
Abstract
Cisplatin is a platinum-based first-line drug for treating ovarian cancer. However, chemotherapy tolerance has limited the efficacy of cisplatin for ovarian cancer patients. Research has demonstrated that cisplatin causes changes in cell survival and death signaling pathways through its interaction with macromolecules and organelles, which indicates that investigation into the DNA off-target effects of cisplatin may provide critical insights into the mechanisms underlying drug resistance. The multifunctional protein p62 works as a signaling hub in the regulation of pro-survival transcriptional factors NF-κB and Nrf2 and connects autophagy and apoptotic signals, which play important roles in maintaining cell homeostasis. In this review, we discuss the role of p62 in cisplatin resistance by exploring p62-associated signaling pathways based on current studies and our work. Insights into these resistance mechanisms may lead to more effective therapeutic strategies for ovarian cancer by targeting p62.
Similar content being viewed by others


Background
Ovarian cancer is a gynecologic cancer with a high mortality rate. In developed countries, the mortality rate of ovarian cancer is three times that of breast cancer and the 5-year survival rate of patients with stage IV ovarian cancer is only 28% [1]. In 1965, the platinum-containing drug cisplatin (cis-diamminedichloroplatinum) was found to exhibit antimicrobial activity, and subsequent studies demonstrated that platinum compounds have strong antitumor activity [2, 3]. In the 1980s, the first-line drugs for ovarian cancer were cisplatin and cyclophosphamide. Carboplatin, a second-generation platinum drug, showed equivalent therapeutic effects as cisplatin but with fewer toxic side effects [4, 5]. At present, the standard treatment for patients with advanced ovarian cancer is surgery, followed by six cycles of paclitaxel and carboplatin neoadjuvant chemotherapy [6]. However, most ovarian cancer patients relapse after treatment and eventually show no sensitivity to platinum drugs. Although several clinical trials have been conducted in recent years to improve the efficacy of platinum-based therapies (Table 1), chemotherapy resistance to platinum drugs remains an obstacle that limits the clinical application and efficacy of these drugs.
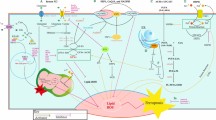
Platinum is an electrophilic reagent characterized by its ability to form covalent linkages with nucleophilic residues of nucleobases such as guanine and adenine. Because a variety of cellular macromolecules contain nucleophilic residues, platinum drugs have the potential to interact with various cellular components, such as ribosomes, spliceosomes, and the RNA in telomerase, as well as proteins through Met, His and free Cys side chains [10, 11]. Galluzzi et al. [12] proposed that cisplatin can accumulate in mitochondria, lysosomes, endoplasmic reticulum, nucleus, cell membrane, cytoskeleton and cytosol, which causes cell stress. These findings indicate that cisplatin may exhibit far more effects on tumor cells than only through its interaction with DNA. And it may not only induce death signals, but also adaptive response including autophagy, the unfolded protein response and other pro-survival signals while disturbing organelles and proteins in the cytoplasm [13,14,15] (Fig. 1).

Cytoplasm effects induced by cisplatin in ovarian cancer cells. Cisplatin interacts with mitochondria, lysosomes, endoplasmic reticulum and cytoplasmic proteins, leading to cell stress and the activation of both death and pro-survival signals in ovarian cancer cells
Although the signaling networks that determine cell survival and death are extensive, only a small number of molecules have been identified that function in coordinating these signaling pathways. The multifunctional protein p62/SQSTM1 (also known as sequestosome-1, hereinafter referred to as p62) integrates both survival and death signaling by regulating the ubiquitination of key cell signaling molecules that control survival and death [16,17,18,19]. p62 contains multiple protein-binding domains: the N-terminal PB1 (Phox and Bem1p) domain that binds to the atypical kinase (aPKC) and mediates p62 self-oligomerization; the central zinc finger (ZZ) domain that promotes NF-κB pathway activation; a TB module (motif) that binds TNF receptor associated factor 6 (TRAF6); the KIR (Keap1-interacting region) domain that competes with NRF2 for Keap1; a UBA (ubiquitin-associated) domain that recruits ubiquitin-linked proteins and mediates their degradation through autophagy or the ubiquitin–proteasome system; and the LIR (LC3-interacting region), which recognizes a specific sequence in the autophagosome membrane protein LC3. These multiple domains make p62 an important player in the regulation of selective autophagy [20, 21].
In this review, we discuss the changes in p62-mediated signaling pathways in ovarian cancer during cisplatin treatment based on our work and current research. We describe a role for p62 in cisplatin resistance of ovarian cancer, providing a theoretical basis for potential strategies for overcoming chemotherapy resistance in ovarian cancer.
p62 participates in drug resistance of ovarian cancer by regulating autophagy
Macroautophagy
During macroautophagy, hereinafter referred to as autophagy, autophagosomes isolate components targeted for autophagy by forming a closed membrane structure and transporting them to lysosomes for degradation. Autophagy serves a protective function against malignant transformation and maintains homeostasis in normal tissues. However, once cells undergo transformation, autophagy provides cancer-protective functions to deal with stress from the worse survival environment [22, 23]. Previous reports demonstrated that cisplatin activates autophagy through the MEK/ERK pathway in ovarian cancer, which may lead to cisplatin resistance [24].
p62 is an autophagy receptor involved in the recognition of ubiquitin-labeled substrates targeted for autophagy [25]. Matsumoto et al. [26] suggested that p62-mediated selective autophagy is a compensatory pathway for protease degradation. The UBA domain structure in p62 forms a compact triple-spiral stalk with a hydrophobic surface, which may be a targeted contact site for protein interactions [27, 28]. Additionally, p62 binds the autophagy membrane protein Atg8/LC3 through the LIR. Current studies suggest D335, D336, D337, and W338 mutations eliminate the binding of p62 to LC3 [29]. We previously found that the levels of mature LC3II were increased in cisplatin-resistant SKOV3/DDP ovarian cancer cells compared with parental cells [30]. When we suppressed autophagy in ovarian cancer cells with 3-MA or chloroquine, cisplatin showed increased efficacy; furthermore, p62 expression was increased in SKOV3/DDP cells. Upon cisplatin treatment, p62 and LC3 puncta were co-localized. RNAi-mediated downregulation of p62 also increased the sensitivity of ovarian cancer cells to cisplatin [31]. These results indicate that p62-mediated autophagy induced by cisplatin may function as a protective mechanism in ovarian cancer cells.
Cha-Molstad et al. [32] found since ZZ domain may lock p62 in a close state, promoted the combination of ZZ domain and Nt-Arg leading to autophagy upregulation. Our study suggested that the LIR and UBA domains in p62 may modulate autophagic flux. We transfected SKOV3 cells with a vector encoding the L417V mutant (UBA mutant) that lost binding to ubiquitinated proteins and found increased autophagolysosomes in the UBA mutant-expressing cells, suggesting upregulated autophagic flux [33]. MTT assays revealed that UBA mutant-expressing cells showed reduced sensitivity to cisplatin compared with parental cells. Additionally, post-translational modifications of p62 also influence autophagy levels. Keap1/Cullin3 increased the co-localization of p62 and LC3 by ubiquitinating p62 at lysine 420 in the UBA domain and promotes the autophagy degradation pathway [34]. CK2 (casein kinase 2) increased the affinity of p62 to ubiquitin through phosphorylation of serine 403 in the UBA domain and promotes the clearance of ubiquitinated proteins by autophagy [26]. Specific mutations in the PB1 domain (C105A and C113A mutations) also significantly inhibited autophagy [35]. The effective mutations and post-translational modification sites reported to affect autophagy were shown in Fig. 2. Therefore, developing small molecule drugs that inhibit autophagy by targeting p62 may provide new strategies for combination treatment with cisplatin in ovarian cancer.

Schematic representation of p62 functional domains involved in autophagy regulation
Mitophagy
Mitochondrial autophagy (mitophagy) maintains mitochondria mass and function by clearing damaged or overloaded mitochondria. Kingnate et al. [36] proposed that increased mitochondrial fusion and reduced mitochondrial fission may be one of the mechanisms of cisplatin resistance in ovarian cancer cells. Williams et al. [37] suggested that inhibition of mitochondrial fission leads to mitophagy suppression. The PINK1 (PTEN-induced putative kinase protein 1)/Parkin pathway is one of the regulators of mitophagy. The PINK1 serine/threonine kinase enters the mitochondrial membrane space through the translocase outer membrane complex. In healthy cells, the mitochondrial intramembrane rhomboid protease PARL mediates cleavage and inactivation of PINK1. However, in response to abnormal mitochondrial membrane potential, PINK1 accumulates in the mitochondrial outer membrane and recruits the E3 ubiquitin ligase Parkin to initiate mitophagy [38, 39].
Recent studies have indicated that p62 is not only found in the outer mitochondrial membrane but also localized to the inner mitochondrial membrane, which may be involved in mitochondrial function maintenance and morphology regulation [40]. VDAC (voltage-dependent anion channel), an important component of the mitochondrial permeability transition pore (mPTP), is a cysteine-containing protein that binds cisplatin [41]. VDAC maintains an open mPTP at low holding potentials but shows anion-selectivity upon increase of holding potential, which is associated with apoptosis regulation [42]. Geisler et al. [43] demonstrated that Parkin polyubiquitinates VDAC, and p62 recognizes ubiquitinated VDAC and mediates the degradation of damaged mitochondria. Whether p62 regulation of PINK and VDAC is involved in the cisplatin resistance of ovarian cancer cells is unknown and should be examined in future studies.
p62 participates in pro-survival signaling regulation induced by cisplatin treatment in ovarian cancer
NF-κB signaling
NF-κB is one of the classical pro-survival signaling factors in cells. Phosphorylation of IκB kinase (IKK) promotes degradation of the NF-κB inhibitor IκB through the proteasome pathway, which subsequently activates NF-κB signaling. Many studies showed that the activated NF-κB pathway functions in promoting cell survival in ovarian cancer. Yang et al. [44] found that the E3 ligase TRIM52 (the tripartite motif 52) increased the expression of IKKβ and IKBα and promoted NF-κB subunit p65 nuclear translocation to activate the transcription of downstream MAPK9 (mitogen-activated protein kinase 9), BCL2 (B-cell lymphoma 2), CXCL8 (C-X-C motif chemokine ligand 8) and TNF (tumor necrosis factor) genes in SKOV3 and Caov3 ovarian cancer cells. Mabuchi et al. found that chemotherapy increased the levels of p-IκB and NF-κB transcriptional activity to higher levels in Caov-3 cisplatin-resistant ovarian cancer cells compared with A2780 cells. Furthermore, inhibiting NF-κB activity by BAY 11-7085 enhanced the sensitivity of Caov-3 cells to cisplatin [45, 46]. These studies indicate that NF-κB may be involved in the mechanism of cisplatin resistance in ovarian cancer.
Recent studies found that p62 inhibition significantly suppressed activation of the NF-κB pathway [47]. We found that NF-κB signaling was activated in SKOV3/DDP cisplatin-resistant ovarian cancer cells. Inhibition of p62 by RNAi significantly reduced the translocation of p50/p65 into the nucleus and inhibited the transcriptional activity of NF-κB, which suggests that p62 may control cisplatin resistance in ovarian cancer cells by regulating NF-κB signaling [31]. Recent studies showed that TRAF6 and receptor-interacting protein 1 (RIP1) are both involved in p62-related NF-κB activation.
The TRAF6/NF-κB pathway was originally identified in immune cells [48]. The E3 ubiquitin ligase TRAF6 enhances the ubiquitination of IKKβ and the phosphorylation and degradation of IκB, leading to increased DNA-binding of NF-κB [49]. Moscat et al. demonstrated that RAS promoted the transcription of p62 through the ERK and PI3K pathways, which increase the oligomerization and polyubiquitination of TRAF6 and expressions of IKKα and IKKβ, the major upstream activators of the NF-κB pathway [17, 50, 51]. Another study showed that the p62 TB domain specifically binds to the TRAF domain in TRAF6, which results in autoubiquitination of TRAF6 [52]. Ubiquitination of TRAF6 was also inhibited in p62-knockout mice. Furthermore, a p62 UBA deletion mutant (F406V mutation) also inhibited the ubiquitination of TRAF6, which suggests that the C-terminal UBA domain of p62 is involved in TRAF6 regulation [53].
The RIP1 serine/threonine protein kinase is involved in the regulation of inflammatory signaling and various cell death pathways such as apoptosis and programmed necrosis [54, 55]. RIP1 function is tightly regulated by ubiquitination and deubiquitination. When cells are stimulated by tumor necrosis factor (TNF), TNF receptor 1 (TNFR1) forms trimers to recruit tumor necrosis factor receptor type 1-associated DEATH domain protein (TRADD), RIP1, E3 ubiquitin ligase TNFR-related factor 2 (TRAF2), cIAP1/2 (cellular inhibitor of apoptosis 1/2) as well as the linear ubiquitin chain assembly complex (LUBAC). RIP1 is rapidly polyubiquitinated with Lys63-linked and linear Met1-linked ubiquitin chains and activates TGFβ-activated kinase 1 (TAK1) and the IKK complex. Phosphorylated IκB is degraded by the ubiquitin–proteasome system, resulting in activation of NF-κB [56,57,58]. p62 directly binds RIP1 but not TRAF2, and the 117–439 residues in p62, which include the ZZ domain, are essential for binding to RIP1 [59]. We found that cisplatin activated the NF-κB pathway and increased K63-linked ubiquitination of RIP1 in SKOV3/DDP cells. Abolishing the regulation of RIP1 K63-linked ubiquitination by p62 increased the sensitivity of SKOV3 cells to cisplatin; deleting the ZZ domain (RIP1 interacting region) in p62 markedly decreased K63-linked ubiquitination of RIP1 in SKOV3 cells and inhibited NF-κB signaling activation [31]. These results suggested that the ZZ domain of p62 not only directly binds RIP1, but also participates in the regulation of RIP1 ubiquitination. NF-κB signaling regulates at least 400 genes encoding proteins involved in proliferation, apoptosis and inflammation [60]. When we blocked NF-κB signaling using p62 inhibition in ovarian cancer cells treated with cisplatin, the expressions of proliferation-related genes such as CCL2, IL6, TGFb and CSF3 genes were significantly suppressed and DNA synthesis was reduced [31].
Keap1/Nrf2 signaling
Mitochondria are the main location of cellular reactive oxygen species (ROS) production. Previous studies showed that cisplatin enters into cells and directly binds to mitochondria, leading to cytochrome C release, calcium-dependent mitochondrial swelling and production of ROS, which induces oxidative stress and reduces genomic stability [12, 61]. The antioxidant pathway and glutathione (GSH) are the main ways to clear ROS [62]. Current studies have shown that the Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (Nrf2) pathway is one of the important antioxidant pathways. Under basal conditions, Keap1 interacts with Nrf2 and mediates its degradation. Exposure to electrophilic reagents (such as cisplatin) causes Keap1-Nrf2 complex disruption, leading to Nrf2 translocation into the nucleus to promote the expression of downstream antioxidant genes such as NQO1 and Hmox1 genes [63]. NRF2 also controls the gene transcription of several important enzymes involved in GSH synthesis and oxidation, maintaining the mitochondrial GSH pool [64]. The genes encoding ATP-binding cassette (ABC) transporters (known as efflux pumps) ABCC2 and ABCF2, which transport molecules across cellular membranes, are also target genes of NRF2 [65, 66]. Bao et al. [72] showed that Nrf2 knockdown inhibited the expression of ABCF2 and increased the sensitivity of ovarian cancer cells to cisplatin. Wu et al. [67] found that Nrf2 is highly expressed in A2780/DDP and COC1/DDP cisplatin-resistant ovarian cancer cells; inhibition of Nrf2 translocation into the nucleus significantly increased the gene expression of transferrin SLC401, a known iron exporter, which reversed the cisplatin resistance of ovarian cells caused by iron overload. These studies confirmed that the Keap1-Nrf2 pathway is involved in the cisplatin resistance mechanism of ovarian cancer cells. Some reports showed that Nrf2 interacts with the p62 promoter, which indicates that p62 may also be a target gene of Nrf2 [68]. Jena et al. [69] found that the E3 ubiquitin-protein ligase TRIM16 promotes p62-mediated autophagy degradation of ubiquitinated proteins by up-regulating Nrf2 under oxidative stress.
Recent studies have also confirmed that p62 is involved in regulation of the Keap1-Nrf2 pathway. Phosphorylation of p62 at serine 349 in the KIR domain increased its binding affinity to Keap1 and promoted Nrf2 activation [70]. Consistent with this data, other studies showed that Keap1/Cullin3 mediates p62 ubiquitination and increases the sequestration activity of Keap1, resulting in Nrf2 activation through non-canonical pathways [34]. We observed that SKOV3/DDP cells produced less ROS compared with SKOV3 cells upon cisplatin treatment, which suggests that cisplatin-resistant cells may have stronger antioxidant capacity. Furthermore, co-localization of p62 and Keap1 was observed in SKOV3/DDP cells and inhibition of p62 expression significantly attenuated the transcriptional activity of Nrf2 [71]. These results indicated that highly expressed p62 in SKOV3/DDP cells may protect ovarian cancer cells from oxidative damage caused by cisplatin by competing with Nrf2 for binding to Keap1.
Stępkowski et al. [72] showed that p62 plays an important role in apoptosis and autophagy by integrating the Keap1-Nrf2 and NF-κB signaling pathways. First, p62 promotes activation of the NF-κB signaling pathway; however, increased binding of p62 to Keap1 leads to release of Nrf2 from Keap1, which may affect the degradation of IKKβ by Keap1, resulting in NF-κB activation. This indicates a complicated role for p62 in pro-survival signal regulation. Additionally, PGAM5 (phosphoglycerate mutase family member 5), which is located in mitochondria and is a chaperone of Keap1, functions in apoptosis, programmed necrosis, and mitophagy. Mealey et al. [73] found that Keap1-Nrf2 interacted with PGAM5, and this complex contributed to mitochondrial retrograde trafficking. Furthermore, co-depleting p62 and Nrf2 inhibited mitochondrial clustering induced by the proteasome inhibitor MG132, which indicates that p62 may be involved in mitochondrial dynamics through Keap1-Nrf2 signaling regulation (Fig. 3). Together these findings indicate that investigating the role of p62 in the pro-survival signaling crosstalk may be a promising approach to develop strategies to overcome cisplatin resistance in ovarian cancer.

Pro-survival signaling regulation by p62 in ovarian cancer cells. a Highly expressed p62 activates NF-κB through RIP1 and TRAF6. p62 also competes with Nrf2 for binding to Keap1, which promotes the transcriptional activity of Nrf2. b p62 is recruited to function in PINK1/Parkin-mediated mitophagy; p62 may also be involved in regulation of the PGAM5-Keap1-Nrf2 complex, which is responsible for mitochondrial dynamics
Death signals recruited by p62 are involved in cisplatin resistance of ovarian cancer
Cisplatin causes cell death by activating apoptosis and programmed necrosis [74, 75]. Annunziata et al. found that ovarian cancer patients with tumors expressing low levels of the pro-apoptotic molecule caspase 8 showed shorter overall survival compared with those with high caspase 8 expression [76, 77]. Furthermore, another report demonstrated that p62 regulates caspase 8 activation induced by TNF [78]. Huang et al. [79] found that p62 accumulation caused by autophagy inhibitors chloroquine and bortezomib promoted the activity of caspase 8 in human colon carcinoma cells. Wang et al. [80] showed that caspase 8 activation may be regulated by intracellular death-inducing signaling complex (iDISC) on autophagosomal membranes. Furthermore, inhibition of ATG5 (autophagy-related gene 5) suppressed the formation of the autophagy membrane and reduced caspase 8 activation. Iurlaro proposed that persistent endoplasmic reticulum stress may promote the formation of iDISC, the autophagosome-associated platform, to activate caspase 8 [81]. Our group demonstrated that the combination of the autophagy inhibitor chloroquine and cisplatin significantly inhibited ovarian cancer growth in vivo, with accumulated p62 in tumor tissue and activated caspase 8. We previously found that deleting the p62 UBA domain, which is responsible for ubiquitin binding and self-oligomerization [82], inhibited cisplatin-induced caspase 8 activation, leading to chemoresistance in ovarian cancer cells. Further studies showed that the L417V mutation in the UBA domain, which reduced ubiquitin-binding activity, also suppressed caspase 8 activation and the co-localization with LC3 induced by cisplatin, which suggests that p62-mediated autophagy may participate in cisplatin resistance through caspase 8 regulation in ovarian cancer [33].
Cellular FLICE-inhibitory protein (cFLIP) is structurally similar to caspase 8 and forms a dimer with caspase 8 to inhibit its activity. cFLIP is highly expressed in ovarian cancer [83, 84]. Li et al. found that cFLIP knockdown significantly increased TRAIL-induced apoptosis in SKOV3 cells. Nazim et al. showed that increased autophagy flux inhibited the expression of cFLIP and enhanced TRAIL-induced apoptosis [85, 86]. Further studies are needed to clarify the role of cFLIP in p62-mediated caspase 8 activation in ovarian cancer with cisplatin treatment.
In the presence of RIP3, cisplatin induces formation of the necrosome containing the RIP1/RIP3/MLKL complex as core, which initiates programmed necrosis via ROS production [87]. Liu et al. [88] showed that the accumulation of p62 caused by autophagy inhibition promoted the formation of necrosomes. A recent study indicated that p62 induced programmed necrosis by recruiting RIP1 to assemble RIP3/MLKL on the autophagosome membrane in mouse prostate cells lacking Map3k7 [89]. These results suggest that p62 also works as a switch to determine the transition between apoptosis and necrosis. Targeting p62 to promote necrosis may be an alternative therapeutic strategy in ovarian cancers resistant to cisplatin-mediated apoptosis (Fig. 4).

Death signals recruited by p62 in ovarian cancer cells. p62 induces apoptosis and programmed necrosis by recruiting pro-death partners on the autophagosome membrane while blocking autophagy flux
Conclusion and future perspectives
While early studies suggested that DNA was the main target of cisplatin [90], later reports demonstrated that cisplatin also triggers multiple changes in signals involved in proliferation, apoptosis and anti-oxidation by binding to macromolecular proteins and organelles [12]. In this review, we summarize our current understanding about the role of p62 in the mechanisms of cisplatin resistance in ovarian cancer cells.
p62 functions as a key receptor for autophagy. Increasing studies have demonstrated that autophagy is involved in chemotherapy resistance and several compounds have been identified that regulate autophagy in ovarian cancer (Table 2). We suggest that highly expressed p62 in ovarian cancer cells not only mediates selective autophagy to degrade excessive accumulated ubiquitinated proteins, but also exerts functions beyond autophagy. p62 also activates NF-κB by enhancing K63-linked ubiquitination of RIP1, promotes Nrf2 nucleus translocation to counteract oxidative damage caused by cisplatin by interacting with Keap1 and initiates cell death signals from autophagy flux blockage. These functions may explain why an autophagy inhibitor increased the efficacy of cisplatin in ovarian cancer cells. Together these studies indicate that p62 is involved in cisplatin resistant mechanisms by operating as a signal hub that regulates critical proteins in key signaling pathways that determine cell survival and death (Table 3).
Notably, clarifying how p62 functions in chemotherapy may provide benefits for ovarian cancer clinical diagnosis and prognosis. Most ovarian cancer patients treated with cisplatin eventually develop resistance and show poor outcome. Iwadate examined p62 expression in tumor tissues of 266 patients with primary ovarian cancer and found that patients with high expression of p62 had poor prognosis [99]. Another study found that ovarian cancer patients with high expression of p62 had longer survival [100]. This paradox indicates that the prognosis of ovarian cancer patients may not be easily defined by p62 expression alone. Our study showed that patients with high expression of p62 and caspase 8 had longer survival and were negatively correlated with tumor-node-metastasis stage and relapse risk compared with patients with high p62 and low caspase 8 expression [33]. Considering the role of p62 in caspase 8 activation mentioned above, these findings suggest that patients with both overexpressed p62 and caspase 8 may be more sensitive to platinum-based chemotherapy; autophagy inhibitors may increase the sensitivity to cisplatin in patients with high p62 expression but low caspase 8 expression. These findings suggest that evaluation of p62 and its effector molecules could increase the accuracy of prognosis and optimize therapeutic strategies in ovarian cancer.
Several questions remain to be answered. For example, the role of accumulated p62 caused by autophagy blockage in NF-κB or Keap-1/Nrf2 signaling activation is unclear. Furthermore, the function of accumulated p62 induced by increasing transcription or degradation suppression is also unknown. Future studies should focus on alterations in the binding partners of p62 and the post-translational modifications that enhance p62 functions in ovarian cancer cells with cisplatin treatment. Together these findings may provide new strategies to overcome cisplatin resistance in ovarian cancer by targeting p62.
Availability of data and materials
Not applicable
Abbreviations
- ABC transporters:
-
ATP-binding cassette transporters
- ABCC2:
-
ATP binding cassette subfamily C member 2
- ABCF2:
-
ATP binding cassette subfamily F member 2
- aPKC:
-
Atypical kinase
- ATG5:
-
Autophagy-related gene 5
- BCL2:
-
B cell lymphoma 2
- CCL2:
-
CC-chemokine Ligand 2
- cFLIP:
-
Cellular FLICE-inhibitory protein
- cIAP:
-
Cellular inhibitor of apoptosis
- Cisplatin:
-
Cis-diamminedichloroplatinum
- CK2:
-
Casein kinase 2
- CXCL8:
-
C-X-C motif chemokine ligand 8
- DISC:
-
Death-inducing signaling complex
- DR:
-
Death receptor
- GSH:
-
Glutathione
- iDISC:
-
Intracellular death-inducing signaling complex
- IKK:
-
IκB kinase
- IL6:
-
Interleukin 6
- Keap1:
-
Kelch-like ECH-associated protein 1
- KIR:
-
Keap1-interacting region
- LIR:
-
LC3-interacting region
- MAP3K7:
-
Mitogen-activated protein kinase kinase kinase 7
- MAPK9:
-
Mitogen-activated protein kinase 9
- MLKL:
-
Mixed lineage kinase domain-like protein
- mPTP:
-
Mitochondrial permeability transition pore
- NQO1:
-
NAD (P) H quinone dehydrogenase 1
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- PINK1:
-
PTEN-induced putative kinase protein 1
- RIP1:
-
Receptor-interacting protein 1
- SLC401:
-
Solute carrier 401
- TAK1:
-
TGFβ-activated kinase 1
- TGF:
-
Transforming growth factor
- TNF:
-
Tumor necrosis factor
- TOM:
-
Translocase outer membrane
- TRADD:
-
Tumor necrosis factor receptor type 1-associated DEATH domain protein
- TRAF6:
-
TNF receptor associated factor 6
- TRIM52:
-
The tripartite motif 52
- UBA:
-
Ubiquitin-associated
- VDAC:
-
Voltage-dependent anion channels
- ZZ:
-
Zinc finger
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66(1):7–30.
Rosenberg B, Vancamp L, Krigas T. Inhibition of cell division in Escherichia Coli by electrolysis products from a platinum electrode. Nature. 1965;205:698–9.
Peng H, Jin H, Zhuo H, Huang H. Enhanced antitumor efficacy of cisplatin for treating ovarian cancer in vitro and in vivo via transferrin binding. Oncotarget. 2017;8(28):45597–611.
Pfisterer J, Vergote I, Du Bois A, Eisenhauer E, Ago O, Ncic CTG, Eortc GCG. Combination therapy with gemcitabine and carboplatin in recurrent ovarian cancer. Int J Gynecol Cancer. 2005;15(Suppl 1):36–41.
Ozols RF, Bundy BN, Greer BE, Fowler JM, Clarke-Pearson D, Burger RA, Mannel RS, DeGeest K, Hartenbach EM, Baergen R, et al. Phase III trial of carboplatin and paclitaxel compared with cisplatin and paclitaxel in patients with optimally resected stage III ovarian cancer: a Gynecologic Oncology Group study. J Clin Oncol. 2003;21(17):3194–200.
Aebi S, Castiglione M, Group EGW. Newly and relapsed epithelial ovarian carcinoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009;20(Suppl 4):21–3.
McGuire WP, Hoskins WJ, Brady MF, Kucera PR, Partridge EE, Look KY, Clarke-Pearson DL, Davidson M. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6.
Gonzalez-Martin A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR, McCormick C, Lorusso D, Hoskins P, Freyer G, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–402.
Bible KC, Peethambaram PP, Oberg AL, Maples W, Groteluschen DL, Boente M, Burton JK, Gomez Dahl LC, Tibodeau JD, Isham CR, et al. A phase 2 trial of flavopiridol (Alvocidib) and cisplatin in platin-resistant ovarian and primary peritoneal carcinoma: MC0261. Gynecol Oncol. 2012;127(1):55–62.
Melnikov SV, Soll D, Steitz TA, Polikanov YS. Insights into RNA binding by the anticancer drug cisplatin from the crystal structure of cisplatin-modified ribosome. Nucleic Acids Res. 2016;44(10):4978–87.
Russo Krauss I, Ferraro G, Merlino A. Cisplatin-protein interactions: unexpected drug binding to N-terminal amine and lysine side chains. Inorg Chem. 2016;55(16):7814–6.
Gatti L, Cassinelli G, Zaffaroni N, Lanzi C, Perego P. New mechanisms for old drugs: insights into DNA-unrelated effects of platinum compounds and drug resistance determinants. Drug Resist Updat. 2015;20:1–11.
Galluzzi L, Vitale I, Michels J, Brenner C, Szabadkai G, Harel-Bellan A, Castedo M, Kroemer G. Systems biology of cisplatin resistance: past, present and future. Cell Death Dis. 2014;5:e1257.
Thakur B, Ray P. Cisplatin triggers cancer stem cell enrichment in platinum-resistant cells through NF-kappaB-TNFalpha-PIK3CA loop. J Exp Clin Cancer Res. 2017;36(1):164.
Shen DW, Pouliot LM, Hall MD, Gottesman MM. Cisplatin resistance: a cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol Rev. 2012;64(3):706–21.
Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, Hansen M, Moscat J, Diaz-Meco MT. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. 2011;44(1):134–46.
Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–4.
Reina-Campos M, Shelton PM, Diaz-Meco MT, Moscat J. Metabolic reprogramming of the tumor microenvironment by p62 and its partners. Biochem Biophys Acta. 2018;1870(1):88–95.
Ju LL, Zhao CY, Ye KF, Yang H, Zhang J. Expression and clinical implication of Beclin1, HMGB1, p62, survivin, BRCA1 and ERCC1 in epithelial ovarian tumor tissues. Eur Rev Med Pharmacol Sci. 2016;20(10):1993–2003.
Sanchez-Martin P, Saito T, Komatsu M. p62/SQSTM1: ‘Jack of all trades’ in health and cancer. FEBS J. 2019;286(1):8–23.
Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61(6):609–24.
He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Ann Rev Genet. 2009;43:67–93.
Galluzzi L, Pietrocola F, Bravo-San Pedro JM, Amaravadi RK, Baehrecke EH, Cecconi F, Codogno P, Debnath J, Gewirtz DA, Karantza V, et al. Autophagy in malignant transformation and cancer progression. EMBO J. 2015;34(7):856–80.
Zhang XY, Zhang M, Cong Q, Zhang MX, Zhang MY, Lu YY, Xu CJ. Hexokinase 2 confers resistance to cisplatin in ovarian cancer cells by enhancing cisplatin-induced autophagy. Int J Biochem Cell Biol. 2018;95:9–16.
Periyasamy-Thandavan S, Jiang M, Schoenlein P, Dong Z. Autophagy: molecular machinery, regulation, and implications for renal pathophysiology. Am J Physiol Renal Physiol. 2009;297(2):F244–56.
Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011;44(2):279–89.
Geetha T, Wooten MW. Structure and functional properties of the ubiquitin binding protein p62. FEBS Lett. 2002;512(1–3):19–24.
Johansen T, Lamark T. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011;7(3):279–96.
Wurzer B, Zaffagnini G, Fracchiolla D, Turco E, Abert C, Romanov J, Martens S. Oligomerization of p62 allows for selection of ubiquitinated cargo and isolation membrane during selective autophagy. Elife. 2015;4:e08941.
Ma L, Xu Y, Su J, Yu H, Kang J, Li H, Li X, Xie Q, Yu C, Sun L, et al. Autophagic flux promotes cisplatin resistance in human ovarian carcinoma cells through ATP-mediated lysosomal function. Int J Oncol. 2015;47(5):1890–900.
Yan XY, Zhang Y, Zhang JJ, Zhang LC, Liu YN, Wu Y, Xue YN, Lu SY, Su J, Sun LK. p62/SQSTM1 as an oncotarget mediates cisplatin resistance through activating RIP1-NF-kappaB pathway in human ovarian cancer cells. Cancer Sci. 2017;108(7):1405–13.
Cha-Molstad H, Yu JE, Feng Z, Lee SH, Kim JG, Yang P, Han B, Sung KW, Yoo YD, Hwang J, et al. p62/SQSTM1/Sequestosome-1 is an N-recognin of the N-end rule pathway which modulates autophagosome biogenesis. Nat Commun. 2017;8(1):102.
Yan XY, Zhong XR, Yu SH, Zhang LC, Liu YN, Zhang Y, Sun LK, Su J. p62 aggregates mediated Caspase 8 activation is responsible for progression of ovarian cancer. J Cell Mol Med. 2019;23(6):4030–42.
Lee Y, Chou TF, Pittman SK, Keith AL, Razani B, Weihl CC. Keap1/Cullin3 modulates p62/SQSTM1 activity via UBA domain ubiquitination. Cell Rep. 2017;20(8):1994.
Carroll B, Otten EG, Manni D, Stefanatos R, Menzies FM, Smith GR, Jurk D, Kenneth N, Wilkinson S, Passos JF, et al. Oxidation of SQSTM1/p62 mediates the link between redox state and protein homeostasis. Nat Commun. 2018;9(1):256.
Kingnate C, Charoenkwan K, Kumfu S, Chattipakorn N, Chattipakorn SC. Possible roles of mitochondrial dynamics and the effects of pharmacological interventions in chemoresistant ovarian cancer. EBioMedicine. 2018;34:256–66.
Williams JA, Ding WX. Mechanisms, pathophysiological roles and methods for analyzing mitophagy—recent insights. Biol Chem. 2018;399(2):147–78.
Sekine S, Youle RJ. PINK1 import regulation; a fine system to convey mitochondrial stress to the cytosol. BMC Biol. 2018;16(1):2.
Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y, Ishihara N, et al. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem. 2012;287(41):34635–45.
Seibenhener ML, Du Y, Diaz-Meco MT, Moscat J, Wooten MC, Wooten MW. A role for sequestosome 1/p62 in mitochondrial dynamics, import and genome integrity. Biochim Biophys Acta. 2013;1833(3):452–9.
Cullen KJ, Yang Z, Schumaker L, Guo Z. Mitochondria as a critical target of the chemotheraputic agent cisplatin in head and neck cancer. J Bioenergy Biomembr. 2007;39(1):43–50.
Ponnalagu D, Singh H. Anion channels of mitochondria. Handb Exp Pharmacol. 2017;240:71–101.
Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol. 2010;12(2):119–31.
Yang W, Liu L, Li C, Luo N, Chen R, Li L, Yu F, Cheng Z. TRIM52 plays an oncogenic role in ovarian cancer associated with NF-kB pathway. Cell Death Dis. 2018;9(9):908.
Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Saito M, Kawagoe J, Takahashi K, Yada-Hashimoto N, et al. Inhibition of NFkappaB increases the efficacy of cisplatin in in vitro and in vivo ovarian cancer models. J Biol Chem. 2004;279(22):23477–85.
Mabuchi S, Ohmichi M, Nishio Y, Hayasaka T, Kimura A, Ohta T, Kawagoe J, Takahashi K, Yada-Hashimoto N, Seino-Noda H, et al. Inhibition of inhibitor of nuclear factor-kappaB phosphorylation increases the efficacy of paclitaxel in in vitro and in vivo ovarian cancer models. Clin Cancer Res. 2004;10(22):7645–54.
Wei H, Wang C, Croce CM, Guan JL. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014;28(11):1204–16.
Martin P, Moscat J. Th1/Th2 differentiation and B cell function by the atypical PKCs and their regulators. Front Immunol. 2012;3:241.
Meads MB, Li ZW, Dalton WS. A novel TNF receptor-associated factor 6 binding domain mediates NF-kappa B signaling by the common cytokine receptor beta subunit. J Immunol. 2010;185(3):1606–15.
Moscat J, Karin M, Diaz-Meco MT. p62 in cancer: signaling adaptor beyond autophagy. Cell. 2016;167(3):606–9.
Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, Moscat J. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13(4):343–54.
Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19(7):1576–86.
Wooten MW, Geetha T, Seibenhener ML, Babu JR, Diaz-Meco MT, Moscat J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J Biol Chem. 2005;280(42):35625–9.
Ofengeim D, Yuan J. Regulation of RIP1 kinase signalling at the crossroads of inflammation and cell death. Nat Rev Mol Cell Biol. 2013;14(11):727–36.
Liu XY, Lai F, Yan XG, Jiang CC, Guo ST, Wang CY, Croft A, Tseng HY, Wilmott JS, Scolyer RA, et al. RIP1 kinase is an oncogenic driver in melanoma. Cancer Res. 2015;75(8):1736–48.
Li H, Kobayashi M, Blonska M, You Y, Lin X. Ubiquitination of RIP is required for tumor necrosis factor alpha-induced NF-kappaB activation. J Biol Chem. 2006;281(19):13636–43.
Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, Cohen P. Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci USA. 2013;110(38):15247–52.
Christofferson DE, Li Y, Hitomi J, Zhou W, Upperman C, Zhu H, Gerber SA, Gygi S, Yuan J. A novel role for RIP1 kinase in mediating TNFalpha production. Cell Death Dis. 2012;3:e320.
Sanz L, Sanchez P, Lallena MJ, Diaz-Meco MT, Moscat J. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18(11):3044–53.
Cai PC, Shi L, Liu VW, Tang HW, Liu IJ, Leung TH, Chan KK, Yam JW, Yao KM, Ngan HY, et al. Elevated TAK1 augments tumor growth and metastatic capacities of ovarian cancer cells through activation of NF-kappaB signaling. Oncotarget. 2014;5(17):7549–62.
Wangpaichitr M, Wu C, Li YY, Nguyen DJM, Kandemir H, Shah S, Chen S, Feun LG, Prince JS, Kuo MT, et al. Exploiting ROS and metabolic differences to kill cisplatin resistant lung cancer. Oncotarget. 2017;8(30):49275–92.
Roh JL, Jang H, Kim EH, Shin D. Targeting of the glutathione, thioredoxin, and Nrf2 antioxidant systems in head and neck cancer. Antioxid Redox Signal. 2017;27(2):106–14.
Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45–9.
Ryoo IG, Kwak MK. Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol Appl Pharmacol. 2018;359:24–33.
Canet MJ, Merrell MD, Harder BG, Maher JM, Wu T, Lickteig AJ, Jackson JP, Zhang DD, Yamamoto M, Cherrington NJ. Identification of a functional antioxidant response element within the eighth intron of the human ABCC3 gene. Drug Metab Dispos. 2015;43(1):93–9.
Bao L, Wu J, Dodson M, Rojo de la Vega EM, Ning Y, Zhang Z, Yao M, Zhang DD, Xu C, Yi X. ABCF2, an Nrf2 target gene, contributes to cisplatin resistance in ovarian cancer cells. Mol Carcinog. 2017;56(6):1543–53.
Wu J, Bao L, Zhang Z, Yi X. Nrf2 induces cisplatin resistance via suppressing the iron export related gene SLC40A1 in ovarian cancer cells. Oncotarget. 2017;8(55):93502–15.
Jain A, Lamark T, Sjottem E, Larsen KB, Awuh JA, Overvatn A, McMahon M, Hayes JD, Johansen T. p62/SQSTM1 is a target gene for transcription factor NRF2 and creates a positive feedback loop by inducing antioxidant response element-driven gene transcription. J Biol Chem. 2010;285(29):22576–91.
Jena KK, Kolapalli SP, Mehto S, Nath P, Das B, Sahoo PK, Ahad A, Syed GH, Raghav SK, Senapati S, et al. TRIM16 controls assembly and degradation of protein aggregates by modulating the p62-NRF2 axis and autophagy. EMBO J. 2018;37(18):e98358.
Ichimura Y, Komatsu M. Activation of p62/SQSTM1-Keap1-nuclear factor erythroid 2-related factor 2 pathway in cancer. Front Oncol. 2018;8:210.
Xia M, Yu H, Gu S, Xu Y, Su J, Li H, Kang J, Cui M. p62/SQSTM1 is involved in cisplatin resistance in human ovarian cancer cells via the Keap1-Nrf2-ARE system. Int J Oncol. 2014;45(6):2341–8.
Stepkowski TM, Kruszewski MK. Molecular cross-talk between the NRF2/KEAP1 signaling pathway, autophagy, and apoptosis. Free Radic Biol Med. 2011;50(9):1186–95.
O’Mealey GB, Plafker KS, Berry WL, Janknecht R, Chan JY, Plafker SM. A PGAM5-KEAP1-Nrf2 complex is required for stress-induced mitochondrial retrograde trafficking. J Cell Sci. 2017;130(20):3467–80.
Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol. 2014;740:364–78.
Koo GB, Morgan MJ, Lee DG, Kim WJ, Yoon JH, Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al. Methylation-dependent loss of RIP3 expression in cancer represses programmed necrosis in response to chemotherapeutics. Cell Res. 2015;25(6):707–25.
Kim M, Hernandez L, Annunziata CM. Caspase 8 expression may determine the survival of women with ovarian cancer. Cell Death Dis. 2016;7:e2045.
Hernandez L, Kim MK, Noonan AM, Sagher E, Kohlhammer H, Wright G, Lyle LT, Steeg PS, Anver M, Bowtell DD, et al. A dual role for Caspase8 and NF-kappaB interactions in regulating apoptosis and necroptosis of ovarian cancer, with correlation to patient survival. Cell Death Discov. 2015;1:15053.
Jin Z, Li Y, Pitti R, Lawrence D, Pham VC, Lill JR, Ashkenazi A. Cullin3-based polyubiquitination and p62-dependent aggregation of caspase-8 mediate extrinsic apoptosis signaling. Cell. 2009;137(4):721–35.
Huang S, Okamoto K, Yu C, Sinicrope FA. p62/sequestosome-1 up-regulation promotes ABT-263-induced caspase-8 aggregation/activation on the autophagosome. J Biol Chem. 2013;288(47):33654–66.
Young MM, Takahashi Y, Khan O, Park S, Hori T, Yun J, Sharma AK, Amin S, Hu CD, Zhang J, et al. Autophagosomal membrane serves as platform for intracellular death-inducing signaling complex (iDISC)-mediated caspase-8 activation and apoptosis. J Biol Chem. 2012;287(15):12455–68.
Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016;283(14):2640–52.
Peng H, Yang J, Li G, You Q, Han W, Li T, Gao D, Xie X, Lee BH, Du J, et al. Ubiquitylation of p62/sequestosome1 activates its autophagy receptor function and controls selective autophagy upon ubiquitin stress. Cell Res. 2017;27(5):657–74.
El-Gazzar A, Wittinger M, Perco P, Anees M, Horvat R, Mikulits W, Grunt TW, Mayer B, Krainer M. The role of c-FLIP(L) in ovarian cancer: chaperoning tumor cells from immunosurveillance and increasing their invasive potential. Gynecol Oncol. 2010;117(3):451–9.
Clarke P, Tyler KL. Down-regulation of cFLIP following reovirus infection sensitizes human ovarian cancer cells to TRAIL-induced apoptosis. Apoptosis. 2007;12(1):211–23.
Li LC, Jayaram S, Ganesh L, Qian L, Rotmensch J, Maker AV, Prabhakar BS. Knockdown of MADD and c-FLIP overcomes resistance to TRAIL-induced apoptosis in ovarian cancer cells. Am J Obstet Gynecol. 2011;205(4):362 e312-325.
Nazim UM, Moon JH, Lee JH, Lee YJ, Seol JW, Eo SK, Lee JH, Park SY. Activation of autophagy flux by metformin downregulates cellular FLICE-like inhibitory protein and enhances TRAIL—induced apoptosis. Oncotarget. 2016;7(17):23468–81.
Xu Y, Ma HB, Fang YL, Zhang ZR, Shao J, Hong M, Huang CJ, Liu J, Chen RQ. Cisplatin-induced necroptosis in TNFalpha dependent and independent pathways. Cell Signal. 2017;31:112–23.
Liu S, Li Y, Choi HMC, Sarkar C, Koh EY, Wu J, Lipinski MM. Lysosomal damage after spinal cord injury causes accumulation of RIPK1 and RIPK3 proteins and potentiation of necroptosis. Cell Death Dis. 2018;9(5):476.
Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, Green DR, Morgan M, Cramer SD, Thorburn A. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Devel Cell. 2016;37(4):337–49.
Pilie PG, Tang C, Mills GB, Yap TA. State-of-the-art strategies for targeting the DNA damage response in cancer. Nature reviews Clinical oncology. 2019;16(2):81–104.
Gao L, Wang Z, Lu D, Huang J, Liu J, Hong L. Paeonol induces cytoprotective autophagy via blocking the Akt/mTOR pathway in ovarian cancer cells. Cell Death Dis. 2019;10(8):609.
Liu B, Huang X, Li Y, Liao W, Li M, Liu Y, He R, Feng D, Zhu R, Kurihara H. JS-K, a nitric oxide donor, induces autophagy as a complementary mechanism inhibiting ovarian cancer. BMC Cancer. 2019;19(1):645.
Zhang X, Hou G, Liu A, Xu H, Guan Y, Wu Y, Deng J, Cao X. Matrine inhibits the development and progression of ovarian cancer by repressing cancer associated phosphorylation signaling pathways. Cell Death Dis. 2019;10(10):770.
Bhattacharjee A, Hasanain M, Kathuria M, Singh A, Datta D, Sarkar J, Mitra K. Ormeloxifene-induced unfolded protein response contributes to autophagy-associated apoptosis via disruption of Akt/mTOR and activation of JNK. Sci Rep. 2018;8(1):2303.
Young AN, Herrera D, Huntsman AC, Korkmaz MA, Lantvit DD, Mazumder S, Kolli S, Coss CC, King S, Wang H, et al. Phyllanthusmin derivatives induce apoptosis and reduce tumor burden in high-grade serous ovarian cancer by late-stage autophagy inhibition. Mol Cancer Ther. 2018;17(10):2123–35.
Li BX, Wang HB, Qiu MZ, Luo QY, Yi HJ, Yan XL, Pan WT, Yuan LP, Zhang YX, Xu JH, et al. Novel smac mimetic APG-1387 elicits ovarian cancer cell killing through TNF-alpha, ripoptosome and autophagy mediated cell death pathway. J Exp Clin Cancer Res. 2018;37(1):53.
Kao C, Chao A, Tsai CL, Chuang WC, Huang WP, Chen GC, Lin CY, Wang TH, Wang HS, Lai CH. Bortezomib enhances cancer cell death by blocking the autophagic flux through stimulating ERK phosphorylation. Cell Death Dis. 2014;5:e1510.
Mao J, Ma L, Shen Y, Zhu K, Zhang R, Xi W, Ruan Z, Luo C, Chen Z, Xi X, et al. Arsenic circumvents the gefitinib resistance by binding to P62 and mediating autophagic degradation of EGFR in non-small cell lung cancer. Cell Death Dis. 2018;9(10):963.
Iwadate R, Inoue J, Tsuda H, Takano M, Furuya K, Hirasawa A, Aoki D, Inazawa J. High expression of p62 protein is associated with poor prognosis and aggressive phenotypes in endometrial cancer. Am J Pathol. 2015;185(9):2523–33.
Wang J, Garbutt C, Ma H, Gao P, Hornicek FJ, Kan Q, Shi H, Duan Z. Expression and role of autophagy-associated p62 (SQSTM1) in multidrug resistant ovarian cancer. Gynecol Oncol. 2018;150(1):143–50.
Acknowledgements
We thank Liwen Bianji, Edanz Editing China (http://www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.
Funding
This work is supported by the National Natural Science Foundation of China (81672948, 81772794, 81572927); Jilin Provincial Industrial Innovation Project (2018C052-7); Jilin Provincial Research Foundation for the Development of Science and Technology Projects (20191004004TC, 20170623093-03TC).
Author information
Authors and Affiliations
Contributions
Conceptualization, LKS and JS; writing—original draft preparation, LX, RT and XRZ; visualization, SHY; writing—review and editing, XYY and XZQ. All the authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Yan, XY., Qu, XZ., Xu, L. et al. Insight into the role of p62 in the cisplatin resistant mechanisms of ovarian cancer. Cancer Cell Int 20, 128 (2020). https://doi.org/10.1186/s12935-020-01196-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12935-020-01196-w