
Abstract
Background
l-Alanyl-l-glutamine (AQ) is a functional dipeptide with high water solubility, good thermal stability and high bioavailability. It is widely used in clinical treatment, post-operative rehabilitation, sports health care and other fields. AQ is mainly produced via chemical synthesis which is complicated, time-consuming, labor-intensive, and have a low yield accompanied with the generation of by-products. It is therefore highly desirable to develop an efficient biotechnological process for the industrial production of AQ.
Results
A metabolically engineered E. coli strain for AQ production was developed by over-expressing l-amino acid α-ligase (BacD) from Bacillus subtilis, and inactivating the peptidases PepA, PepB, PepD, and PepN, as well as the dipeptide transport system Dpp. In order to use the more readily available substrate glutamic acid, a module for glutamine synthesis from glutamic acid was constructed by introducing glutamine synthetase (GlnA). Additionally, we knocked out glsA–glsB to block the first step in glutamine metabolism, and glnE–glnB involved in the ATP-dependent addition of AMP/UMP to a subunit of glutamine synthetase, which resulted in increased glutamine supply. Then the glutamine synthesis module was combined with the AQ synthesis module to develop the engineered strain that uses glutamic acid and alanine for AQ production. The expression of BacD and GlnA was further balanced to improve AQ production. Using the final engineered strain p15/AQ10 as a whole-cell biocatalyst, 71.7 mM AQ was produced with a productivity of 3.98 mM/h and conversion rate of 71.7%.
Conclusion
A metabolically engineered strain for AQ production was successfully developed via inactivation of peptidases, screening of BacD, introduction of glutamine synthesis module, and balancing the glutamine and AQ synthesis modules to improve the yield of AQ. This work provides a microbial cell factory for efficient production of AQ with industrial potential.

Similar content being viewed by others

Background
Glutamine (l-Gln) plays an important role in maintaining intestinal function [1,2,3,4], promoting immune function [5, 6], maintaining homeostasis of the internal environment [7] and improving the adaptability of organism to stress [8]. During disease or stress conditions, demand outpaces supply, and glutamine becomes conditionally essential [9]. The supply of exogenous l-glutamine or glutamyl dipeptide is an important nutritional solution to reduce glutamine deficiency in vivo, and can be applied in clinical treatment. However, some characteristics of glutamine such as low solubility in water, easy decomposition and poor thermal stability, as well as production of toxic pyroglutamate during heat sterilization restricted its application in medicine. Otherwise, as a result of its unstable nature, the low yield of glutamine during the purification process is a great challenge [10].
l-Alanyl-l-glutamine (abbreviated as AQ) is a dipeptide of glutamine and alanine, which is more stable and water-soluble than glutamine. AQ is hydrolyzed to release glutamine and alanine in vivo. While it acts as a source of glutamine, AQ has higher bioavailability and a short half-life [11], and does not cause cumulative damage to the body, so it is used as a substitute for glutamine in clinical practice.
The commercial demand for AQ is increasing with the expansion of new applications and the development of new products based on AQ. Chemical and biotechnological methods for AQ production have been developed [12,13,14,15,16]. AQ can be synthesized by chemical condensation of benzyloxycarbonyl-l-alanine and methyl glutamate via activated ester [17] or triphenyl phosphine/hexachloroethane condensation, which used to require complex steps such as amino acid activation, subunit protection, and removing the protective groups. Enzymatic processes for AQ production have been developed using an α-amino acid ester acyltransferase [18, 19]. Tabata and Hashimoto engineered Escherichia coli by expressing l-amino acid α-ligase (BacD), which catalyzes the formation of AQ in an ATP-dependent manner, and produced more than 100 mM AQ in 47 h of fermentation [20]. Whole-cell biocatalysis can be used to perform enzyme cascade reaction, improve catalytic efficiency, and simplify the preparation process.
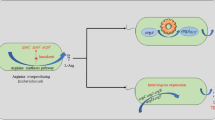
A metabolically engineered E. coli strain for AQ production was developed by over-expressing l-amino acid α-ligase (BacD) from Bacillus subtilis and inactivating native peptidases. In order to use a more readily available substrate, the glutamine synthesis module based on introducing glutamine synthetase (GlnA) was constructed and optimized. Then glutamine synthesis module was combined with the AQ synthesis module to use glutamic acid and alanine for AQ production. The protein expression of BacD and GlnA was further balanced to improve the AQ production. Finally, we used the engineered strain to develop a whole-cell biocatalytic process for AQ production (Fig. 1). This work provides an environmentally friendly, highly efficient and cost-effective process for industrial biosynthesis of AQ.

Schematic presentation of the AQ metabolic pathway in engineered E. coli. The GlnA and BacD were co-expressed. Discontinued arrows represent the enzymatic activities that have been eliminated. GlsA glutaminase 1, GlsB glutaminase 2, GlnE fused glutamine synthetase deadenylase/adenylyltransferase, GlnB nitrogen regulatory protein PII-1, PepA aminopeptidase A/I, PepB aminopeptidase B, PepD peptidase D, PepN aminopeptidase N, Dpp dipeptide ABC transporter DppABCDF
Results
Construction of the AQ synthesis module
The E. coli strain p01/BW25113, overexpressing BacD from Bacillus subtilis (BsBacD) which catalyzes the formation of AQ from alanine and glutamine, was constructed for the production of AQ. Extracellular AQ concentration was measured by HPLC, and 2.0 mM AQ was obtained (Fig. 2).

Extracellular titers of AQ produced by engineered strains overexpressing BsBacD. WT wild type; BW, AQ06, AQ09, AQ10 were transformed with plasmid p01. The engineered strains were induced and suspended in a reaction mixture containing 50 mM glutamine, 50 mM alanine, and 10 mM magnesium chloride. The bioconversion reactions were performed at 30 °C and 200 rpm for 18 h. Glucose was supplemented at a concentration of 10 mM every 3 h
In E. coli, peptidases encoded by pepA, pepB, pepD and pepN, have been reported to degrade a broad spectrum of dipeptides [20, 21], and inactivating them might reduce AQ degradation. It was reported that deletion of dpp, encoding a dipeptide ABC transporter increased AQ accumulation [20, 22, 23]. By knocking out the genes pepN, pepA, pepB, pepD and dpp, the degradation of AQ was alleviated. In the starting host BW25113, 20 mM AQ was completely degraded after 3 h, compared to only 1.3 mM in the chassis AQ09 (BW25113ΔpepN, ΔpepA, ΔpepB, ΔpepD, Δdpp) after 6 h. A whole-cell biocatalysis system with the strain (p01/AQ09) yielded 3.3 mM AQ after 18 h (Fig. 2). These results demonstrated that inactivation of peptidases and the dipeptide transporter Dpp reduced the degradation of AQ, and thus increased AQ production.
Screening of BacD homologues
BacD is the key enzyme for AQ synthesis. We examined the known sequences annotated as l-amino acid α-ligase (BacD) in the NCBI database. According to the sequences of known or predicted BacD homologs, we selected a set of related sequences from different species, and constructed a phylogenetic tree of the previously reported BacD homologs and those used in this study (Additional file 1: Fig. S1). The genes encoding BacD homologs from different species were codon-optimized by Nanjing Generay (China) and cloned into strain AQ10 (BW25113, ∆glnEB, ∆glsAB, ∆lpxM, ∆pepABDN, ∆dpp). The performance of different BacD proteins was investigated in vivo, using the respective strains as a whole-cell biocatalysts. The result showed that the strain overexpressing BaBacD (from Bacillus altitudinis) produced higher amount of AQ (19.2 mM) than strains with other BacD homologs. By comparison, 7.9 mM AQ was obtained using the strain overexpressing BsBacD (from Bacillus subtilis) (Fig. 3). Although there was soluble expression of BvBacD (from Beta vulgaris), VcBacD (from Vibrio campbellii), and SrBacD (from Streptomyces rubrolavendulae), only 3.0, 1.8, 0.5 mM AQ was respectively obtained (Fig. 3). BsaBacD (from Bacillus safensis), BloBacD (from Bifidobacterium longum subsp. infantis), PmBacD (from Perkinsus marinus), and PfBacD (from Pseudomonas fluorescens) were expressed as inclusion bodies (Additional file 1: Fig. S2), and only a low amount of AQ was detected.

AQ production by engineered strains over-expressing BacD homologs from different species. Ba, BacD from Bacillus altitudinis; Bs, BacD from Bacillus subtilis; Bv, BacD from Beta vulgaris; Vc, BacD from Vibrio campbellii; Pm, BacD from Perkinsus marinus; Sr, BacD from Streptomyces rubrolavendulae; Bsa, BacD from Bacillus safensis; Blo, BacD from Bifidobacterium longum subsp. Infantis; Pf, BacD from Pseudomonas fluorescens
Construction of a glutamine synthesis module
To use the more readily available substrate glutamic acid, glutamine synthetase from Corynebacterium glutamicum (CgGlnA), which convert glutamic acid to glutamine was cloned into E. coli, resulting in the strain p00/BW25113. A final glutamine titer of 22.4 mM was obtained in the whole-cell bioconversion. During growth nitrogen replete growth conditions, glutamine synthetase adenylyltransferase/deadenylase (encoded by glnE) interacts with PII-1 protein encoded by glnB, which reduces the activity of glutamine synthetase, and glnE–glnB-deficiency was reported to lead to increased glutamine accumulation [24,25,26]. Therefore, a glnE–glnB-deficient strain expressing GlnA (p00/AQ02) was constructed, and a glutamine titer of 27.8 mM was achieved, which was 24.1% higher than that of the strain p00/BW25113 (Fig. 4). In E. coli, glutamine was converts into glutamic acid by glutaminases GlsA (encoded by glsA) and GlsB (encoded by glsB) [27], and further into α-ketoglutarate. Consequently, the glsA and glsB genes were deleted, resulting in strain AQ04 (ΔglsAΔglsB), which was then transformed with the plasmid p00, and the resulting strain p00/AQ04 produced 33.8 mM of glutamine after 18 h of bioconversion. Further, glnE, glnB, glsA, glsB and lpxM were sequentially knocked out and 46.5 mM glutamine was accumulated by the resulting strain p00/AQ06, with a conversion rate of 93.0% (Fig. 4). Consequently, the strain p00/AQ06 was used for further engineering.

Extracellular glutamine titers produced by engineered strains overexpressing GlnA. WT wild type; BW, AQ02, AQ04, and AQ06 were transformed with plasmid p00. The engineered strains were induced and suspended in a reaction mixture containing 50 mM sodium glutamate, 10 mM magnesium chloride, and 100 mM ammonium chloride. The bioconversion reactions were performed at 30 °C and 200 rpm for 18 h. Glucose was supplemented at a concentration of 10 mM every 3 h
Combination the AQ and glutamine synthesis modules
To achieve AQ production from glutamic acid and alanine, the AQ and glutamine synthesis modules were combined. The strain AQ10 was obtained by knocking out the genes pepN, pepA, pepB, pepD, dpp, glnE, glnB, glsA, glsB and lpxM. In AQ10, the degradation of AQ was alleviated, glutamine catabolism was effectively weakened as well. After introducing BsBacD, the resulting strain p01/AQ10 produced 7.9 mM AQ, which was four times more than the production of the original strain p01/BW25113 (Fig. 2).
The engineered strains with the plasmid p11 (pYB1s-CgglnA-BsbacD), co-expressing BsBacD and CgGlnA was used as a whole-cell biocatalyst for AQ production from alanine and glutamic acid. Removal of the peptidases PepA, PepB, PepD, and PepN, together with knocking out the transporter Dpp significantly increased AQ production, and a titer of 17.9 mM was produced by the strain AQ09 harboring p11 (Fig. 5). Due to the deletion of glnE–glnB and glsAglsB, the biosynthesis of glutamine was enhanced, which resulted in increased AQ production, leading to a product titer of 29.8 mM in the strain AQ10 harboring p11 (Fig. 5). Inactivation of peptidases alleviated AQ degradation, and removing the transporter Dpp promoted the efflux of AQ. The results showed that combination of the strategies of peptidases inactivation, knocking out the transporter Dpp, and enhancing the glutamine supply by deletion of glnE–glnB and glsA–glsB greatly enhanced AQ production.

Production of AQ by engineered strains co-expressing CgGlnA and BsBacD. WT wild type; BW, AQ02, AQ04, and AQ06 were transformed with plasmid p11. The engineered strains were induced and suspended in a reaction mixture containing 100 mM sodium glutamate, 100 mM alanine, 200 mM ammonium chloride, and 10 mM magnesium chloride. The bioconversion reactions were performed at 30 °C and 200 rpm for 18 h. Glucose was supplemented at a concentration of 10 mM every 3 h
Balance of the two synthesis modules by regulating protein expression
To balance flux in the two synthesis module for the purpose of increasing AQ production, the expression of BacD and GlnA proteins was studied. To co-express BaBacD or BsBacD with CgGlnA in different order, four plasmids p11 (pYB1s-CgglnA-BsbacD), p12 (pYB1s-BsbacD-CgglnA), p13 (pYB1s-CgglnA-BabacD), and p14 (pYB1s-BabacD-CgglnA) were constructed (Fig. 6c), and used to individually transform the host AQ10. Either l-amino acid α-ligase or glutamine synthetase was poorly expressed when BaBacD was co-expressed with CgGlnA (Additional file 1: Fig. S3), leading to decreased AQ production. However, when CgGlnA was co-expressed with BsBacD, both proteins were expressed at high levels, and contributed to an increased yield of AQ after 18 h of bioconversion. The AQ titer reached 29.8 mM when CgglnA was inserted in front of BsbacD (p11/AQ10), compared to 22.3 mM when CgglnA was expressed behind BsbacD (p12/AQ10) (Fig. 6a). The concentration of the intermediate metabolite, glutamine, in p11/AQ10 (22.8 mM) was higher than in p12/AQ10 (12.0 mM) (Fig. 6a). SDS-PAGE analysis of protein expression (Additional file 1: Fig. S3) and the concentration of glutamine suggested that higher soluble expression of CgGlnA enhanced the supply of glutamine, and increasing the expression of BsBacD might further improve the synthesis of AQ.

Regulation of the two synthesis modules by balancing the protein expression. a Production of AQ in whole-cell bioconversions using engineered strains with bacD and CgglnA expressed different order. The abbreviations are: p11, pYB1s-CgglnA-BsbacD; p12, pYB1s-BsbacD-CgglnA; p13, pYB1s-CgglnA-BabacD; p14, pYB1s-BabacD-CgglnA. p11, p12, p13, and p14 were individually introduced into strain AQ10. The engineered strains were induced and suspended in a reaction mixture containing 100 mM sodium glutamate, 100 mM alanine, 200 mM ammonium chloride, and 10 mM magnesium chloride. The bioconversion reactions were performed at 30 °C and 200 rpm for 18 h. b Extracellular AQ titer produced by strains with different RBS in the front of BsbacD CDS. The reaction was performed in a reaction mixture containing 100 mM sodium glutamate, and 100 mM alanine for 6 h. CK, p11/AQ10. c Modular expression of CgglnA and bacD genes in different order in a ara-operon configuration
In order to enhance the expression of BsBacD, its native RBS was replaced to upregulate the mRNA translation initiation rate in the recombinant strain. The translation rate prediction and design of new RBS was done using RBS Calculator 2.0 [28,29,30]. The strain p15/AQ10 expressed more BsBacD protein (Additional file 1: Fig. S4), and its AQ production increased by 76.1% compared to p11/AQ10 (Fig. 6b).
Optimization of the conditions for whole-cell biocatalysis
After successfully constructing an engineered E. coli strains for AQ production by metabolic engineering, we investigated its applicability as whole-cell biocatalyst for the biotechnological production of AQ. Bioconversion parameters that affect the activity of the biocatalyst, such as temperature and pH, were investigated. AQ production reached maximal values at 30 °C (Fig. 7a) and pH 9.0 (Fig. 7b). A decreased in pH was observed as the bioconversion proceeded, which affected the biosynthesis of AQ. It should be noted that glucose was supplemented in the reaction mixture to supply ATP for the reactions catalyzed by GlnA and BacD, it was reported that excess glucose can lead to acetate accumulation. Consequently, we measured the concentration of acetate and found that it was accumulated. To alleviate this, different glucose feeding strategies were applied to reduce acetate accumulation in the bioconversion process, including (1) 50 mM glucose at once; (2) 10 mM every 3 h; and (3) 20 mM every 3 h. When a low concentration of glucose (10 mM) was fed every 3 h (Fig. 7c), glucose was fully utilized (Additional file 1: Fig. S5a), and only a small amount of acetic acid accumulated (Additional file 1: Fig. S5b), indicating that 10 mM glucose fed every 3 h matched AQ productivity. The time profiles of the bioconversion indicated that alanine was exhausted first, and the ratio of glutamic acid to alanine was investigated (Fig. 7d). Under feeding with 10 mM glucose every 3 h at 30 °C and pH 9.0, the strain p15/AQ10 produced 71.7 mM AQ, from 100 mM glutamic acid and 125 mM alanine, after 18 h of reaction, corresponding to a productivity of 3.98 mM/h. Moreover, a conversion rate of 71.7% was achieved for glutamic acid, representing a 100% increase compared to the conversion rate before the optimization.

Optimization of the conditions for whole-cell biocatalysis. a Effects of pH on AQ production by strain p15/AQ10. b Effects of temperature on AQ production by p15/AQ10. c Effects of different glucose feeding strategies on AQ production. Feeding strategy: (1) 50 mM glucose was added at once; (2) 10 mM glucose was added every 3 h; (3) 20 mM glucose was added every 3 h. d Effects of the initial alanine concentration on AQ production. Initial alanine concentrations of 100–200 mM were combined with a fixed glutamic acid concentration at 100 mM, and the reaction was performed for 12 h. e Time profiles of the AQ, glutamine, glutamic acid and alanine concentrations. The bioconversion was performed in a reaction mixture containing 100 mM sodium glutamate, 125 mM alanine, 200 mM ammonium chloride, and 10 mM magnesium chloride at 30 °C and pH 9.0
Discussion
In this study, we constructed a metabolically engineered E. coli strain for AQ production by overexpressing l-amino acid α-ligase from Bacillus subtilis, knocking out peptidases PepA, PepB, PepD, and PepN, as well as the transporter Dpp, which effectively weakened AQ degradation. To use a more readily available and cheaper substrate, a module for the synthesis of glutamine from glutamic acid was constructed by overexpressing glutamine synthetase from Corynebacterium glutamicum. It was reported that the glutaminases GlsA–GlsB convert glutamine to glutamic acid, which is the main pathway of glutamine catabolism, while GlnE interacts with GlnB to induce the adenylation of glutamine synthetase under nitrogen-rich growth, leading to a reduction of the activity of glutamine synthetase. Thus, GlsA, GlsB, GlnE, and GlnB were blocked, which resulted in increased glutamine supply with a 93.0% bioconversion of glutamic acid. Then AQ and glutamine synthesis modules were coupled, and 29.8 mM AQ production was achieved by co-expressing BsBacD and CgGlnA. To balance the flux through two modules, the expression of BacD and GlnA was fine-tuned by modifying the RBS, and AQ production was further increased by 76.1%. Finally, the reaction conditions for the whole-cell biocatalysis were optimized, and 71.7 mM AQ was obtained using strain p15/AQ10 after 18 h of reaction, with productivity of 3.98 mM/h and a conversion rate of 71.7% from glutamic acid to AQ was achieved.
Both fermentation and enzymatic processes for AQ production were reported. We compared the AQ biosynthesis results obtained in this study with previous studies, as shown in Table 1. Enzymatic processes were reported for AQ production from l-alanine methyl ester hydrochloride (AlaOMe) and glutamine using amino acid ester acyltransferase [18, 31, 32]. The substrates used in the process, AlaOMe and glutamine are expensive, making the process not economically feasible. Tabata and Hashimoto reported a fermentative process for AQ production from glucose using a engineered E. coli strain co-expressing l-amino acid α-ligase (Lal) and l-alanine dehydrogenase (Ald) in the strain background of JKYPQ3 (pepA pepB pepD pepN dpp glnE glnB putA) [20]. The amino acids needed for AQ production were biosynthesized from glucose. However, it was challenging to balance the ratio of glutamine and alanine, which might affect AQ production. Moreover, the synthesis of amino acids was generally tightly regulated. Therefore, AQ accumulation was slow, and the productivity of AQ was low in the fermentation process. In this study, AQ was synthesized from glutamic acid and alanine using whole-cell biocatalytic process. Alanine was added to the reaction mixture as substrate, and glutamine was synthesized from externally added glutamic acid. A very small amount of glucose was used to supply ATP. The concentrations of the substrates (glutamate and alanine) can be easily controlled. The productivity of AQ in this study was 3.98 mM/h which was much higher than previously reported [20].
It was reported that expression of l-amino acid α-ligase exerted a negative effect on cell growth in the fermentation [20]. In the whole-cell bioconversion, cell growth (the enzyme manufacturing phase) and the AQ production phase were separated. Substrates were converted to AQ by resting cells in the production phase. Therefore, the growth inhibitory effect of l-amino acid α-ligase was reduced.
BacD enzymes have been reported to have insufficient substrate specificity and form by-products, such as different dipeptides or longer oligopeptides [31], which undoubtedly increases the cost of downstream separation and purification of target products. In this study, the extracellular concentrations of Ala–Ala reached 12.4 mM after 18 h of reaction, and no other dipeptides or longer oligopeptides were detected. The concentration of alanine decreased faster than that of glutamic acid, which suggested that the alanine catabolism should be reduced. Further studies are underway to improve the system including screening of BacD homologs with higher enzymatic activity and substrate specificity, enhancing AQ efflux using dipeptide efflux pump, reducing the degradation of alanine by blocking its catabolism, and scale-up of the bioconversion in fermenters.
Conclusions
In this study, we conducted systematic metabolic engineering of E. coli to develop a whole-cell biocatalysis for the synthesis of AQ from glutamic acid and alanine. Inactivation of peptidases and the dipeptide transport system, combined with screening of BacD homologs improved the yield of AQ. The supply of glutamine from glutamic acid was improved by overexpression of glutamine synthetase (GlnA), reduction of glutamine degradation by inactivating of glutaminases (ΔglsAΔglsB), and deregulation of glutamine biosynthesis (ΔglnEΔglnB). The final engineered E. coli strain p15/AQ10 produced 71.7 mM AQ with a conversion rate of 71.7% for glutamic acid. This study offers new opportunities for the bio-industrial production of AQ. The metabolic engineering strategies developed in this study can be applied in the synthesis of other high-value-added dipeptides and oligopeptides.
Materials and methods
Construction of plasmids and strains
Escherichia coli K12 (BW25113) was used for protein expression. All bacteria strains and plasmids used in this study are listed in Table 2. Target genes (CgglnA, BsbacD, BabacD, BlabacD, BvbacD, VcbacD, BlabacD, PfbacD, BlobacD, PmbacD, BsabacD, and SrbacD) were codon-optimized and synthesized by Generay (Shanghai, China), and then ligated into pYB1a between the Xho I and EcoR I sites via Gibson assembly [33]. Inactivation of genes was conducted using the CRISPR–Cas9 system [34].
Culture condition
Strains were grown in LB medium (10 g/L tryptone, 5 g/L yeast extract, 10 g/L NaCl) at 37 °C and 220 rpm. Antibiotics (ampicillin 100 µg/mL, or streptomycin 40 µg/mL) were added as required. For protein expression, auto-inducing ZYM medium (per liter: tryptone 10 g, yeast extract 5 g, glycerol 5 g, glucose 0.5 g, l-arabinose 2 g, Na2HPO4 25 mM, KH2PO4 25 mM, NH4Cl 50 mM, Na2SO4 5 mM, MgSO4 2 mM, and trace elements including 0.05 mM FeCl3, 0.02 mM CaCl2, 0.01 mM MnCl2, 0.01 mM ZnSO4, and 0.002 mM each of CoCl2, NiCl2, Na2Mo4, Na2SeO3, and H3BO3) was used [35], and the strains were allowed to auto-induced at 30 °C for 12–16 h.
Whole‑cell biocatalysis conditions
Cells were harvested after induction by centrifugation at 5000×g for 10 min, washed once with 0.85% NaCl solution, and then used for the production of AQ via whole-cell biocatalytic conversion [36]. For AQ synthesis from glutamine and alanine, the conversion system contained 50 mM MOPS (morpholine propane sulfonic acid) buffer pH7.0, 50 mM glutamine, 50 mM alanine, 10 mM magnesium chloride, and 50 mM glucose with a starting OD600nm = 30. The bioconversion reaction was performed at 30 °C and 220 rpm in a test tube. Glucose was supplemented at a concentration of 10 mM every 3 h. For glutamine synthesis, the cells were suspended in 1 mL bioconversion medium containing 50 mM MOPS buffer pH7.0, 50 mM sodium glutamate, 100 mM ammonium chloride, 10 mM magnesium chloride, 50 mM glucose to form a cell suspension with a starting OD600nm = 30. For AQ production from glutamic acid and alanine, the conversion system contained 50 mM MOPS buffer pH9.0, 100 mM sodium glutamate, 100 mM alanine, 100 mM ammonium chloride, 10 mM magnesium chloride, and 50 mM glucose.
In the process of optimizing the whole-cell catalytic conditions, the bioconversion reaction was performed at different temperatures (20–45 °C) and pH values (6.0–11.0). When optimizing the strategy of glucose feeding, three different strategies were investigated: (1) 50 mM glucose at one time; (2) 10 mM glucose was added every 3 h; and (3) 20 mM glucose was added every 3 h. When the concentration of substrate was studied, 100 to 200 mM alanine was added at once, together with 100 mM sodium glutamate.
Analytical methods
Biomass was estimated by measuring the optical density at 600 nm. Proteins expression was analyzed by SDS-PAGE. The concentrations of glucose and acetate in the supernatant were determined by HPLC on a Bio-Rad Aminex HPX-87H Ion Exclusion column (7.8 × 300 mm; Hercules, CA, USA), with a refractive index detector. Analysis was performed at 55 °C with a mobile phase of 5 mM H2SO4 at a flow rate of 0.5 mL/min.
The AQ and amino acids including glutamine, glutamic acid, and alanine, were derivatized using 9-fluorenylmethoxy carbonyl chloroformate and measured by HPLC as described by Kazuhiko Tabata [37], with minor modifications as follows. The mobile phase components A and B were acetonitrile and 50 mM sodium acetate, and the gradient program was slightly modified as follows: 0 min, solvent A-solvent B at 10:90; 0 to 20 min, a linear increase in solvent A to A–B at 60:40; 20 to 24 min, a linear increase to A–B at 100:0; 24 to 27 min, held at A–B at 100:0; 27 to 28 min, a linear decrease in solvent B to A–B at 10:90. The column temperature was set at 30 °C, the injection volume was 5 μL, and the flow rate was 0.6 mL/min.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional file.
References
Stehle P, Kuhn KS. Glutamine: an obligatory parenteral nutrition substrate in critical care therapy. Biomed Res Int. 2015;2015:545467.
Labow BI, Souba WW. Glutamine. World J Surg. 2014;24:1503–13.
Fuentes-Orozco C, Anaya-Prado R, González-Ojeda A, Arenas-Márquez H, Cabrera-Pivaral C, Cervantes-Guevara G, Barrera-Zepeda LM. l-Alanyl-l-glutamine-supplemented parenteral nutrition improves infectious morbidity in secondary peritonitis. Clin Nutr. 2004;23:13–21.
Lasierra JLF, Carmona TG. l-Alanyl-l-glutamine dipeptide-supplemented total parenteral nutrition. Diet Nutr Crit Care. 2014;2014:1–11.
Moore SR, Guedes MM, Costa TB, Vallance J, Maier EA, Betz KJ, Aihara E, Mahe MM, Lima AA, Oria RB, Shroyer NF. Glutamine and alanyl-glutamine promote crypt expansion and mTOR signaling in murine enteroids. Am J Physiol Gastrointest Liver Physiol. 2015;308:G831–9.
Mundi MS, Shah M, Hurt RT. When is it appropriate to use glutamine in critical illness? Nutr Clin Pract. 2016;31:445–50.
van der Hulst RRWJ, Meyenfeldt MFV, Soeters PB. Glutamine: a gut essential amino acid.
Zhou Y, Zhang P, Deng G, Liu X, Lu D. Improvements of immune status, intestinal integrity and gain performance in the early-weaned calves parenterally supplemented with l-alanyl-l-glutamine dipeptide. Vet Immunol Immunopathol. 2012;145:134–42.
Furst P, Pogan K, Stehle P. Glutamine dipeptides in clinical nutrition. Nutrition. 1997;13(7–8):731–7.
Li SL, Li C, Liu YS, Wang XL, Cao ZA. Separation of (l-glutamine from fermentation broth by nanofiltration. J Membr Sci. 2003;222(1–2):191–201.
Tan B, Liu H, He G, Xiao H, Xiao D, Liu Y, Wu J, Fang J, Yin Y. Alanyl-glutamine but not glycyl-glutamine improved the proliferation of enterocytes as glutamine substitution in vitro. Amino Acids. 2017;49:2023–31.
Sano T, Sugaya T, Inoue K, Mizutaki S I, Kasai M. Cheminform abstract: process research and development of l-alanyl-l-glutamine, a component of parenteral nutrition. ChemInform. 2000;31(39).
Neuh User-Berthold M, Eufinger H. Utilization of alanyl-l-glutamine and glycyl-l-glutamine during long-term parenteral nutrition in the growing rat. Amino Acids. 1992;3(3):293–302.
Yokozeki K, Hara S. A novel and efficient enzymatic method for the production of peptides from unprotected starting materials. J Biotechnol. 2005;115:211–20.
Fields GB, Noble RL. Solid phase peptide synthesis utilizing 9-fluorenylmethoxycarbonyl amino acids. Int J Pept Protein Res. 1990;35(3):161–214.
Wang M, Qi W, Yu Q, Su R, He Z. Kinetically controlled enzymatic synthesis of dipeptide precursor of l-alanyl-l-glutamine. Biotechnol Appl Biochem. 2011;58:449–55.
Akabori S, Sakakibara S, Shimonishi Y. Protection of amidenitrogen for peptide synthesis. A novel synthesis of peptide containing Cterminal glutamine. Bull Chem Soc Jpn. 1961;34:739.
Li Y, Yuan W, Gao J, Fan C, Wu W, Bai F. Production of l-alanyl-l-glutamine by recycling E. coli expressing alpha-amino acid ester acyltransferase. Bioresour Technol. 2017;245:1603–9.
Tanaka T, Takagi K, Saddam HM, Takeda Y, Wakayama M. Purification and characterization of Elizabethkingia L-amino acid esterase: an enzyme useful for enzymatic synthesis of the dipeptide, Valyl-Glycine. Appl Biochem Biotechnol. 2017;183:362–73.
Tabata K, Hashimoto S. Fermentative production of l-alanyl-l-glutamine by a metabolically engineered Escherichia coli strain expressing l-amino acid alpha-ligase. Appl Environ Microbiol. 2007;73:6378–85.
Lutz W, Jazdzewski B. Degradation of intracellular proteins. Pol Tyg Lek. 1980;35(33):1275–8.
Smith MW, Tyreman DR, Payne GM, Marshall NJ, Payne JW. Substrate specificity of the periplasmic dipeptide-binding protein from Escherichia coli: experimental basis for the design of peptide prodrugs. Microbiology. 1999;145(Pt 10)(10):2891.
Abouhamad WN, Manson MD. The dipeptide permease of Escherichia coli closely resembies other bacterial transport systems and shows growth-phase-dependent expression. Mol Microbiol. 1994;14:1077–92.
Jiang P, Ninfa AJ. Escherichia coli PII signal transduction protein controlling nitrogen assimilation acts as a sensor of adenylate energy charge in vitro. Biochemistry. 2007;46(45):12979–96.
Jaggi R. The two opposing activities of adenylyl transferase reside in distinct homologous domains, with intramolecular signal transduction. EMBO J. 1997;16(18):5562–71.
Pahel G, Tyler B. A new glnA-linked regulatory gene for glutamine synthetase in Escherichia coli. Proc Natl Acad Sci USA. 1979;76:4544–8.
Brown G, Singer A, Proudfoot M, Skarina T, Kim Y, Chang C. Functional and structural characterization of four glutaminases from Escherichia coli and bacillus subtilis. Biochemistry. 2008;47(21):5724–35.
Espah Borujeni A, Channarasappa AS, Salis HM. Translation rate is controlled by coupled trade-offs between site accessibility, selective RNA unfolding and sliding at upstream standby sites. Nucleic Acids Res. 2014;42:2646–59.
Espah Borujeni A, Salis HM. Translation initiation is controlled by RNA folding kinetics via a ribosome drafting mechanism. J Am Chem Soc. 2016;138:7016–23.
Espah Borujeni A, Cetnar D, Farasat I, Smith A, Lundgren N, Salis HM. Precise quantification of translation inhibition by mRNA structures that overlap with the ribosomal footprint in N-terminal coding sequences. Nucleic Acids Res. 2017;45:5437–48.
Yokozeki K, Hara S. A novel and efficient enzymatic method for the production of peptides from unprotected starting materials. J Biotechnol. 2005;115(2):211–20.
Hirao Y, Mihara Y, Kira I, Abe I, Yokozeki K. Enzymatic production of l-alanyl-l-glutamine by recombinant E. coli expressing α-amino acid ester acyltransferase from Sphingobacterium siyangensis. J Agric Chem Soc Jpn. 2013;77(3):618–23.
Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–5.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR–Cas9 system. Nat Protoc. 2013;8:2281–308.
Studier FW. Protein production by auto-induction in high-density shaking cultures. Protein Expr Purif. 2005;41(1):207–34.
Lin B, Tao Y. Whole-cell biocatalysts by design. Microb Cell Fact. 2017;16:106.
Tabata K, Ikeda H, Hashimoto S. ywfE in Bacillus subtilis codes for a novel enzyme, l-amino acid α-ligase. J Bacteriol. 2005;187:5195–202.
Acknowledgements
This work was supported by the National Natural Science Foundation of China Grant (31670051) and the National Key Research and Development Program of China (2018YFA0901400).
Funding
National Natural Science Foundation of China Grant (31670051) and the National Key Research and Development Program of China (2018YFA0901400).
Author information
Authors and Affiliations
Contributions
J-MZ carried out the main work, collected and analyzed the data, and drafted the manuscript. WY, B-HW, QL, X-TZ, Q-XG, J-ZL participated in the research. B-XL supervised the work, design of the study, participated in data analysis, and revised the manuscript. J-ZH and YT participated in the conception and finalized the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Fig. S1.
Phylogenetic tree of BacD homologs. Fig. S2. SDS-PAGE analysis of different BacD proteins in E. coli. Fig. S3. SDS-PAGE analysis of strains which co-expressing CgGlnA and BacD. Fig. S4. SDS-PAGE analysis of strains by RBS optimization. Fig. S5. Concentration of glucose and acetic acid in the conversion.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhu, J., Yang, W., Wang, B. et al. Metabolic engineering of Escherichia coli for efficient production of l-alanyl-l-glutamine. Microb Cell Fact 19, 129 (2020). https://doi.org/10.1186/s12934-020-01369-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-020-01369-2