passive force. The active force induces the action of the cycling crossbridge, and the passive force induces the complete muscle response with viscoelastic elements. Mass prevents instantaneous changes in muscle shortening velocity for quick-release protocols, whereas a linear elastic element is intended to simulate the effects of compliant end connections that take place in real muscle preparations.
The electrophysiological part of the model is based on the ion model proposed by Ten Tusscher et al. [12]. This model, which reproduces the conduction phenomenon of APs in myocardial cells, was applied to the electrical conduction equation based on continuum mechanics:
where Vm is the membrane potential, t is time,\({\text{I}}_{\text{ion}}\) is the sum of all transmembrane ionic currents, Istim is the current due to the external stimulus, and Cm is membrane capacitance.
To represent electrical propagation by means of conduction in three dimensional space, the partial differential equation expressing the electric conduction phenomenon in myocardial tissue and the ordinary differential equation for the electrical wave propagation of the ionic channel were derived [12,13,14].
where ρx, \(\uprho_{\text{y}}\), and \(\uprho_{\text{z}}\) denote cell resistance in x, y, and z directions, respectively. Sx, \({\text{S}}_{\text{y}}\), and Sz represent the ratio of the volume to the surface in x, y, and z directions, respectively. In the model of Ten Tusscher et al. [12], Iion was calculated as follows:
where INa is the Na+ current. IKi, Ito, IKr, and IKs respectively represent K+ currents: the inward rectifier K+ current, transient outward K+ current, rapid delayed rectifier K+ current, and slow delayed rectifier K+ current. ICa,L is the L-type inward Ca2+ current, INa,Ca is the Na+–Ca2+ exchange current, INa,K denotes the Na+–K+ exchange current,\({\text{I}}_{{{\text{p}},{\text{Ca}}}}\) is the current of the Ca2+ pump, Ip,K represents the current of the K+ pump, Ib,Ca is the background Ca2+ current, and Ib,Na denotes the background Na+ current.
We used transient Ca2+ data from electrophysiological simulation for the electromechanical simulation. We employed Ten Tusscher’s equation to express the Ca2+ dynamics, which induce contraction of the thin filament via the Ca2+ induced Ca2+ released (CICR) current and generates tension.
where Ileak, Iup, and Irel denote leakage current from the sarcoplasmic reticulum (SR) to the cytoplasm, pump current taking up calcium in SR, and CICR current, respectively. Vleak is maximal Ileak, Vmaxup is maximal Iup. Cai- and Casr represent the free calcium concentration in the cytoplasm and in the SR. Kup is the half-saturation constant of Iup. arel, brel, and crel are maximal Casr-dependent Irel, Casr half-saturation constant of Irel, and maximal Casr-independent Irel, respectively. d is the activation gate of Irel, and g is the calcium-dependent inactivation gate of Irel. Caitotal represents total Ca2+ in the cytoplasm, and Casrtotal is the total amount of Ca2+ in the SR. Vc and VSR are the cytoplasm and SR volumes, respectively. F is the Faraday’s constant.
The Ca2+-binding mechanism in the canine cardiac muscle model proposed by Rice et al. was utilized here to describe the crossbridge, which means contraction in the sarcomere [15]. Thus, the length of the sarcomere (SL) affected by the crossbridge is expressed as follows:
where IntegralForce represents the sum of the normalized forces integrated by time, and viscosity is the viscous factor in the crossbridge. This equation implies contraction or expansion of the sarcomere (not isosarcometric conditions). In the case of isosarcometric conditions, \(\frac{\text{d}}{{\text{dt}}}{\text{SL}}= 0\), and SL takes initial value SL0.
where Factive (x) is defined as active force, and Fpassive (x) represents passive force. The term Fpreload is a constant force. As this would induce an initial sarcomere length that is larger than the resting length, Fpreload corresponds to Fpassive (SL0). Fafterload term is differently used for isotonic contraction and isometric contraction. Under the isotonic contraction condition, this term is fixed after the release. On the other hand, under the isometric contraction, this term is used to simulate compliant ends of the muscle as shown Eq. (11). Here, x is sarcomere length, and KSE denotes the stiffness in units of normalized force per μm.
Three dimensional ventricular electromechanical model
To achieve our goal, we adapted an image-based electromechanical model of the human ventricular heart from Johns Hopkins University [16]. Human ventricular geometries were generated using the methodology described by Gurev et al. [17,18,19,20]. The three-dimensional human ventricular finite-element model used in this study consists of a lumped-parameter model of the physiological circulatory system [20, 21]. Our ventricular model for the electrophysiological simulation consisted of 214,319 tetrahedral finite elements. In contrast, the ventricular model for electromechanical simulation consisted of 14,720 Hermite-based finite elements to represent a natural three-dimensional curve of the cardiac surface [14, 22, 23]. Three dimensional simulation was carried out using a human arrhythmic ventricle model including the Purkinje fiber mesh, in which propagation of electrical stimuli during electrophysiological simulation starts from the atrioventricular node and spreads throughout the ventricle by means of the Purkinje networks based on Berenfeld et al. [24]. In addition, considering the differences in the structural characteristics and ventricle thickness, we assumed that all tissues of the ventricles were heterogeneous, and the conductance varied depending on the part of the ventricle.
Simulation protocol
In this study, we observed APD variation in single-cell and three-dimensional ventricular models. First, we used a cellular electrophysiological simulation to observe the electrical changes due to APD variation and to clarify the relationship between APD and calcium concentration. Second, we used a three-dimensional electromechanical simulation to compare cardiac pumping efficiency quantitatively under sinus rhythm conditions.
The single-cell simulation was performed using the ventricular cell model suggested by Ten Tusscher et al. [12]. The formulation of the IKs current equation was modified to examine the variation with the change in APD.
where gKs is conductance of the K+ channel, xs is the activated gate parameter of the K+ channel, and EKs denotes the equilibrium potential of K+. In this study, the initial values of gKs were set to 0.392 × 1.3 mS/μF. This value is based on other studies [12, 25]. Next, it was simulated by increasing the initial values 2-, 4-, 6-, 8-, and 10-fold to induce changes in APD.
The three dimensional ventricular simulation was conducted by setting the basic cycle length (BCL) to 600 ms. The change in APD during 1 cycle and the conduction wavelength were measured.
where CV is the conduction velocity in myocardial cells. APD90 means the time point where ventricular cells become excited and 90% repolarized. For calculation of conduction velocity, we specified the node (down) located on the lower side of the ventricular model surface and the node (up) on the upper side of its vertically positioned surface. The conduction velocity was calculated by dividing the distance along the straight line between these two nodes by the difference in the time of the response to a stimulus between the two nodes.
Three-dimensional electromechanical simulations used data on transient Ca2+ concentration from electrophysiological simulation results. The contraction of the ventricles was caused by active tension in the dynamic model of a myosin filament proposed by Rice et al. [15]. To numerical APD on cardiac pumping efficacy, we compared changes in stroke volume, ejection fraction, stroke work, and ATP consumption rate for the six conditions (including the normal group) according to APD variation. Each value was measured in only the last cycle (600 ms) of the simulation to observe changes in the steady state [26, 27].
Results
Cellular electrophysiological responses
We performed a single cellular electrophysiological simulation by increasing electrical conductivity of the IKs channel, which affects the APD of myocardial cells. The results are presented in Fig. 2.

Cellular simulation results for ventricular tissue according to electrical conductivity conditions of the IKs channel. a–c IKs graphs as a function of electrical conductivity g Ks of the IKs channel. d–f Action potential shape under the influence of variation of electrical conductivity. g–i Ca2+ concentration. j Ca2+ integration depending on APD variation. “norm” indicates the case where g ks has a normal value. ‘gks2’, ‘gks4’, ‘gks6’, ‘gks8’, and ‘gks10’ represent electrical conductivity when g ks is increased 2-, 4-, 6-, 8-, and 10-fold, respectively. ‘Endo’ is the endocardial cell, ‘M’ is the mid-myocardial cell, and ‘Epi’ is the epicardial cell. ‘APD’ denotes action potential duration
Figure 2a–c shows intensity of the IKs current depending on the electrical conductivity of the IKs channel, where A is an “endocardium cell,” B is a “midmyocardium cell,” and C is an “epicardium cell.” In each graph, “normal” is the case where the electrical conductivity of the IKs channel is normal. Labels “gks2” to “gks10” indicate that the electrical conductivity of the IKs channel is 2, 4, 6, 8, or 10 times the normal value, respectively. In Fig. 2a–c, the current flowing through the IKs channel is increased due to the increased electrical conductivity of the IKs channel. The IKs current, which was slowly delayed at normal electrical conductivity, was rapidly delayed as the electrical conductivity of the IKs channel increased.
In the three ventricular tissues, APD90 shortened as the electrical conductivity of the IKs channel increased (Fig. 2d–f). In the results of ventricular cellular simulation, the APD90 measured in an endocardium cell was 279 ms when the electrical conductivity of the IKs channel was normal. APD90 was 228 ms when the electrical conductivity was doubled, 182 ms when it was quadrupled, 158 ms when multiplied by 6, 141 ms when multiplied by 8, and 123 ms when multiplied by 10. The APD90 measured in a midmyocardium cell was 353, 301, 249, 222, 202, and 189 ms, corresponding to electrical conductivity of the IKs channel when it was normal or increased 2-, 4-, 6-, 8-, and 10-fold. The APD90 measured in an epicardium cell was 280, 230, 185, 162, 147, and 135 ms, corresponding to the electrical conductivity of the IKs channel when it was normal or increased 2-, 4-, 6-, 8-, and 10-fold. APD90 is the highest in the midmyocardium cell compared to the other two cell types in all cases owing to the change in electrical conductivity of the IKs channel.
Figure 2g–i show a graph of the intracellular Ca2+ concentration as a function of electrical conductivity of the IKs channel in the ventricular tissue during a BCL of 600 ms. As the electrical conductivity of the IKs channel increased in all three ventricular cell types, the wavelength of the Ca2+ concentration graph was almost constant, but the amplitude significantly diminished. The amplitude of Ca2+ in the normal case was ~ \(0.8 \times 10^{ - 4}\) mM in the endocardium and epicardium cells (Fig. 2g and i). When the conductivity of the IKs channel was increased tenfold, the concentration was \(0.2 \times 10^{ - 4}\) to \(0.3 \times 10^{ - 4}\) mM. This result is similar to the data from the midmyocardium cell; the amplitude of the Ca2+ concentration graph in the normal case was \(1.1 \times 10^{ - 4}\) mM, but the value decreased as the electrical conductivity of the IKs channel increased. When the electrical conductivity increased tenfold, Ca2+ concentration decreased to ~ \(0.45 \times 10^{ - 4}\) mM.
Figure 2j showed the integration of Fig. 2d–i, which is the changes of intracellular calcium concentration in ventricular cells due to APD variation according to the change of electrical conductivity of the IKs channel during a 1-cycle period. The shortened APD lower the intracellular calcium concentration than normal group. There was a linear relationship between APD90 and intracellular calcium concentration in each ventricular cell.
Three dimensional ventricular electrophysiological responses
Figure 3 shows the electrophysiological simulation results from the three dimensional ventricular model. Simulation results during the BCL of 600 ms are snapshots taken at intervals of 100 ms on the horizontal axis, as a function of the variation of APD90 on the vertical axis. The vertical axis of Fig. 3 indicates that the APD90 measured by means of the three dimensional electrophysiological model was 299 ms (normal), 256 ms (gks2), 212 ms (gks4), 188 ms (gks6), 171 ms (gks8), and 150 ms (gks10). As APD90 decreased, the excitement resulting from the electrical stimulation delivered to ventricular tissue by the Purkinje networks was terminated faster. In the ventricular tissue, the conduction wavelength was 17 cm when APD90 was 299 ms, but it decreased as APD90 was shortened under the heterogeneous condition. When APD90 was 150 ms, the conduction wavelength became 9 cm by decreasing to 44% compared to normal group.

Snapshots and conduction wavelengths for three-dimensional electrophysiological simulations with APD variation. ‘APD’ denotes action potential duration
Figure 4 showed the calcium contour due to APD variation in three dimensional electrophysiological simulations. As APD90 decreased, the period of Ca2+ concentration change on the ventricular surface at BCL 600 ms stayed almost the same. In contrast, in accordance with the shortening of APD90, a color change of the Ca2+ contour occurred at the time point 2500 ms. Although the Ca2+ contour at 2500 ms appears as the brightest yellow for 299 APD , at 150 APD , it was almost gray.

Calcium contour according to three-dimensional electrophysiological simulations with APD variation. ‘APD’ denotes action potential duration
Three dimensional ventricular mechanical responses
The effects of APD variation on the pumping efficiency depending on the electrical conductivity of the IKs channel were analyzed next. Figure 5 depicts a three-dimensional ventricular contour that shows the muscle tension, adenosine triphosphate (ATP) status at the end-systolic volume (ESV) time point, strain at the end-diastolic volume (EDV) time point, and a graph of each variable (tension, strain, and ATP) at BCL 600 ms by ventricular cell type.

Changes in tension, strain, and ATP consumption rate depending on APD variation. Graphs showing variation in tension (a), strain (b) and ATP consumption rate (c) with time in each ventricular tissue: the endocardium (Endo), mid-myocardium (M), and epicardium (Epi). Snapshots of end-systolic tension, ATP consumption rate, and end-diastolic strain in a three-dimensional sinus pacing simulation. ‘ATP CR’ denotes ATP consumption rate, and ‘BCL’ denotes basic cycle length, which is 600 ms
The shortening of APD90 decreased the contraction force of the muscle at the ESV time point and increased the strain of the ventricles at the EDV time point. In addition, ATP consumption decreased in ventricular cells at the ESV time point.
The total consumption of ATP at the ESV time point was ~ 136 s−1 at 299 APD , and the average consumption was ~ 0.000594 s−1. In this case, total tension was 3,468,332 kPa and contracted to an average of 15.15 kPa. At 256 APD , the total amount of ATP consumed at the ESV time point was ~ 95 s−1, and the average was 0.000414 s−1. End systolic tension at APD90 of 256 ms was 2,350,950 kPa in total and decreased to an average of 10.27 kPa The total consumption and the average of ATP at 212 APD were found to be 64.9 and 0.000284 s−1, respectively. The corresponding total tension was 1,533,506 kPa, and the average was 6.7 kPa. Similarly, the total consumption and average ATP consumption at 188 APD , 171 APD , and 150 APD were 53.2 and 0.000232 s−1 (188 APD ), 45.2 and 0.000198 s−1 (171 APD ), and 42.9 and 0.000187 s−1 (150 APD ), respectively. The total amount of and average tension in each case were calculated and found to be 1,211,614 and 5.29 kPa (188 APD ), 992,201 kPa and 4.33 kPa (171 APD ), and 927,608 and 5.05 kPa (150 APD ), respectively.
In addition, during 600 ms, the difference in the change of ATP and tension was noticeable due to shortened APD in each ventricular tissue. The amplitude of the tension change in the endocardium and midmyocardium at 299 APD was ~ 60 kPa, but at 150 APD , it was ~ 30 kPa. The tension graph of the epicardium shows a smaller amplitude in all cases depending on APD variation in comparison with the other two ventricular tissues. These results were also obtained from the graph of ATP consumption by ventricular tissue. The graph of amplitude of ATP consumption in the endocardium and midmyocardium at 299 APD shows ~ 0.0025 s−1, but the amplitude in the epicardium was smaller. The amplitude of the ATP graph was smaller as APD90 decreased, and the change in ATP consumption at 150 APD during BCL of 600 ms was reduced by 50% as compared to 299 APD .
On the other hand, the degree of deformation of the ventricles during BCL of 600 ms decreased as APD90 was shortened, and the graph of the strain change shifted upward. The strain variable was smaller in the epicardium than in the other ventricular tissues. As APD90 decreased, the difference in strain graphs between the two ventricular tissues and the epicardium increased.
Analysis of the changes in the mechanical responses and cardiac pumping efficiency of the ventricle depending on APD variation is shown in Figs. 6, 7. Figure 6 depicts a dynamic response graph of the three dimensional electromechanical simulation. The pressure in the left ventricle and in systemic arteries decreased for the sinus rhythm. EDV and ESV increased due to shortening of APD90. The pressure–volume (PV) loop was shifted to the left as APD90 was shortened (Fig. 6a, b). As a result, the pulse pressure at 299 APD was 35.1 mmHg, but the value decreased with the shortening of APD90. In each case, the pulse pressure was 30.7 mmHg (256 APD ), 24.2 mmHg (212 APD ), 19.3 mmHg (188 APD ), 15.3 mmHg (171 APD ), and 12.2 mmHg (150 APD ).

Mechanical-response graphs for the electromechanical simulation involving the three dimensional ventricular-tissue model with APD variation. a Pressure in the left ventricle and systemic arteries. b Pressure–volume loop (PV loop) of the left ventricle. ‘BCL’ is basic cycle length, which is 600 ms

APD-dependent cardiac pumping efficacy changes in electromechanical simulation using a 3D ventricular-tissue model. a Energy consumption of the left ventricle in the form of ATP. b Stroke work of the left ventricle. c Rate of stroke work for ATP consumption. Amount of work done by the left ventricle relative to energy consumption. d Stroke volume-ejection fraction for the left ventricle
EDV is the maximal volume, and ESV is the minimal volume of the PV loop in Fig. 6b. At 299 APD , EDV of the left ventricle was 80.6 mL, and ESV was 44.1 mL. On the other hand, EDV and ESV were 92.3 and 79.1 mL, respectively, at 150 APD . As APD90 decreased, the pressure in the left ventricle diminished, and the volume increased. Accordingly, the internal area of the PV loop (which moved to the left in each case) decreased. EDV and ESV were 83.6 and 51.2 mL (256 APD ), 87.1 and 61.7 mL (212 APD ), 89.2 and 69 mL (188 APD ), and 91 and 74.6 mL (171 APD ), respectively.
Figure 7 shows changes in the cardiac pumping efficiency depending on APD variation in the three dimensional ventricular-tissue model. Figure 7a depicts quantification of total ATP consumed to produce contraction from the crossbridge of a myofilament during BCL of 600 ms, and its value decreased as APD90 was shortened. In addition, the stroke work in the ventricle decreased for 1 cycle. Its value, which was 4403 mmHg mL at 299 APD , decreased by ~ 80% at 880 mmHg mL (150 APD ; Fig. 7b). It was computed from the internal area of the PV loop in Fig. 6b.
This change reduced the amount of ventricular work per unit ATP consumed by the myofilament during the sinus rhythm in accordance with shortened APD90 (Fig. 7c). This means that as APD90 is shortened, energy efficiency of the ventricle during 1 cycle decreased. The energy efficiency at 299 APD was 41, and the efficiency at 39, 34, 28, and 23 is respectively shown at 256 APD , 212 APD , 188 APD , and 171 APD . At 150 APD , this efficiency was the lowest: 18.
Figure 7d is a graph of stroke volume and the ejection fraction calculated from EDV to ESV in the PV loop of Fig. 6b. The stroke volume was measured by means of the difference between EDV and ESV in the PV loop, and the ejection fraction was obtained by means of the stroke volume ratio for EDV. These data refer to the amount and efficiency of blood that is sent from the heart to the aorta and the pulmonary artery. Stroke volume and ejection fractions at 299 APD were measured and found to be 36 mL and 45%, respectively. Nonetheless, these values decreased with decreasing APD90, resulting in 13 mL and 14% at 150 APD . The stroke volume and the ejection fraction of the left ventricle decreased as APD90 was shortened. The pumping efficiency when APD90 became 150 ms was 68% lower relative to the value at 299 APD .
Discussion
In this simulation study, we analyzed the ventricular pacing efficiency under APD variation. The main findings of the study are the following:
-
1.
In the cellular electrophysiological simulation, as APD is shortened owing to the increase in the electrical conductivity of the K+ channel, the intracellular Ca2+ concentration decreases. That is, the APD and the sum of the intracellular Ca2+ concentrations showed a positive correlation.
-
2.
The shortened APD reduced the conduction wavelength in the three-dimensional ventricular tissue by shortening the plateau and early repolarization in myocardial cells.
-
3.
In addition, the shortened APD reduced cardiac pumping efficiency by more than 60% compared with the normal group.
Increasing the electrical conductivity (g Ks ) of the IKs channel makes the flow of IKs currents faster owing to K+ flowing out of the cells. The fast IKs current induces rapid repolarization of the APs, leading to rapid return to the resting cell membrane potential. This event reduces APD90 and shortens the opening time of the L-type Ca2+ channel. Finally, because the period for extracellular Ca2+ to flow into the intracellular area decreases, the amount of Ca2+ entering the cell during one cycle decreases. That is, as APD decreases, the concentration of Ca2+ in the cells decreases. This finding is consistent with the result of Ten Tusscher et al. [12, 28]. From these results, we can conclude that there is a linear relationship between APD and intracellular Ca2+ concentration within the range of our experimental conditions (Fig. 2j).
In addition, cardiac tissues composed of cells with shortened APD have short wavelengths even if the conduction velocity is the same (Fig. 3). These results are consistent with those of Roden et al. [6]: in cell types with shortened APD, the period of return to the resting phase is reduced, even though the whole tissue is excited at the same time in one cycle of the sinus rhythm [29]. This finding indicates that the cells with short APD decrease the depolarization period, thereby reducing the opening period of the voltage-dependent L-type Ca2+ channel and reducing the intracellular Ca2+ concentration (Fig. 4).
Intracellular Ca2+ includes Ca2+ ions that are stored in the SR and released into the cells (the Ca2+-induced Ca2+-released current). Ca2+ released into the cell binds to troponin, causing a conformational change in tropomyosin and formation of the crossbridge where the myosin head binds to the actin filament. In other words, the strength of the tension generated during the crossbridge of the myofilament is proportional to the concentration of Ca2+ in the cell. However, in tissues with short APD, the amount of Ca2+ released into the cells from the SR is lower. Therefore, the Ca2+ concentration in the cells decreases, and the strength of the myocardial tension diminishes (Figs. 4 and 5a). This state weakens the contraction force of the ventricular muscle, thereby reducing heart rate and inhibiting blood circulation (Fig. 6a, b) [30, 31]. In addition, the end-diastolic strain of the ventricle increases as the contraction force decreases (Fig. 5b), and the amount of ATP consumed in the ventricle during one cycle decreases as the crossbridge generation diminishes under conditions of short APD (Fig. 5c) [29, 32].
Therefore, in the tissue with shortened APD, the strength of the tension is low because of the low Ca2+ concentration, which reduces the myocardial contractility, thus reducing the stroke work produced by the ventricle during one cardiac cycle (Fig. 7b) [29]. Additionally, myocardial contractility weakened due to a reduction in tension also results in a decrease in the stroke volume of the ventricle (Fig. 7d). The decline in the stroke volume, which is the volume of blood pumped from the left ventricle per beat, leads to a decrease in the pressure in the left ventricle and lowered aortic pressure (blood pressure) according to Poiseuille’s law (Fig. 6a) [26, 31, 33, 34]. Thus, the amount of work performed by the ventricle, which is the ratio of stroke work to ATP consumption, is lower than that of a ventricular tissue with normal APD (Fig. 7b). The pumping efficiency of the ventricles can be deduced from the ratio of cardiac work rate to energy consumption [32, 33, 35, 36]. These results suggest that ventricular pacing efficiency is lower in tissues with shortened APD than in tissues with normal APD (Fig. 7c, d).
Conclusions
The shortening of APD owing to increased electrical conductivity of a protein channel on myocardial cells likely decreases the wavelength and the pumping efficiency of the ventricles. Additionally, it may increase tissue sensitivity to ventricular fibrillation, including reentry, and cause symptoms such as dyspnea and dizziness.
References
Shih HT. Anatomy of the action potential in the heart. Tex Heart Inst J. 1994;21:30–41.
Tomson TT, Arora R. Modulation of cardiac potassium current by neural tone and ischemia. Card Electrophysiol Clin. 2016;8:349–60.
Hille B. Ion channels of excitable membranes. 507th ed. Sunderland: Sinauer; 2001.
Priebe L, Beuckelmann DJ. Simulation study of cellular electric properties in heart failure. Circ Res. 1998;82:1206–23.
Ravens U, Cerbai E. Role of potassium currents in cardiac arrhythmias. Europace. 2008;10:1133–7.
Roden DM, Hoffman BF. Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine Purkinje fibers. Relationship to potassium and cycle length. Circ Res. 1985;56:857–67.
Gilmour RF, Heger JJ, Prystowsky EN, Zipes DP. Cellular electrophysiologic abnormalities of diseased human ventricular myocardium. Am J Cardiol. 1983;51:137–44.
Nichols C, Ripoll C, Lederer W. ATP-sensitive potassium channel modulation of the guinea pig ventricular action potential and contraction. Circ Res. 1991;68:280–7.
Lim KM, Lee JS, Gyeong M-S, Choi J-S, Choi SW, Shim EB. Computational quantification of the cardiac energy consumption during intra-aortic balloon pumping using a cardiac electromechanics model. J Korean Med Sci. 2013;28:93–9.
Imaniastuti R, Lee HS, Kim N, Youm JB, Shim EB, Lim KM. Computational prediction of proarrhythmogenic effect of the V241F KCNQ1 mutation in human atrium. Prog Biophys Mol Biol. 2014;116:70–5.
Lim KM, Hong S-B, Lee BK, Shim EB, Trayanova N. Computational analysis of the effect of valvular regurgitation on ventricular mechanics using a 3D electromechanics model. J Physiol Sci. 2015;65:159–64.
Ten Tusscher K, Noble D, Noble P, Panfilov A. A model for human ventricular tissue. Am J Physiol Heart Circ Physiol. 2004;286:H1573–89.
Fox JJ, McHarg JL, Gilmour RF. Ionic mechanism of electrical alternans. Am J Physiol Heart Circ Physiol. 2002;282:H516–30.
Zhang H, Ye H, Huang W. A meshfree method for simulating myocardial electrical activity. Comput Math Methods Med. 2012;2012:1–16.
Rice JJ, Wang F, Bers DM, de Tombe PP. Approximate model of cooperative activation and crossbridge cycling in cardiac muscle using ordinary differential equations. Biophys J. 2008;95:2368–90.
Constantino J, Hu Y, Lardo AC, Trayanova NA. Mechanistic insight into prolonged electromechanical delay in dyssynchronous heart failure: a computational study. Am J Physiol Heart Circ Physiol. 2013;305:H1265–73.
Gurev V, Lee T, Constantino J, Arevalo H, Trayanova NA. Models of cardiac electromechanics based on individual hearts imaging data. Biomech Model Mechanobiol. 2011;10:295–306.
Xu C, Xu L, Gao Z, Zhao S, Zhang H, Zhang Y, Du X, Zhao S, Ghista D, Li S. Direct detection of pixel-level myocardial infarction areas via a deep-learning algorithm. In: International conference on medical image computing and computer-assisted intervention. Berlin: Springer; 2017; p. 240–249.
Wong KKL, Wang D, Ko JKL, Mazumdar J, Le T-T, Ghista D. Computational medical imaging and hemodynamics framework for functional analysis and assessment of cardiovascular structures. Biomed Eng Online. 2017;16:35.
Du X, Zhang W, Zhang H, Chen J, Zhang Y, Warrington JC, Brahm G, Li S. Deep regression segmentation for cardiac bi-ventricle MR images. IEEE Access. 2018;6:3828–38.
Wei L, Wan S, Guo J, Wong KKL. A novel hierarchical selective ensemble classifier with bioinformatics application. Artif Intell Med. 2017;83:82–90.
Xu L, Huang X, Ma J, Huang J, Fan Y, Li H, Qiu J, Zhang H, Huang W. Value of three-dimensional strain parameters for predicting left ventricular remodeling after ST-elevation myocardial infarction. Int J Cardiovasc Imaging. 2017;33:663–73.
Zhang H, Gao Z, Xu L, Yu X, Wong KCL, Liu H, Zhuang L, Shi P. A meshfree representation for cardiac medical image computing. IEEE J Transl Eng Health Med. 2018;6:1–12.
Berenfeld O, Jalife J. Purkinje-muscle reentry as a mechanism of polymorphic ventricular arrhythmias in a 3-dimensional model of the ventricles. Circ Res. 1998;82:1063–77.
Kharche S, Adeniran I, Stott J, Law P, Boyett MR, Hancox JC, Zhang H. Pro-arrhythmogenic effects of the S140G KCNQ1 mutation in human atrial fibrillation—insights from modelling. J Physiol. 2012;590:4501–14.
Zhao S, Gao Z, Zhang H, Xie Y, Luo J, Ghista D, Wei Z, Bi X, Xiong H, Xu C, Li S. Robust segmentation of intima-media borders with different morphologies and dynamics during the cardiac cycle. IEEE J Biomed Health Inform. 2017;99:1. https://doi.org/10.1109/JBHI.2017.2776246.
Zhen X, Zhang H, Islam A, Bhaduri M, Chan I, Li S. Direct and simultaneous estimation of cardiac four chamber volumes by multioutput sparse regression. Med Image Anal. 2017;36:184–96.
Ten Tusscher KHWJ, Panfilov AV. Alternans and spiral breakup in a human ventricular tissue model. Am J Phys Heart Circ Physiol. 2006;291:H1088–100.
Wong KKL, Kelso RM, Worthley SG, Sanders P, Mazumdar J, Abbott D. Cardiac flow analysis applied to phase contrast magnetic resonance imaging of the heart. Ann Biomed Eng. 2009;37:1495–515.
Wong KKL, Tu J, Kelso RM, Worthley SG, Sanders P, Mazumdar J, Abbott D. Cardiac flow component analysis. Med Eng Phys. 2010;32:174–88.
Qin Y, Wu J, Hu Q, Ghista DN, Wong KK. Computational evaluation of smoothed particle hydrodynamics for implementing blood flow modeling through CT reconstructed arteries. J X-ray Sci Technol. 2017;25:213–32.
Wong KKL, Cheung SCP, Yang W, Tu J. Numerical simulation and experimental validation of swirling flow in spiral vortex ventricular assist device. Int J Artif Organs. 2010;33:856–67.
Wong KKL, Molaee P, Kuklik P, Kelso RM, Worthley SG, Sanders P, Mazumdar J, Abbott D. Motion estimation of vortical blood flow within the right atrium in a patient with atrial septal defect. In: 2007 IEEE/ICME international conference on complex medical engineering. 2007. p. 862–869.
Wong KKL, Kelso RM, Worthley SG, Sanders P, Mazumdar J, Abbott D. Medical imaging and processing methods for cardiac flow reconstruction. J Mech Med Biol. 2009;09:1–20.
Wong KK, Tu J, Sun Z, Dissanayake DW. Methods in research and development of biomedical devices. Singapore: World Scientific; 2013.
Wong KK, Tu J, Kelso RM. Vortical flow analysis. J Mech Med Biol. 2010;10:191–212.
Authors’ contributions
This work is the product of intellectual work of the entire team. All the members contributed (to varying degrees) to the analytical methods used, research concept, simulation design, simulation source code, simulation process, and writing of the manuscript. Simulation, data analysis, writing-original draft: DUJ. Methodology, writing-review and editing: KML. Both authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
All of the data and material is written in the manuscript and available during request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This research was partially supported by the MSIT (Ministry of Science, ICT), under the ITRC (Information Technology Research Center) support program (IITP-2018-2014-0-00639) supervised by the IITP, and NRF (National Research Foundation) under basic engineering research project (2016R1D1A1B0101440) and the EDISON (NRF-2011-0020576) Programs.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Jeong, D.U., Lim, K.M. The effect of myocardial action potential duration on cardiac pumping efficacy: a computational study. BioMed Eng OnLine 17, 79 (2018). https://doi.org/10.1186/s12938-018-0508-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12938-018-0508-2