
Abstract
Background
Inclusion bodies (IBs) are protein aggregates in recombinant bacterial cells containing mainly the target recombinant protein. Although it has been shown that IBs contain functional proteins along with protein aggregates, their direct application as pharmaceuticals is hindered by their heterogeneity and hazardous contaminants with bacterial origin. Therefore, together with the production of soluble species, IBs remain the main source for manufacture of recombinant proteins with medical application. The quality and composition of the IBs affect the refolding yield and further purification of the recombinant protein. The knowledge whether nucleic acids are genuine components or concomitant impurities of the IBs is a prerequisite for the understanding of the IBs formation and for development of optimized protocols for recombinant protein refolding and purification. IBs isolated from Escherichia coli overexpressing human interferon-gamma (hIFNγ), a protein with therapeutic application, were used as a model.
Results
IBs were isolated from E. coli LE392 cells transformed with a hIFNγ expressing plasmid under standard conditions and further purified by centrifugation on a sucrose cushion, followed by several steps of sonication and washings with non-denaturing concentrations of urea. The efficiency of the purification was estimated by SDS-PAGE gel electrophoresis and parallel microbiological testing for the presence of residual intact bacteria. Phenol/chloroform extraction showed that the highly purified IBs contain both DNA and RNA. The latter were studied by UV spectroscopy and agarose gel electrophoresis combined with enzymatic treatment and hybridization. DNA was observed as a diffuse fraction mainly in the range of 250 to 1000 bp. RNA isolated by TRIzol® also demonstrated a substantial molecular heterogeneity. Hybridization with 32P-labelled oligonucleotides showed that the IBs contain rRNA and are enriched of hIFNγ mRNA.
Conclusions
The results presented in this study indicate that the nucleic acids might be intrinsic components rather than co-precipitated impurities in the IBs. We assume that the nucleic acids are active participants in the aggregation of recombinant proteins and formation of the IBs that originate from the transcription and translation machinery of the microbial cell factory. Further studies are needed to ascertain this notion.
Similar content being viewed by others


Introduction
Escherichia coli is one of the most preferable microbial factory for production of recombinant proteins for research, diagnostics and medical use because of its easy, fast and cheap cultivation, well investigated genetics and physiology as well as for the availability of numerous tools for genetic manipulation [1 and references therein]. It is well known that the overexpression of eukaryotic proteins in E. coli often causes formation of inclusion bodies (IBs) [2], containing mainly improperly folded proteins [3]. For a long time, it has been believed that the aggregated proteins are immunogenic and partly or completely devoid of biological activity [4]. At present, increasing evidence show that IBs have amyloid-like structure and comprise aggregated as well as native folded proteins with preserved biological activity [2]. This fact, together with the mechanical stability and high porosity of the IBs has defined them as unconventional functional materials with a wide spectrum of applications in biotechnology and biomedicine [5,6,7]. Recently, first report towards in vitro preparation of tailored and chemically defined IBs with potential for clinical application was published [8]. In spite of these first steps, the large-scale clinical application of IBs is still hindered by their undefined heterogeneous composition and the presence of hazardous contaminants from the bacterial cell, especially endotoxins [9]. Thus IBs remain a main source for production of pure biologically active recombinant proteins for medical purposes that can be isolated upon cell disruption, solubilisation, subsequent refolding and purification [10]. Furthermore, to turn IBs aggregation into greatest use, new combinatorial approaches at each step was proposed [11].
Protein folding and IBs formation is extensively discussed in a number of reviews [1, 12,13,14,15]. For a long time, protein aggregation has been considered as a process driven by hydrophobic interactions between fully-denatured protein molecules, however, increasing evidence indicate that protein aggregates are composed of partially unfolded or misfolded proteins linked by unspecific hydrophobic interactions [14, 16, 17]. Therefore, aggregation seems to be a competitive reaction to folding, depending on specific folding behaviour and conditions [18]. It is influenced by various factors such as protein size, presence of specific hydrophobic compartments in the molecule, pI, protein abundance [19, 20], high local concentration of the polypeptide chains emerging from ribosomes [21], limited amount of bacterial chaperones and proteases, which affect either folding or degradation of the unfolded or misfolded polypeptides [22, 23].
The chemical composition of IBs still remains obscure. It varies in a broad range and depends on the properties of the specific recombinant protein, fermentation conditions, host genetic background, IBs purification procedures, etc. [18]. Apparently, the major component of the bacterial IBs is the target recombinant protein [24,25,26,27]. In addition, they can also contain various contaminants such as lipids, nucleic acids, endogenous cell proteins, chaperones, etc. [28]. Since chaperons assist in protein folding, they are the main cell components “controlling” protein aggregation [29]. Among them, the heat shock proteins IbpA and IbpB [30,31,32] and DnaK and GroEL [32, 33] have been found in bacterial IBs. There is evidence that the IBs contain also plasmid DNA [34], ribosomal RNA, RNA polymerase subunits [35, 36], ribosomal proteins L13 [36], L7 and L12 [24], elongation factor Tu [37], membrane proteins OmpF, OmpC, and OmpA [24], membrane phospholipids and cellular RNAs [38]. Based on these findings some authors assume that the protein aggregation in vivo occurs simultaneously with the protein synthesis [35, 36]. Taking into account that in most of these studies the IBs have not been precisely purified, Rinas and Bailey [24] suggest that the cellular ingredients found in IBs co-precipitate during their isolation.
The quality and composition of the IBs affect the refolding yield and further purification of the recombinant protein. It has been shown that fully denatured mammalian proteins show unusually high solubility in nucleic acid-free pure water [39]. Since the recombinant proteins that are produced as pharmaceuticals are mainly mammalian proteins, the presence of nucleic acids in their preparation is critical for their refolding because the nucleic acids actively participate in the protein aggregation process via direct electrostatic interactions with partially folded or unfolded proteins. The literature survey shows that most of the studies on E. coli IBs carried so far are focused on protein composition and mechanisms of aggregation, whereas the data concerning nucleic acids are scarce and vague. Bearing in mind that the recombinant proteins manufactured for medical applications should be free of nucleic acids, we have focussed in this study on the content and nature of nucleic acids in highly purified E. coli IBs. As a model in this study we use IBs isolated from E. coli LE392 overexpressing human interferon-gamma (hIFNγ).
Results
Purification of hIFNγ IBs
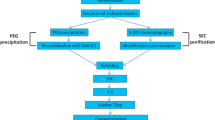
To study the type of nucleic acids co-aggregating with recombinant hIFNγ in E. coli cells we developed a three step procedure for purification of IBs from intact bacterial cells and subcellular components (Fig. 1). The first step included additional sonication of the crude IBs pellet followed by treatment with 100 μg/ml lysozyme. According to literature data, the combination of these two procedures should lead to IBs free from intact bacterial cells [40]. We observed, however, that substantial amount of viable E. coli cells still remained in the pellet (Fig. 1). The latter were successfully removed by centrifugation on a cushion of 20% sucrose that comprised the second step of the purification procedure. Further the IBs were collected by centrifugation of the supernatant at 13,000 rpm for 20 min at 4 °C. The pellet thus obtained contained negligible amount (sporadic) of bacterial cells (Fig. 1). During the third purification step the IBs were washed twice with washing buffer containing EDTA and non-denaturing concentrations of urea (1 M). Between the washing steps the IBs were sonicated and collected by centrifugation. This procedure proved to be essential for the quality and yield of the purified IBs since no bacterial growth was further observed (Fig. 1).

Schematic illustration showing the IBs purification steps and analysis for presence of viable bacterial cells
The IBs electrophoretic pattern was also used as a criterion for their purity. Figure 2 illustrates the significant difference in the protein composition of the IBs prepared by the standard procedure [41] and after the additional purification.

SDS PAGE electrophoretic pattern of hIFNγ IBs. 1—Purified hIFNγ obtained as described in [41] was used as a standard, the molecular weight of the monomer and the covalently bound dimer and tetramer are shown in kDa; 2—Crude IBs; 3—IBs after the last purification step 3
Characteristics of nucleic acid in purified hIFNγ IBs
Nucleic acids were isolated from purified IBs by phenol–chloroform extraction and precipitation with ethanol. Their concentration and UV spectra were analysed by NanoDrop®. The UV spectra showed absorption maxima at 260 nm and typical for the nucleic acids A260/280 and A260/230 ratios (Additional file 1: Fig. S1). The agarose electrophoretic pattern of the isolated total nucleic acids (Fig. 2a) revealed two main fractions—a high-molecular weight fraction (above 10,000 bp) and a more diffusive one in the range of 250 to 1000 bp.
One can speculate that the low mobility fraction (conditionally called high-molecular, Fig. 3a) consists of genomic or multimeric plasmid DNA, or a mixture of both. In order to investigate the origin of this fraction, samples were treated with the restriction endonucleases XhoI and HindIII. They were chosen because of the fact that the E. coli genome has multiple restriction sites for both enzymes, while no XhoI site exists in the expression plasmid pP1SD-hIFNγ. Therefore, any change in the mobility of the high-molecular fraction upon treatment with XhoI would mean that it consists of genomic DNA. Since the expression plasmid bears a unique HindIII site, the treatment with this enzyme would generate a distinguished single fraction from eventual plasmid concatemers.

a Agarose gel-electrophoresis of nucleic acids isolated from purified hIFNγ IBs. 1—Sample, isolated from IBs, b Agarose gel-electrophoresis of nucleic acids isolated from purified hIFNγ IBs treated with restriction endonuclease XhoI and c HindIII. 1—Non-treated sample; 2(b)—Sample treated with XhoI; 2(c)—sample treated with HindIII; d Enzymatic digestion of nucleic acids isolated from purified hIFNγ IBs 1—non treated sample; 2—sample treated with mixture of RNase A and RNase T1; 3—sample treated with DNase I; 4—sample treated with Proteinase K; M—Molecular weight marker, bp
As seen from Fig. 3b, c, the digestion with neither of the enzymes resulted in any significant change in the mobility of the high-molecular fraction. Its resistance to both restriction endonucleases suggested that this fraction might be composed of RNA rather than DNA. In order to check this assumption, samples were further treated with DNase I and a mixture of RNase A and RNase T1. In addition, to test for the presence of residual proteins that could form stable complexes with the nucleic acids, the sample was also treated with Proteinase K.
Figure 3d shows that the mixture of RNase A and RNase T1 resulted in a complete hydrolysis of the high-molecular fraction thus confirming that it consists of RNA fragments that under non-denatured electrophoresis conditions have preserved higher order structures and migrate slow. Surprisingly, DNase I degraded entirely the high mobility fraction (250–1000 bp). This means that it is composed of very heterogeneous in size DNA fragments. Proteinase K did not cause changes in the mobility of any electrophoretic fraction thus proving that the nucleic acids isolated from IBs were devoid of interfering proteins.
Characterization of RNA isolated from purified hIFNγ IBs
To characterize the RNA in highly purified IBs, RNA was isolated using TRIzol® under conditions recommended by the producer [42]. The obtained UV spectrum was typical for RNA and showed absorption maxima at 260 nm (Additional file 1: Fig. S2). The amount of the total RNA related to that of the IBs showed that its average content was 0.2 mg per 1 g wet purified IBs. The electrophoretic pattern of the obtained sample (Fig. 4a) showed that it consists of three main fractions—a slow-mobility one (high molecular mass), similar to the one isolated by the phenol–chloroform extraction (Fig. 3a) and two others corresponding to the bacterial 23S and 16S rRNA. The latter means that whole ribosomes have been entrapped at the time of recombinant hIFNγ aggregation and IBs formation.

a Agarose gel-electrophoresis of RNA, isolated from purified hIFNγ IBs. 1—RNA, isolated from purified hIFNγ IBs; 2—total RNA, isolated from non-transformed E. coli cells. b, c Dot-blot hybridization of RNA isolated from hIFNγ IBs. b RNA was dotted on nitrocellulose filters, hybridized with 32P-labelled oligonucleotide specific for hIFNγ mRNA, striped and c reprobed with oligonucleotide specific for the E. coli 16S rRNA. Total amount of RNA on the dots: A1 and A2—10 µg; B1 and B2—5 µg; C1 and C2—2.5 µg; D1 and D2—1 µg; A3—10 µg RNA pre-treated with DNase; B3—10 µg total RNA isolated from transformed E. coli cells; C3 and D3—10 µg total RNA isolated from non-transformed E. coli cells
To shed light on the origin of the RNA isolated from IBs, hybridization was carried out with two radiolabelled oligonucleotides of which one was complementary to the 5′ end of the hIFNγ mRNA and the second was complementary to the E. coli 16S rRNA. As shown in Fig. 4b, c, both oligonucleotides demonstrated specific hybridization with the RNA sample isolated from purified IBs. The hybridization signal in the DNase-treated sample (Fig. 4b, A3) proved that hIFNγ mRNA, but not plasmid DNA encoding the hIFNγ gene was present in the samples.
Discussion
Inclusion bodies formation in bacterial cells overexpressing eukaryotic proteins is a well-known phenomenon. Although the IBs are routinely used for isolation of recombinant proteins, systematic studies on their chemical composition and mechanism of their formation are sporadic and rare. There is no consensus on whether the IBs formation is a specific process affecting the recombinant protein only or it happens with the participation of other cellular components. Hartley and Kane [35] have identified ribosomal RNA, RNA polymerase subunits and various forms of plasmid DNA in bovine somatotropin IBs. Since the latter have been non-purified, it is not clear whether the observed substances are genuine components of the IBs or co-precipitated cellular impurities. Rinas and Bailey [24] found ribosomal proteins L7/L12 in TEM β-lactamase and β-galactosidase IBs, considering them as co-precipitated protein contaminants. In NMR studies Wasmer and co-workers [38] found RNA in HET-s(218–289) IBs washed 3 times with pure water that has not been further observed after additional purification. Other authors have detected nucleic acids in IBs, but they have regarded them as non-specific impurities rather than as their genuine components [24, 26]. Chaturvedi and co-workers [34] observed that the δ-endotoxin CryIAc exists in E. coli IBs in the form of tight complexes with chromosomal and plasmid DNA. However, this might be due to the presence of a large hydrophobic domain in the molecule of the recombinant δ-endotoxin or to a poor purification of the IBs.
To minimize the impurities in IBs, we developed an elaborated procedure for purification of hIFNγ IBs by introducing a centrifugation step on sucrose cushion followed by twofold ultrasonication and extensive washing between the latter steps with non-denaturing concentrations of urea. Literature data show that sonication of IBs destroys the non-covalent interactions between proteins and nucleic acids thus helping their separation by centrifugation [43]. Based on this study we believe that the extensive ultrasonication followed by washing is critical for the efficient removal of co-precipitated nucleic acids from the IBs. The bacterial growth test (see Fig. 1) shows that these additional steps improve also the disintegration of the residual bacterial cells in the IBs fraction. Our results are supported also by the results of Futami and co-authors [44] who extracted equal amounts of nucleic acids from extensively sonicated IBs and IBs treated with a mixture of DNase and RNase. Thus we assume that the nucleic acids found in highly purified hIFNγ IBs are their genuine components.
As mentioned above, exploring several recombinant proteins Futami and co-authors [44] have obtained DNA and RNA from IBs treated with nucleases and concluded that the inclusion bodies contain nucleic acids that are tightly bound to the expressed unfolded protein. They have suggested that the most important factor causing aggregates during the folding is the electrostatic interaction between unfolded protein and anionic contaminants such as nucleic acids. Our results support this conclusion. Furthermore, we characterized the nature of the RNA obtained from hIFNγ IBs as hIFNγ mRNA and E. coli ribosomal RNA. We explain this by the fact that the processes of replication, transcription and translation are not space and time segregated in bacteria, which allows immediate interactions between the expression plasmid DNA, mRNA, translating ribosomes and newly synthesized polypeptide chains (both completely and partially folded). This is particularly valid for the expression of genes under strong constitutive promoters and strong SD sequences, as is the case with the expression plasmid pP1SD-hIFNγ used in this study. Our assumption is supported by the fact that the initiation of the chromosomal DNA replication in E. coli is dependent on its transcriptional activity [45] where a direct interaction between DnaA (a bacterial replication initiator protein) and RNA polymerase has been found. In addition, we have previously shown that the segregation of the expression plasmid pP1SD-hIFNγ strongly depends on the level of hFNγ-mRNA in E. coli cells [46, 47].
Probably many factors, such as amino acid composition, solubility, affinity to the cell membrane and other properties of the recombinant protein might affect the formation of inclusion bodies and the captivation of nucleic acids and even whole ribosomes. We speculate however, that the natural platform for this phenomenon is the lack of compartmentalization in bacteria due to which the genetic processes occur in one “test tube” contributing to the electrostatic interaction between unfolded protein and nucleic acids. Therefore, we assume that nucleic acids might be components of IBs formed by different target proteins. The aggregation of proteins (including recombinant hIFNγ) in E. coli cells and therefore the IBs formation could be further enhanced by the non-enzymatic glycosylation (glycation) that occurs in vivo [48,49,50]. The advanced glycation end products (AGEs) promote protein–protein and protein-nucleic acids cross-linking [51] that might contribute to an even more complex structure of the E. coli IBs.
Conclusions
Based on the results presented in this study we conclude that the highly purified IBs isolated from E. coli cells overexpressing recombinant hIFNγ, contain tightly bound nucleic acids, which might be regarded as an integral part of their structure. They cannot be removed by extensive sonication and washing with non-denaturing urea solutions. They represent both sheared DNA and RNA that is composed of ribosomal and target protein mRNA. Although the role of nucleic acids in the formation of IBs remains to be clarified, we suppose that they come from gene expressing complexes (expression plasmid, mRNA and translating ribosomes) and actively participate in the aggregation process by electrostatic interactions with unfolded or partially folded proteins. Further experiments are needed to show whether these finding are valid for other recombinant proteins aggregating in IBs. Besides its fundamental significance, this finding is important for the biotechnological practice. It could serve as basis for development of new technologies for manufacturing of recombinant proteins based on preliminary removal or suppression of the co-aggregation of nucleic acids in IBs. The proteins thus obtain is expected to have improved purity and stability in water solutions.
Methods
Isolation of crude hIFNγ IBs fraction
Escherichia coli LE391 cells were transformed with a plasmid for constitutive expression of hIFNγ. The cells were grown and lysed in 200 ml of 1 M urea, 0.4 M guanidinium hydrochloride, 20 mM Tris–HCl, pH 8.8 by ultrasonic disintegration as described by Petrov et al. [41]. The pellet was collected by centrifugation at 14,000 rpm for 30 min and stored at 4 °C.
Purification of hIFNγ IBs
The crude pellet of hIFNγ IBs was suspended in TE buffer pH 7.4, sonicated (3 cycles of 1 min at amplitude 50%) and 100 μg/ml lysozyme was added at a 1/10 v/v ratio. The suspension was incubated at 37 °C for 1 h, overlaid on a 20% sucrose cushion at 1/3 v/v ratio and centrifuged at 4 °C for 20 min at 5000 rpm. The cell pellet was removed and the supernatant above the sucrose layer was collected and centrifuged at 4 °C for 15 min at 13,000 rpm. The pellet was suspended in washing buffer (0.02 M Tris, pH 8,8; 1 M urea and 0.01 M EDTA) at 1/10 v/v ratio, homogenized on a vortex mixer and centrifuged at 4 °C for 15 min at 14,000 rpm. After centrifugation, the supernatant was discarded and the pellet was re-suspended again in washing buffer. After sonication (3 cycles of 1 min at amplitude 50%) and centrifugation under the same conditions, the pellet was suspended in 1/10 v/v washing buffer.
Analysis of the IBs for presence of viable bacterial cells
The presence of viable bacterial cells was monitored by seeding on a solid LB agar (1.7%), supplemented with tetracycline (12.5 mg/ml) and ampicillin (100 mg/ml), at each purification step.
Analysis of the IBs protein composition
The protein pattern of the IBs fraction was monitored by Laemmli SDS-polyacrylamide gel electrophoresis. Prior loading on the 15% SDS-polyacrylamide gel, the IBs sample was resuspended in loading buffer (4× buffer: 400 mg SDS, 5 ml 0.5 M Tris pH, 6.8, 5 ml 100% glycerol, bromo-phenol blue), incubated for 5 min at 95 °C, and centrifuged for 5 min at 12,000 rpm.
Isolation of nucleic acids from IBs
Purified IBs were suspended in 10 v/v washing buffer, incubated at 37 °C for 15 min followed by 5 min at 60 °C and cooled on ice. The isolation of nucleic acids with phenol–chloroform was carried out according to Sambrook and Russell [52]. The obtained nucleic acids were precipitated by 1/3 v/v ethanol at − 20 °C [53]. The pellet was collected by centrifugation and dissolved in 50 μl TE-buffer (pH 7.4). Nucleic acids concentration was measured by Nanodrop® ND-1000 (NanoDrop Technologies, Inc., USA) at λ = 260 nm and the electrophoretic pattern was determined by standard agarose gel electrophoresis.
Enzymatic treatment of nucleic acids isolated from IBs
Nucleic acids extracted from purified IBs were treated with RNAse A (Thermo Scientific™, 10 mg/ml), RNase T1 (Thermo Scientific™, 1000 U/ml), DNAse I, restriction endonucleases XhoI and HindIII (New England BioLabs) and Proteinase K (Roche, 10 μg/ml) following the manufacturers’ protocols.
Isolation of RNA from hIFNγ IBs
RNA was extracted from purified IBs by TRIzol® reagent (Invitrogen™) following the manufacturer’s protocol. After the last step, RNA was dissolved in 50 μl DEPC treated H2O (AppliChem) and stored at − 70 °C.
Determination of hIFNγ-mRNA and E. coli 16S RNA in purified IBs
hIFNγ-mRNA and E. coli 16S RNA were determined by hybridization with 19 nt and 20 nt long 32P-labelled oligonucleotides specific for hIFNγ gene and E. coli 16S rRNA respectively as previously described [54].
Availability of data and materials
All data generated or analyzed during this study are included in this article.
Abbreviations
- IBs:
-
Inclusion bodies
- hIFNγ:
-
Human interferon-gamma
- PAAG electrophoresis:
-
Polyacrylamide gel electrophoresis
- AGEs:
-
Advanced glycation end products
- LB:
-
Luria-Bertani (LB) broth
- DEPC:
-
Diethylpyrocarbonate
References
De Marco A, Ferrer-Miralles N, Garcia-Fruitós E, Mitraki A, Peternel S, Rinas U, Trujillo-Roldán MA, Valdez-Cruz NA, Vázquez E, Villaverde A. Bacterial inclusion bodies are industrially exploitable amyloids. FEMS Microbiol Rev. 2019;43(1):53–72. https://doi.org/10.1093/femsre/fuy038.
Rinas U, Garcia-Fruitós E, Corchero JL, Vázquez E, Seras-Franzoso J, Villaverde A. Bacterial inclusion bodies: discovering their better half. Trends Biochem Sci. 2017;42(9):726–37. https://doi.org/10.1016/j.tibs.2017.01.005.
Baneyx F, Mujacic M. Recombinant protein folding and misfolding in Escherichia coli. Nat Biotechnol. 2004;22(11):1399. https://doi.org/10.1038/nbt1029.
Wang W, Nema S, Teagarden D. Protein aggregation—pathways and influencing factors. Int J Pharm. 2010;390(2):89–99. https://doi.org/10.1016/j.ijpharm.2010.02.025.
Slouka C, Kopp J, Spadiut O, Herwig C. Perspectives of inclusion bodies for bio-based products: curse or blessing? Appl Microbiol Biotechnol. 2019;103(3):1143–53. https://doi.org/10.1007/s00253-018-9569-1.
Gifre-Renom L, Seras-Franzoso J, Rafael D, Andrade F, Cano-Garrido O, Martinez-Trucharte F, Ugarte-Berzal E, Martens E, Boon L, Villaverde A, Opdenakker G. The biological potential hidden in inclusion bodies. Pharmaceutics. 2020;12(2):157. https://doi.org/10.3390/pharmaceutics12020157.
Carratalá JV, Cano-Garrido O, Sánchez J, Membrado C, Pérez E, Conchillo-Solé O, Daura X, Sánchez-Chardi A, Villaverde A, Arís A, Garcia-Fruitós E. Aggregation-prone peptides modulate activity of bovine interferon gamma released from naturally occurring protein nanoparticles. New Biotechnol. 2020;25(57):11–9. https://doi.org/10.1016/j.nbt.2020.02.001.
Sánchez JM, López-Laguna H, Álamo P, Serna N, Sánchez-Chardi A, Nolan V, Cano-Garrido O, Casanova I, Unzueta U, Vazquez E, Mangues R. Artificial inclusion bodies for clinical development. Adv Sci. 2019. https://doi.org/10.1002/advs.201902420.
Slouka C, Kopp J, Hutwimmer S, Strahammer M, Strohmer D, Eitenberger E, Schwaighofer A, Herwig C. Custom made inclusion bodies: impact of classical process parameters and physiological parameters on inclusion body quality attributes. Microb Cell Fact. 2018;17(1):148. https://doi.org/10.1186/s12934-018-0997-5.
Gatti-Lafranconi P, Natalello A, Ami D, Doglia SM, Lotti M. Concepts and tools to exploit the potential of bacterial inclusion bodies in protein science and biotechnology. FEBS J. 2011;278(14):2408–18. https://doi.org/10.1111/j.1742-4658.2011.08163.x.
Singhvi P, Saneja A, Srichandan S, Panda AK. Bacterial inclusion bodies: a treasure trove of bioactive proteins. Trends Biotechnol. 2020;38(5):474–86. https://doi.org/10.1016/j.tibtech.2019.12.011.
Philo JS, Arakawa T. Mechanisms of protein aggregation. Curr Pharm Biotechnol. 2009;10(4):348–51. https://doi.org/10.2174/138920109788488932.
Morris AM, Watzky MA, Finke RG. Protein aggregation kinetics, mechanism, and curve-fitting: a review of the literature. Biochim Biophys Acta Proteins Proteom. 2009;1794(3):375–97. https://doi.org/10.1016/j.bbapap.2008.10.016.
Weiss WF IV, Young TM, Roberts CJ. Principles, approaches, and challenges for predicting protein aggregation rates and shelf life. J Pharm Sci. 2009;98(4):1246–77. https://doi.org/10.1002/jps.21521.
Hamada H, Arakawa T, Shiraki K. Effect of additives on protein aggregation. Curr Pharm Biotechnol. 2009;10(4):400–7. https://doi.org/10.2174/138920109788488941.
Andrews JM, Roberts CJ. A Lumry−Eyring nucleated polymerization model of protein aggregation kinetics: 1. Aggregation with pre-equilibrated unfolding. J Phys Chem B. 2007;111(27):7897–913. https://doi.org/10.1021/jp070212j.
Buck PM, Kumar S, Wang X, Agrawal NJ, Trout BL, Singh SK. Computational methods to predict therapeutic protein aggregation. In: Voynov V, Caravella JA, editors. Therapeutic proteins. Totowa: Humana Press; 2012. p. 425–51. https://doi.org/10.1007/978-1-61779-921-1_26.
Jürgen B, Breitenstein A, Urlacher V, Büttner K, Lin H, Hecker M, Schweder T, Neubauer P. Quality control of inclusion bodies in Escherichia coli. Microb Cell Fact. 2010;9(1):41. https://doi.org/10.1186/1475-2859-9-41.
Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S. AGGRESCAN: a server for the prediction and evaluation of” hot spots” of aggregation in polypeptides. BMC Bioinform. 2007;8(1):65. https://doi.org/10.1186/1471-2105-8-65.
de Groot NS, Pallarés I, Avilés FX, Vendrell J, Ventura S. Prediction of” hot spots” of aggregation in disease-linked polypeptides. BMC Struct Biol. 2005;5(1):18. https://doi.org/10.1186/1472-6807-5-18.
Ventura S. Sequence determinants of protein aggregation: tools to increase protein solubility. Microb Cell Fact. 2005;4(1):11. https://doi.org/10.1186/1475-2859-4-11.
Villaverde A, Carrió MM. Protein aggregation in recombinant bacteria: biological role of inclusion bodies. Biotechnol Lett. 2003;25(17):1385–95. https://doi.org/10.1023/A:1025024104862.
Espargaró A, Castillo V, de Groot NS, Ventura S. The in vivo and in vitro aggregation properties of globular proteins correlate with their conformational stability: the SH3 case. J Mol Biol. 2008;378(5):1116–31. https://doi.org/10.1016/j.jmb.2008.03.020.
Rinas U, Bailey JE. Protein compositional analysis of inclusion bodies produced in recombinant Escherichia coli. Appl Microbiol Biotechnol. 1992;37(5):609–14. https://doi.org/10.1007/BF00240735.
Rinas U, Boone TC, Bailey JE. Characterization of inclusion bodies in recombinant Escherichia coli producing high levels of porcine somatotropin. J Biotechnol. 1993;28(2–3):313–20. https://doi.org/10.1016/0168-1656(93)90179-Q.
Valax P, Georgiou G. Molecular characterization of beta-lactamase inclusion bodies produced in Escherichia coli. 1. Composition. Biotechnol Prog. 1993;9(5):539–47. https://doi.org/10.1021/bp00023a014.
Carrio MM, Corchero JL, Villaverde A. Dynamics of in vivo protein aggregation: building inclusion bodies in recombinant bacteria. FEMS Microbiol Lett. 1998;169(1):9–15. https://doi.org/10.1111/j.1574-6968.1998.tb13292.x.
Singh SM, Panda AK. Solubilization and refolding of bacterial inclusion body proteins. J Biosci Bioeng. 2005;99(4):303–10. https://doi.org/10.1263/jbb.99.303.
Veinger L, Diamant S, Buchner J, Goloubinoff P. The small heat-shock protein IbpB from Escherichia coli stabilizes stress-denatured proteins for subsequent refolding by a multichaperone network. J Biol Chem. 1998;273(18):11032–7. https://doi.org/10.1074/jbc.273.18.11032.
Allen SP, Polazzi JO, Gierse JK, Easton AM. Two novel heat shock genes encoding proteins produced in response to heterologous protein expression in Escherichia coli. J Bacteriol. 1992;174(21):6938–47. https://doi.org/10.1128/jb.174.21.6938-6947.1992.
Hoffmann F, Rinas U. Kinetics of heat-shock response and inclusion body formation during temperature-induced production of basic fibroblast growth factor in high-cell-density cultures of recombinant Escherichia coli. Biotechnol Prog. 2000;16(6):1000–7. https://doi.org/10.1021/bp0000959.
Carrio MM, Villaverde A. Construction and deconstruction of bacterial inclusion bodies. J Biotechnol. 2002;96(1):3–12. https://doi.org/10.1016/S0168-1656(02)00032-9.
Jürgen B, Lin HY, Riemschneider S, Scharf C, Neubauer P, Schmid R, Hecker M, Schweder T. Monitoring of genes that respond to overproduction of an insoluble recombinant protein in Escherichia coli glucose-limited fed-batch fermentations. Biotechnol Bioeng. 2000;70(2):217–24. https://doi.org/10.1002/1097-0290(20001020)70:2%3c217:AID-BIT11%3e3.0.CO;2-W.
Chaturvedi R, Bhakuni V, Tuli R. The δ-endotoxin proteins accumulate in Escherichia coli as a protein–DNA complex that can be dissociated by hydrophobic interaction chromatography. Protein Expr Purif. 2000;20(1):21–6. https://doi.org/10.1006/prep.2000.1270.
Hartley DL, Kane JF. Properties of inclusion bodies from recombinant Escherichia coli. Biochem Soc Trans. 1988;16(2):101–2.
Kane JF, Hartley DL. Properties of recombinant protein-containing inclusion bodies in Escherichia coli. Bioprocess Technol. 1991;12:121–45.
Hart RA, Rinas U, Bailey JE. Protein composition of Vitreoscilla hemoglobin inclusion bodies produced in Escherichia coli. J Biol Chem. 1990;265(21):12728–33.
Wasmer C, Benkemoun L, Sabaté R, Steinmetz MO, Coulary-Salin B, Wang L, Riek R, Saupe SJ, Meier BH. Solid-state NMR spectroscopy reveals that E. coli inclusion bodies of HET-s (218–289) are amyloids. Angew Chem Int Ed. 2009;48(26):4858–60. https://doi.org/10.1002/anie.200806100.
Futami J, Fujiyama H, Kinoshita R, Nonomura H, Honjo T, Tada H, Matsushita H, Abe Y, Kakimi K. Denatured mammalian protein mixtures exhibit unusually high solubility in nucleic acid-free pure water. PLoS ONE. 2014;9(11):e113295. https://doi.org/10.1371/journal.pone.0113295.
Rodríguez-Carmona E, Cano-Garrido O, Seras-Franzoso J, Villaverde A, García-Fruitós E. Isolation of cell-free bacterial inclusion bodies. Microb Cell Fact. 2010;9(1):71. https://doi.org/10.1186/1475-2859-9-71.
Petrov S, Nacheva G, Ivanov I. Purification and refolding of recombinant human interferon-gamma in urea–ammonium chloride solution. Protein Expr Purif. 2010;73(1):70–3. https://doi.org/10.1016/j.pep.2010.03.026.
Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol–chloroform extraction. Anal Biochem. 1987;162(1):156–9. https://doi.org/10.1016/0003-2697(87)90021-2.
Neerathilingam M, Mysore S, Gandham SH. Soni-removal of nucleic acids from inclusion bodies. Biochem Biophys Res Commun. 2014;448(1):45–9. https://doi.org/10.1016/j.bbrc.2014.04.049.
Futami J, Tsushima Y, Tada H, Seno M, Yamada H. Convenient and efficient in vitro folding of disulfide-containing globular protein from crude bacterial inclusion bodies. J Biochem. 2000;127(3):435–41. https://doi.org/10.1093/oxfordjournals.jbchem.a022625.
Baker TA, Kornberg A. Transcriptional activation of initiation of replication from the E. coli chromosomal origin: an RNA–DNA hybrid near oriC. Cell. 1988;55(1):113–23. https://doi.org/10.1016/0092-8674(88)90014-1.
Popov M, Petrov S, Kirilov K, Nacheva G, Ivanov I. Segregational instability in E. coli of expression plasmids carrying human interferon gamma gene and its 3′-end truncated variants. Biotechnol Biotechnol Equip. 2009;23(sup1):840–3. https://doi.org/10.1080/13102818.2009.10818553.
Popov M, Petrov S, Nacheva G, Ivanov I, Reichl U. Effects of a recombinant gene expression on ColE1-like plasmid segregation in Escherichia coli. BMC Biotechnol. 2011;11(1):18. https://doi.org/10.1186/1472-6750-11-18.
Mironova R, Niwa T, Hayashi H, Dimitrova R, Ivanov I. Evidence for non-enzymatic glycosylation in Escherichia coli. Mol Microbiol. 2001;39(4):1061–8. https://doi.org/10.1046/j.1365-2958.2001.02304.x.
Mironova R, Niwa T, Handzhiyski Y, Sredovska A, Ivanov I. Evidence for non-enzymatic glycosylation of Escherichia coli chromosomal DNA. Mol Microbiol. 2005;55(6):1801–11. https://doi.org/10.1111/j.1365-2958.2005.04504.x.
Mironova R, Niwa T, Dimitrova R, Boyanova M, Ivanov I. Glycation and post-translational processing of human interferon-gamma expressed in Escherichia coli. J Biol Chem. 2003;278(51):51068–74. https://doi.org/10.1074/jbc.m307470200.
Boteva E, Mironova R. Maillard reaction and aging: can bacteria shed light on the link? Biotechnol Biotechnol Equip. 2019;33(1):481–97. https://doi.org/10.1080/13102818.2019.1590160.
Sambrook J, Russell DW. Purification of nucleic acids by extraction with phenol: chloroform. Cold Spring Harbor Protoc. 2006;2006(1):pdb-rot4455. https://doi.org/10.1101/pdb.prot4455.
Green MR, Sambrook J. Precipitation of DNA with ethanol. Cold Spring Harb Protoc. 2016. https://doi.org/10.1101/pdb.prot093377.
Nacheva G, Todorova K, Boyanova M, Berzal-Herranz A, Karshikoff A, Ivanov I. Human interferon gamma: significance of the C-terminal flexible domain for its biological activity. Arch Biochem Biophys. 2003;413(1):91–8. https://doi.org/10.1016/S0003-9861(03)00113-9.
Funding
This work was supported by Grant DN-11/20/2017 from Bulgarian Science Fund and by the National Research Program “Young scientists and postdoctoral student” DCM # 577/2018 from Ministry of Education and Science, Bulgaria.
Author information
Authors and Affiliations
Contributions
II and GN designed the experiments. EK performed the experiments. GN and EK analyzed and interpreted the data. EK wrote the manuscript. GN and II revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Fig. S1.
UV spectra of nucleic acids isolated from purified IBs by phenol–chloroform extraction and precipitation with ethanol. The probe was analysed by NanoDrop®. Fig. S2. UV spectra of RNA isolated from purified IBs by TRIzol® and analysed by NanoDrop®.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Krachmarova, E., Ivanov, I. & Nacheva, G. Nucleic acids in inclusion bodies obtained from E. coli cells expressing human interferon-gamma. Microb Cell Fact 19, 139 (2020). https://doi.org/10.1186/s12934-020-01400-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01400-6