
Abstract
Actinobacteria are characterized as the most prominent producer of natural products (NPs) with pharmaceutical importance. The production of NPs from these actinobacteria is associated with particular biosynthetic gene clusters (BGCs) in these microorganisms. The majority of these BGCs include polyketide synthase (PKS) or non-ribosomal peptide synthase (NRPS) or a combination of both PKS and NRPS. Macrolides compounds contain a core macro-lactone ring (aglycone) decorated with diverse functional groups in their chemical structures. The aglycon is generated by megaenzyme polyketide synthases (PKSs) from diverse acyl-CoA as precursor substrates. Further, post-PKS enzymes are responsible for allocating the structural diversity and functional characteristics for their biological activities. Macrolides are biologically important for their uses in therapeutics as antibiotics, anti-tumor agents, immunosuppressants, anti-parasites and many more. Thus, precise genetic/metabolic engineering of actinobacteria along with the application of various chemical/biological approaches have made it plausible for production of macrolides in industrial scale or generation of their novel derivatives with more effective biological properties. In this review, we have discussed versatile approaches for generating a wide range of macrolide structures by engineering the PKS and post-PKS cascades at either enzyme or cellular level in actinobacteria species, either the native or heterologous producer strains.
Similar content being viewed by others


Background
Natural products (NPs) from plants or microorganism, native or structurally modified have been utilized for the treatment of infections and ailment of disease conditions [1, 2]. Among all microorganisms, actinobacteria isolated from terrestrial or marine sources are characterized prominent producer of such NPs of pharmaceutical values, such as antibacterial, anticancer agents, anti-parasite, and immunosuppressant [3,4,5]. Actinobacteria are Gram-positive filamentous bacteria containing a high G+C content. They generally produce mycelium and reproduce by sporulation similar to filamentous fungi. However, they possess prokaryotic nucleoid and peptidoglycan cell wall significantly different from fungi [6]. Hence, the name “actinobacteria” is derived from Greek whereas; “aktis or aktin” meaning “ray” and “mukes” meaning “fungi”. The production of NPs from these actinobacteria is associated with diverse biosynthetic gene clusters (BGCs) present in these microorganisms. Each BGC contain a defined set of genes sufficient for biosynthesis of particular chemical structure. The majority of these BGC include polyketide synthase (PKS) or non-ribosomal peptide synthase (NRPS) or combination of PKS and NRPS.
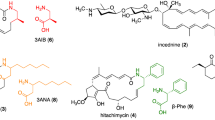
Macrolides include diversified chemical structures containing a core macro-lactone ring (aglycone) decorated with diverse functional groups, most commonly deoxy-sugars and amino-sugars [7]. They are biologically important for their uses in therapeutics as antibiotics (erythromycin, pikromycin, nargenicin, oleandomycin), anti-tumor agents (epothilone), immunosuppressant (rapamycin, FK506) and anti-parasites (avermectin) (Fig. 1) [8, 9]. Macrolides are categorized into different groups according to the number of atoms in the lactone ring, for example, 12-, 14-, 15-, 16-, 17-(ivorenolide B) or 18-(tedanolide C) membered macrolides [10]. They are generally biosynthesized by type I polyketide synthase (PKS) and modified by tailoring enzymes such as glycosyltransferase (GT), methyltransferase (MT) and oxidation enzymes as monooxygenase (MO), cytochrome P450 (CYP450) and oxidoreductase (OR).

Structures of different macrocyclic compounds with their primary producer strain and potential use of these molecules
The alarmingly increase in drug-resistant bugs or drug-tolerant cancer conditions have vitalized the need for bioactive molecules with new or improved pharmacological properties. Therefore, there is immersing interest on isolation and characterization of novel macrolides. The clinically useful macrolides have been traditionally derived from the screening of natural sources, and still the isolation of novel macrolides and evaluating their bioactivities has been an active area of research [11]. Most of the NPs, including macrolides, exhibit a broad range of pharmacophore, but still, they need to be structurally modified to ameliorate their chemical and biological properties for clinical uses [5, 12]. The total synthesis of macrolide derivative or chemical modification to generate semi-synthetic derivative have been an important tool for the development of novel molecules with enhanced pharmacological activities. However, such chemical approaches have limitations as many sites on the macrolactone ring and/or the sugar moiety are not amenable to desired chemical modifications [9, 13]. Another promising approach has been the biological engineering of the production hosts by rational metabolic engineering and synthetic biology tools. The major advantage of such biological process over chemical approach is cost-effectiveness, environmental friendliness, and easy scale-up possibilities [14, 15].
In this review, the progress on engineering of actinomycetes for deriving novel macrolides by engineering of aglycon biosynthesis and post-PKS tailoring enzymes has been summarized. The front part of the review includes early approaches of precursor-directed biosynthesis and mutasynthesis. Finally, the application of the advanced synthetic biological tools assisted by different “–omics” based approaches for combinatorial biosynthesis of macrolides have been discussed.
Outline of biosynthesis of macrolides
The type I PKS generally consist of multifunctional, multi-modular enzymes with non-iterative catalysis of one cycle of polyketide chain elongation. These enzymes are responsible for successive condensation of activated coenzyme A (CoA) thioesters (most commonly acetyl-, propionyl-, malonyl- or methylmalonyl-CoA) for such polyketide chain elongation. Each module contains a set of functional domains of acyl carrier protein (ACP), acyltransferase (AT), and, β-ketoacyl synthase (KS), which are essential for polyketide elongation [16, 17]. The CoAs used as substrate is selected and activated by an acyl transferase (AT) domain and transferred to the 4′-phosphopantetheinylated acyl carrier protein (ACP) domain. The ketosynthase (KS) domain catalyzes the decarboxylative Claisen-like condensation between the substrate and the growing polyketide, to form a carbon–carbon bond between the alpha carbon of the extender unit and the thioester carbonyl of the ACP-bound acyl chain [9, 18, 19]. Besides these minimal domains, additionally, there are β-keto processing domains, ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) that act sequentially to reduce the β-keto group into a fully saturated acyl chain [20]. The synthesized polyketide is off-loaded by thioesterase (TE) domain [21, 22]. Figure 2 shows the schematic organization of modular PKS as exemplified by tylosin (Tyl) PKS, which is 16-membered macrolide produced by Streptomyces fradiae. The tylosin PKS includes one loading module and seven extension modules terminating in thioesterase (TE) domain to generate tylactone [23,24,25]. The aglycone is produced by the cordial action of the megasynthase enzymes encoded by five different genes (TylGI-TylGV). Four different thioester-CoAs are selected by AT domains of each module. Each selected extender units are modified to a certain degree by respective reducing enzymes, except the module 4 in which KR domain is null functional. In the end, the PKS product is off-loaded by the action of TE domain present in the last module 7, encoded by TylGV gene. Once the PKS product is released, the biosynthesis is further extended by post-PKS tailoring enzymes such as cytochrome P450 monooxygenase (CYP450), glycosyltransferases (GTs) and methyltransferases (MTs) [26].

Biosynthetic gene assembly of tylosin biosynthesis gene cluster illustrating condensation and modification of different extender units to form tylactone aglycone which is further decorated with different post-modification enzymes to form tylosin
Despite the numerous reports on the “canonical” organization and co-linearity in most of the type I PKSs, non-canonical examples of type I PKS are popular [27]. The skipping over of the extraneous functional domains or modules leading to the formation of unusual intermediates or final compounds have been reported [28, 29]. Similarly trans-acting domains have been uncovered from various polyketide biosynthesis. These trans-enzymes are usually physically separate from the usual modifying domains in the modular PKSs, but they act at specific points during the processing of the nascent acyl chains. One of the prominent examples is trans-AT or AT-less biosynthesis of polyketides whereas AT activity is provided at each elongation step in trans by free-standing AT usually encoded in the biosynthetic gene clusters [30]. Trans-ER has been characterized from lovastatin polyketide biosynthesis [31] and switchable ER domain has been reported in azalomycin F biosynthesis [32]. Similarly, some trans-acting thioesterases (TE type II) are responsible for editing function by hydrolytic removal of aberrant residues blocking the megasynthase, participation in substrate selection, intermediate, and product release [33]. Similarly broadly selective acyltransferase has been characterized from the polyketide synthase of splenocin [34]. The detailed information on extended unit promiscuity and orthogonal protein interaction of ACP and trans-AT has been also availed [35, 36]. So, these type of new information are not only providing novel insight on the biosynthetic mechanism of polyketides but also providing opportunities for plausible bio-engineering of these macrolides. Basically, the diversification of macrolides can be tuned at four major steps throughout biosynthesis through (1) choice of the building block and chain length, (2) reduction and stereochemical arrangement of β-keto intermediates, (3) rearrangements and secondary cyclizations and (4) post-PKS tailoring [37]. Recent information on the protein structure of the enzymes and advanced genetic manipulation techniques provides enormous opportunity for fine tuning the post-PKS steps to generate novel or structurally diversified macrolides [38, 39]. Figure 3 depicts the plausible modification centers in macrolide that can be altered by utilizing different engineering aspects in the actinobacteria either by rational modification on PKS or post-PKS steps.

Structure of tylosin showing possible modification/engineering sites for engineering/diversification of tylosin and related molecules
Precursor-directed biogenesis of novel antibiotics
The prolific generation of mutant bugs to the many pre-existing drugs has caused pitfall in efficiency of enhancement approach as a panacea for addressing the issues of drug resistance. Nevertheless, precursor directed biogenesis can still substantiate as a promising technique by providing a fertile platform for structurally diversifying the antibiotics, whereas the altered natural product precursors are subjected to the microorganisms and subsequently incorporated to reconstitute the product of interest [40]. The alternative precursor-directed biosynthesis can be utilized as an approach where the chemical and biological inputs are assembled for generation of novel natural products. For example, an orally active β-lactam antibiotic, penicillin V (phenoxymethylpenicillin), was produced by adding phenoxyacetic acid to fermentations of Penicillium chrysogenum [41].
But the major drawbacks of this approach can be inherent competition between alternative precursors and natural precursors, rendering the yield of novel derivatives to be low. However, this shortcoming can be overruled by blocking the synthesis of natural precursors, either by mutating key genes in the respective bio-synthetic pathway or by adding specific inhibitors of biosynthetic enzymes [42]. This approach in which the biosynthetic pathway is coupled with the feeding of synthetic starter precursors, where the precursors are reorganized in the biosynthetic pathway to remold the final compound can be otherwise be termed “chemosynthetic biogenesis”. Hence utilizing the benefits of this approach, various novel analogs of erythromycin and other polyketides are generated [43,44,45,46,47].
Fundamentally for generating diversities of analogs of erythromycin, a designed mutant of a modular PKS by inactivation were generated, that lacked early-stage enzyme activities. DEBS (KS10), a mutant of DEBS that was inactivated by site-directed mutagenesis of the β-ketoacyl-ACP synthase domain of module 1 (KS1) was used for precursor mediated structural diversification strategy [42]. The native KS1 domain catalyzes the first condensation step of 6-deoxyerythronolide B (6-dEB) biosynthesis (Fig. 4a) but the DEBS (KS10) mutant is incapable of carrying out the first round of polyketide chain elongation from the propionyl-CoA and methylmalonyl-CoA causing incapability for the formation of the macrolide 6-dEB. In turn, by the introduction of synthetic diketide and triketide such as the N-acetyl cysteamine (SNAC) thioester, which are analogs of the natural diketide intermediate to Streptomyces coelicolor CH999/pJRJ2, an engineered strain harboring DEBS (KS10), various novel analogs of erythromycin were generated (Fig. 4b) [48].

a Biosynthesis pathway assembly of erythromycin. b Biosynthesis of diverse erythromycin derivatives using Streptomyces coelicolor deficient in KS10. Different activated synthetic diketides and triketides were supplemented to the culture
Mutasynthesis
The precursor directed biogenesis has been utilized and still remains as a promising strategy for structurally diversifying and generating versatile chemical identities with superior properties or activities. But, the major drawback clinging to these approaches, most important being that mixture of natural and nonnatural products with similar physical properties are often produced, leading to complex downstream purification procedures; high concentrations of synthetic precursor are often required to compete with the preferred natural precursor and; a limited range of intermediates are efficiently incorporated into the final product. So, the precursor-directed biosynthesis has been complemented with mutasynthesis approach whereas the naturally occurring precursor pathways are inactivated by mutation to remove the competition with natural precursors.
Streptomyces avermitilis mutant (S. avermitilis Δbkd) was generated wherein the enzymes, branched chain fatty acid dehydrogenase complex Bkd required for generating the precursors 2-methylbutyryl-CoA and isobutyryl-CoA were inactivated. Different precursor analogs were fed to the mutant and a cyclohexyl-containing avermectin derivative (later named doramectin) was generated with increased antiparasitic activity [49]. Similarly, the deletion of 5-chlorodeoxyadenosine (a precursor for chloroethylmalonyl-CoA) biosynthetic genes in Salinispora tropica [50] and exogenous supplementation of 5-fluorodeoxyadenosine resulted in the generation of fluorosalinosporamide.
Combinatorial biosynthesis for diversification of antibiotics
“Combinatorial biosynthesis” is one of the promising strategies for the genesis of novel analogs of predominant antibiotics or modifying their structural aspects for better pharmacological properties. This approach intervenes with the strategies for the genetic engineering of natural product biosynthesis to obtain new molecules, including the use of genetics in medicinal chemistry [51]. It has been an important approach to generate chemical diversity by precise genetic manipulations and thus implies with the possibility of generation of large libraries of complex compounds to feed a modern high-throughput screening operation [51, 52]. Thus, combinatorial biosynthesis is a recent addition to the metabolic engineering toolbox by which genes responsible for individual metabolic reactions from different organisms are combined to generate the metabolic pathways to biosynthesize the desired products [53]. Fundamentally, it can be distinguished from mutasynthesis approach on the bases that a mutasynthesis is an approach where there is inactivation of some key functional genes, thus perturbing the biosynthesis pathway to new products by supplementation of feasible precursors or chemical entities; whereas combinatorial biosynthesis rely on additional gene functions by heterologous expression of functionally similar or diverse genes so that the pathway is tuned for crafting novel analogs. It has been widely applied to achieve new derivatives related to polyketides, coumarins, indolocarbazoles and other types of antibiotics [53,54,55].
Among the compounds that have been developed into medicines, including the antimicrobials and antibiotics, the polyketides occupy significant part. The polyketides are produced as a diverse array of products manifested by the bacterial megaenzyme polyketide synthases (PKSs). The distinct modularity in the genetic architecture of these PKSs provides sufficient ground for expecting feasibility for engineering the enzymes to produce novel drug candidates, by ‘combinatorial biosynthesis’ [56].
The supplementation of the shikimate-derived cyclohexyl-CoA biosynthetic pathway into S. avermitilis Δbkd enabled the production of doramectin without cyclohexanoic acid supplementation [57]. Similarly, supplementation of diverse carboxylic acid starter unit to spinosyn PKS containing loading modules of avermectin or erythromycin could generate diverse spinosyn analogs [58]. Similarly, multiple bioactive macrolides were generated by hybrid modular PKS from pikromycin gene cluster, erythromycin gene cluster, and tylosin gene cluster, whereas rigorous interchange of modules is employed between the PKSs [59]. This work demonstrated the unique capacity of combinatorial biosynthesis for accelerating the creation of novel biologically active natural products.
By utilizing S. venezuelae based combinatorial biosynthesis machinery, many novel analogs of various secondary metabolites with diverse structure and diverse activities have been generated [60,61,62,63,64]. S. venezuelae ATCC 15439 was engineered for deletion of genes responsible for biosynthesis and attachment of TDP-4-keto-6-deoxy-d-glucose and the strain was designated as S. venezuelae YJ003. The sugar gene cassettes encoding for deoxysugar biosynthesis and glycosylation were expressed in the engineered strain to generate olivosyl and quinovosyl derivative of various macrolides [60]. S. venezuelae was genetically engineered for deletion of the entire biosynthetic gene cluster encoding the pikromycin PKS and desosamine biosynthetic enzyme [61]. After crafting the amenable host for combinatorial biosynthesis, it was used multifarious for generation of different analogs of targeted polyketides. The engineered deoxysugar biosynthetic pathways for biosynthesis of thymidine diphosphate (TDP)-d-quinovose or TDP-d-olivose along with substrate flexible glycosyltransferase–auxiliary protein pair DesVII/DesVIII from S. venezuelae were expressed in the mutant strain to which 12-, 14-, and 16-membered ring macrolactones i.e. 10-deoxymethynolide, narbonolide, and tylactone, respectively were feed to generate corresponding quinovose- and olivose-glycosylated macrolides. The conversion of 12-, 14-, and 16-membered ring macrolactones including 10-deoxymethylnolide, narbonolide, and tylactone were achieved in engineered S. venezuelae strain with deoxysugar biosynthetic pathways (TDP-d-quinovose or TDP-d-olivose) together with glycosyltransferase-auxiliary protein (DesVII/DesVIII) and produce their glycosylated scaffolds as quinovosyl and olivosyl macrolactones. The synthesized compounds were YC-17 and narbomycin. Similarly, while replacing the DesVII/DesVIII by substrate-flexible glycosyltransferase TylMII coupled with partner protein TylMIII derived from S. fradiae, a mycaminosyl derivative of tylactone (5-O-mycaminosyl tylactone) was produced [61]. Similarly, expression of complete biosynthetic pathways for the biosynthesis of TDP-3-demethyl-d-chalcose or TDP l-rhamnose together with the glycosyltransferase-auxiliary protein pair DesVII/DesVIII along with subsequent feeding of 16-membered ring macrolactone tylactone was fed to this engineered host, which in turn successfully produced 3-O-demethyl-d-chalcosyl, l-rhamnosyl, and d-quinovosyl derivatives [62]. Similarly, using S. venezuelae YJ003 and expression of complete biosynthetic pathways for the biosynthesis of TDP-3-dimethyl-d-chalcose or TDP-l-rhamnose together with DesVII/DesVIII, novel narbomycin derivative decorated with l-rhamnose or 3-O-demethyl-d-chalcose were generated. These novel analogs exhibited greater antibacterial activity than narbomycin and the clinically relevant erythromycin [62]. In another instance, for another aglycone YC-17, the native d-desosamine was replaced by d-quinovose, l-olivose, l-rhamnose, and d-boivinose to generate YC-17 glycoside analogs as d-quinovosyl-10-deoxymethynolide, l-olivosyl-10-deoxymethynolide, l-rhamnosyl-10-deoxymethynolide, and d-boivinosyl-10-deoxymethynolide respectively by expression of gene cassette responsible for biosynthesis of respective deoxysugars (Fig. 5). The assessment of biological activity indicated that l-rhamnosyl-10-deoxymethynolide exhibited better activities against clinically isolated erythromycin-resistant pathogenic strains, as well as erythromycin-susceptible strains relative to YC-17 and its other analogs [64]. These all studies indicate that the combinatorial biosynthesis mediated structural diversification can be one of the efficient techniques for modifying the structure and thus rendering enhancement in activity mediated by the structure–activity relationship as par distinct structural scaffold contributing for a particular range of activity.

Structures of different sugars conjugated macrolides produced using TDP-sugars biosynthesis pathway engineered Streptomyces recombinant strains
The large multifunctional PKS enzymes synthesize macrolides [65]; which undergoes multiple modifications by post-PKS enzymes mediated reactions such as oxidation, methylation, glycosylation, hydroxylation and many more giving rise to chemically distinct structures. The functional and structural diversity of these compounds is controlled by these post-modifications and most often these modifications are critical for biological activities [38]. The hydroxylation or other modifications contributed by cytochrome P450 monooxygenases are the key steps leading to structural diversity and biological activities to macrolide antibiotics [66]. Hence, by utilizing the substrate flexible cytochromes for combinatorial biosynthesis novel analogs were generated. An engineered S. venezuelae HK954 mutant strain (with deletion of the last module of pikromycin PKS (pikAIV) but intact substrate flexible cytochrome P450, pikC) was used for generating novel hydroxylated analogs of oleandomycin [67]. Similarly, S. venezuelae mutant strain YJ003 blocked in desosamine biosynthesis pathway was used for the expression of substrate flexible cytochrome P450s, EryF from erythromycin gene cluster and OleP from oleandomycin gene cluster to generate novel analogs of 12 and 14 membered macrolactones inherently produced by the host strain (Fig. 6) [67].

Generation of different macrolactones using selected enzymes such as PikC, EryF and OleP
Metabolic engineering approaches in actinomycetes for macrolide biosynthesis
The major constraint for biosynthesis of macrolides in the producer-host has been abundant availability of precursors and cofactors, the lower expression level of biosynthetic genes and regulation of the biosynthetic gene/genes. Generally, the bottlenecks in biosynthetic pathways restrict the substantial production of the natural product in desired titers. This is significantly associated with the limitations of the flux of key precursors from primary metabolism to secondary metabolic pathways [68, 69]. Generally, the precursors/cofactors are derived from primary metabolism including glycolysis, pentose phosphate pathway, tricarboxylic acid cycle and amino acid/nucleic acid metabolism [70]. Thus these types of limitations can be overcomed by amplifying the gene or genes that encode enzymes associated with such bottlenecks resulting in increased enzyme levels to diminish the bottleneck effects and hence, improved titers can be achieved [71, 72]. Fundamentally, regulation and correlation of precursor supply for improving natural product amounts are focused on carbohydrate metabolism, fatty acid precursors, and intracellular cofactor supplies [73, 74]. There are ample of examples illustrating the genetic circuit guided pathway engineering approaches for enhancing the secondary metabolites of importance [75], such as heterologous overexpression of the S -adenosyl-L -methionine (SAM) synthetase metK, improved production of different antibiotics, such as actinorhodin, avermectin, and pikromycin [76, 77]. For example, the engineering of methylmalonyl-CoA biogenesis pathway by generating MCM gene, methylmalonyl-CoA mutase (mutB) knock out mutant of S. erythraea, there was an enhanced level of erythromycin in the carbohydrate-based medium. However, there was an elevated level of erythromycin in S. erythraea recombinant containing MCM operon (meaA, mutB, meaB, mutR) in the oil-based medium [78]. Similarly by supplementation of cheap primary sources which subsequently contribute for specific fatty acid precursors viz. acetyl-CoA, malonyl-CoA, methyl malonyl-CoA and ethyl malonyl-CoA along with precursor redirecting enzyme complexes such as acetyl CoA carboxylase, and propionyl CoA carboxylase different antibiotics such as nargenicin A1, pikromycin, and erythromycin were elevated to significant level [79,80,81,82]. Hence, metabolic engineering combined with redirection of specific precursors can be a rational approach for enhancing the secondary metabolites of importance. Similarly, such approaches for enriching the acyl-CoA precursors have been used for an elevated level of diverse macrolides (Table 1). Recently, various engineering strategies for directing the catabolism of branched-chain amino acids (BCAA) into various acyl-CoA compounds has extended the opportunities for metabolic engineering of acyl-CoA pathways and yield improvement of macrolides [83].
Similarly, the expression of regulatory genes has been promising strategies to enhance the production titer or activation of novel macrolides in diverse actinomycetes. Generally, the introduction of synthetic or natural promoter has been effective for triggering either overproduction or activation of silent/cryptic gene clusters with low or no expression [84,85,86,87,88,89]. The metabolic engineering approaches utilizing overexpression/deletion of various global regulators or pathway-specific positive regulators are utilized for either enhancing the production yield or obtaining novel macrolides from native hosts/heterologous hosts (Table 1).
The availability of the crystal structure of complete module of PKS [16, 90] or its constituent domains as ACP [91], dehydratase [92], thioesterase [93, 94] has provided a better understanding on the mechanism of macrolactone biosynthesis. In case of polyketide biosynthesis, the AT domain is crucial for controlling the recognition of the extender unit. Thus, there is a possibility of changing the building block specificity and the basic backbone of the polyketide by altering the AT domain. Such valuable information has been utilized for rational engineering of macrolactones. The site-directed mutagenesis of the domains or intact domain exchange to change their substrate specificities has been widely used for generating diverse analogs of the macrolides, which have been reviewed elsewhere [13]. The exchange of AT domain by domain swapping can cause impaired protein folding [95]. However, Yuzawa et al. identified the highly conserved boundaries and exhibited the feasibility of AT domain replacement in macrolides [96]. But the direct engineering of innate AT domain can be a more reliable alternative for varying the substrate ranges [97]. For example, the site-directed mutagenesis of AT domain and feeding of 2-propagylmalonyl-SNAC to S. erythraea (containing AT6 of DEBS mutated with Val295 to Ala) to generate 2-propargyl erythromycin [98]. The mutation is not only capable of extending the substrate specificity to natural extender units but also alter the specificity non-natural extender units. For example, the selected mutation at Tyr189 in DEBS3 AT6 domain resulted in the dramatic changes in product distribution by accepting diverse non-natural extender units [99].
Similarly, novel derivatives of NPP have been generated by site-directed mutagenesis in enoyl reductase domain in module 5 of the NPP A1 polyketide synthase NppC. The compound exhibited comparable antifungal activity against Candida albicans with lesser toxicity than antifungal, amphotericin B [100]. However, such kind of domain modification may create inactive proteins or change the chemistry of inter-domain interactions. So entire domain exchange has been a superior option and several analogs of erythromycin have been generated by replacing methylmalonyl-specific acyltransferase (AT) domains of the 6-deoxyerythronolide B synthase (DEBS) with malonyl-, ethylmalonyl-, or methoxymalonyl-specific domains [101].
Similarly, metabolic engineering approach has been performed for complete exchange of the module. For example, the loading module from the rifamycin biosynthetic pathway as a substitution for the original loading domain was incorporated in the PKS so that the altered PKS was amenable for accepting benzoate as starter unit instead of the propionyl-CoA used in native. By this strategy, a novel benzyl-erythromycin analog was generated and by utilizing further precursor flux enhancement and pathway engineering approaches the titer of the novel derivative was substantially increased (Fig. 7). The novel derivative was capable to show comparable activity to the parent compound whereas showing pronounced efficacy against the erythromycin resistant pathogens [102]. Similarly, the domain swapping of pikromycin thioesterase to linear polyketide tautomycetin (TMC) was able to generate the cyclized form of macrolactones [103].

Biosynthesis of benzyoyl erythromycin by replacing loading module of erythromycin from loading module of rifamycin biosynthesis pathway
In general, most of the actinomycetes strain is capable of generating profuse secondary metabolites with different structures or activities. Most often these chemical entities share common scaffold and thus similar precursor régimes. Thus, the precise and efficient manipulation of modular PKSs is often hindered by biological constraints posed by organisms (principally actinomycetes) due to their intricate biological pathways and complex distribution of precursor flux for the constitution of different secondary metabolites. To address this constraint, an heterologous expression host as Escherichia coli was engineered with the introduction of the three DEBS genes from S. erythraea, the sfp phosphopantetheinyl transferase gene from Bacillus subtilis, and the genes encoding a heterodimeric propionyl-CoA carboxylase from S. coelicolor [104]. This engineered E. coli BAP1 strain was utilized for fortifying generation of novel analogs of erythromycin (Fig. 7), as described previously. Further, the production was fine-tuned by reducing the total number of plasmids [105] and utilizing the BAC vector [106]. The metabolic engineering approach by eliminating competitive native pathway for nucleotide-activated sugar biosynthesis pathways (vioA/vioB/wzx, wecD/wecE, and rmID) substantially increased the production titer [106]. The metabolic engineering approach was employed for expanding the formation of novel erythromycin analogs by altering the tailoring enzymes involved in sugar biosynthesis. Among them few of the novel analogs exhibited promising activity against the erythromycin-resistant strain of B. subtilis, which provided a rationale of this diversification strategy for increasing the therapeutic potential of erythromycin [107].
Thus heterologous expression has been an efficient approach of metabolic engineering where a single gene, or a set of genes, or entire biosynthetic pathway genes are introduced in the microbial host to identify and engineer the corresponding natural products [108]. When if the native producer strains are not genetically tractable or not amenable to metabolic engineering approaches, the heterologous hosts provide an efficient approach for gaining access to secondary metabolites encoded by the particular BGCs. In addition, the generation of metabolic engineering by removing the competing biosynthetic pathways to generate clean hosts provides a suitable platform for isolation, characterization, and production of various macrolides [109, 110]. The generation of cluster free clean hosts provides new avenues for redirecting the flow of precursor pathways directly to the target secondary metabolite heterologously expressed [111]. Diverse macrolide biosynthesized through heterologous hosts are presented in Table 2. In some cases, the heterologous expression of the BGCs needs to be tuned by expression of the regulatory genes. For example, the heterologous expression of a tylosin gene cluster in S. venezuelae was enhanced by expression of pikD, a positive regulator from pikromycin biosynthetic pathway of S. venezuelae. In other cases, the tuning of heterologous expression by an engineered precursor pathway can enhance the production titer or generate novel derivatives in heterologous expression systems [112]. The introduction of the biosynthetic pathway for methoxymalonyl-ACP into an S. fradiae strain heterologously expressing the midecamycin pathway led to enhancement of midecamycin and generation of a novel compound as an analog of methoxymalonate-platenolide [112].
Recently, the biocatalytic/chemo-biocatalytic approach employing a heterologous expression of the partial PKS and substantial modification has been employed as an effective approach for generating novel macrolides [113]. For example, the Pik pentaketide precursor was supplemented to the expressed protein of PikAIII-TE or PIKAIII-PikAV to generate the macrolactones, 10-deoxymethynolide, and narbonolide. Further biotransformation with engineered S. venezuelae could generate diverse derivatives related to pikromycin and methymycin. Similarly, activated synthetic hexaketide was further extended with a methyl malonyl and malonyl units followed by lactonization to form tylactone aglycon by in vitro catalysis using tylosin biosynthesis complementary modules (module 6 and 7 encoded by JuvEIV and JuvEV genes) from juvenimicin BGC. The tylactone was further biotransformed to M-4365 G1 using S. fradiae DHS 316 which is deficient of tylactone BGC. This intermediate was modified into ten different macrolide derivatives using CYP450 and chemical oxidation methods [114, 115] (Fig. 8). This approach advanced the use of type I PKS enzymes in vitro for generation of novel antibiotics.

Chemoenzymatic and biocatalytic synthesis of structurally diverse tylactone-based macrolides antibiotics
However, this method suffers from the limitations of scalability whereas the starting materials need to be supplied in sufficient amounts for sustainable production. In addition, there is a requirement of strict control in unwanted reactive centers but enough reaction in desired reaction centers, which makes these synthesis processes tedious and expensive. Thus, biological engineering of macrolides by using sustainable metabolism in the microbial hosts may always remain superior. Recently, synthetic biology approach which utilizes the artificial synthesis of genetic parts in revolutionizing such biological engineering aspects. For example, the promoter engineering by utilizing the regulatory sequences in the promoters can be an effective approach for tuning the production titer of macrolides [116]. The biosynthesis of epothilone was successfully accomplished by modular construction of artificial macrolide pathway [117]. The availability of detailed knowledge of diverse –omics related information can be successfully employed for fine tuning the heterologous expression of such macrolide compounds [118].
Microbial biotransformation for modification of antibiotics
Besides the precise genetic engineering approaches, microbial biotransformation strategies have attracted considerable interest for the post-modification of secondary metabolites in terms of their structural and functional characteristics [119,120,121]. Any changes in parental compounds in a later stage can refer to as post-modification in terms of structure and biotransformation is one of the well-studied and precise approaches to achieve such modifications with an optimal regio- and enantio-selectivity [122, 123]. However, the major modification includes cofactor-dependent hydroxylation/oxidation, dehydrogenation, methylation, glycosylation (O/C/N-glycosylation, deglycosylation), epoxidation, cyclization etc. [124,125,126,127], whereas the intracellular cofactors/enzymes present in the biotransformation host is responsible for desired modification of the supplemented substrate. Such whole-cell biotransformation can be achieved using various microbial species (Streptomyces, Myxobacterium, Bacillus, Corynebacterium, Pseudomonas, etc.) as a host.
A series of erythromycin D analogs conjugated with different sugar scaffolds were produced feeding 6-deoxyerythronolide B or 3-α-mycarosyl-erythronolide B in E. coli overexpressing the specific TDP-L-mycarose and TDP-desosamine sugar biosynthetic pathway [128]. Same biotransformation set-up was used for modification of erythromycin C, 3-α-mycarosyl-erythronolide B, azithromycin, and erythromycin D to modify them into megosamine conjugated products using reconstitutive TDP-l-megosamine biosynthetic pathway in E. coli [129, 130] (Fig. 9a). This experiment also suggested the possible routes for the production of diverse therapeutically important (antiparasitic, antiviral and antibacterial) compound megalomicin A including 12-deoxynucleoside triphosphate megalomicin A [130]. Importantly, sugar appendages present in macrolide antibiotics governs their biological properties and changes in them have a substantial effect [131]. So, deoxy sugar biosynthetic pathways are usually focused in terms of antibiotic modifications. 6-O-megosaminyl-erythromycin A, 6-O-megosaminyl-azithromycin, 6-O-epidigitoxosyl-erythromycin A and 6-O-daunosaminyl-erythromycin A were produced through the whole cell biotransformation. Their evaluation against several clinical isolates; standard and drug-resistant strains of human malarial parasites (Plasmodium falciparum) and liver stages of the rodent malaria parasite (Plasmodium berghei) were found more effective than parental compound [130].


a Biosynthesis of glycosylated macrolides from different TDP-sugars (TDP-L-mycarose, TDP-desosamine, TDP-megosamine) biosynthesis pathway overexpressed recombinant E. coli strains supplemented with corresponding macrolactone/macrolides. b Biotransformation of macrolides using Streptomyces venezuelae strain
Rosamicin, macrolide antibiotics isolated from the culture of soil bacterium was bio-transformed into 10, 11-dihydrorosamicin using S. venezuelae which showed enhanced in vitro antibacterial activity against MRSA [132]. Likewise, natural oligomycin A and semi-synthetic tilmicosin are considered and bio-hydrogenated using S. venezuelae and structurally modify into 2, 3-dihydro-oligomycin A and 10, 11-dihydro-tilmicosin (Fig. 9b). This changes in natural scaffold oligomycin and tilmicosin brought increased activity against S. cerevisiae and Bacillus subtilis respectively [133]. Similarly, macrocyclic lactone antibiotic streptogramin A namely 5, 6-dihydrovirginiamycin M1 was created by feeding virginiamycin M1 into a culture of recombinant S. venezuelae through the mechanism of bio-dehydrogenation catalysis [134]. The generated analog showed enhanced anti-MRSA activity compared to the parent compound.
Conclusions and future perspectives
Actinobacteria have been already established as the promising platform for the production of macrolides and their novel derivatives. Moreover, the recent advances in various “–omics” provides new paths for uncovering the genomic information and their expression levels. Moreover, the availability of various bioinformatics tools for classifying the genome to BGCs and analysis of the products by versatile metabolome analyzing tools helps in connecting the genome to a particular molecule, which is also otherwise called as “genome mining” approach [135,136,137,138,139]. In addition, the current success of enzyme/host engineering has narrowed down the gap between the understanding of PKS biosynthetic logic and its propensity for PKS diversification. The advances in genome sequencing, protein crystallography, and gene-synthesis system have made it feasible for designing, building and testing chimeric PKSs. The advancement in the analytical methods by mass spectrometry and molecular networking have increased our capacity for detecting the products [140]. The feasibility of the optimization of production hosts either native or heterologous by computational and systems biological tools [141] provides an effective or alternative route for fluxing the precursors toward the biosynthesis from such chimeric PKS. Various genome engineering techniques such as Multiplex Automated Genome Engineering (MAGE) [142, 143] and Clustered Regularly Interspaced Short Palindromic Repeats (CRISPRs)/CRISPR associated enzyme (Cas) [144, 145] provides ample of opportunities for precise metabolic engineering of the PKS and post-PKS biosynthetic steps in the native/heterologous hosts. Additionally, diverse BGCs cloning techniques as Bacterial artificial chromosome (BAC) cloning, Gibson assembly, Linear–linear homologous recombination (LLHR), Golden Gate assembly and Transformation-associated recombination (TAR) have played pivotal role in unlocking diverse NP resources [146, 147] The availability of detailed genomic information of producer strains and precise analysis facilitated by efficient cloning methods and utilizing the metabolically engineered host system can be next generation approach for isolation of novel macrolides or enhanced production of existing macrolides with pharmaceutical values.
Availability of data and materials
Not applicable
Abbreviations
- BGCs:
-
biosynthetic gene clusters
- PKSs:
-
polyketide synthases
- NRPS:
-
non-ribosomal peptide synthase
- DEBS:
-
6-deoxyerythronolide B synthase
- NPP:
-
nystatin-like Pseudonocardia polyene
- MRSA:
-
methicillin resistance Staphylococcus aureus
- MAGE:
-
Multiplex Automated Genome Engineering
- CRISPRs:
-
Clustered Regularly Interspaced Short Palindromic Repeats
References
Newman DJ, Cragg GM. Natural products as sources of new drugs from 1981 to 2014. J Nat Prod. 2016;79:629–61.
Dhakal D, Sohng JK. Coalition of biology and chemistry for ameliorating antimicrobial drug discovery. Front Microbiol. 2017;8:734.
Olano C, Méndez C, Salas J. Antitumor compounds from marine actinomycetes. Mar drugs. 2009;7:210–48.
Genilloud O. Actinomycetes: still a source of novel antibiotics. Nat Prod Rep. 2017;34:1203–32.
Dhakal D, Pokhrel AR, Shrestha B, Sohng JK. Marine rare Actinobacteria: isolation, characterization, and strategies for harnessing bioactive compounds. Front Microbiol. 2017;8:1106.
Barka EA, Vatsa P, Sanchez L, Gaveau-Vaillant N, Jacquard C, Meier-Kolthoff JP, Klenk HP, Clément C, Ouhdouch Y, van Wezel GP. Taxonomy, physiology, and natural products of actinobacteria. Microbiol Mol Biol Rev. 2015;80:1–43.
Kirst HA. Introduction to the macrolide antibiotics. In: Schonfeld W, Kirst HA, editors. Macrolide antibiotics. Basel: Birkhauser Verlag; 2002. p. 1–13.
Gaynor M, Mankin AS. Macrolide antibiotics: binding site, mechanism of action, resistance. Curr Top Med Chem. 2003;3:949.
Park SR, Han AR, Ban YH, Yoo YJ, Kim EJ, Yoon YJ. Genetic engineering of macrolide biosynthesis: past advances, current state, and future prospects. Appl Microbiol Biotechnol. 2010;85:1227–39.
Arsic B, Barber J, Čikoš A, Mladenovic M, Stankovic N, Novak P. 16-membered macrolide antibiotics: a review. Int J Antimicrob Agents. 2018;51:283–98.
Demain AL, Sanchez S. Microbial drug discovery: 80 years of progress. J Antibiot. 2009;62:5–16.
Amoutzias GD, Chaliotis A, Mossialos D. Discovery strategies of bioactive compounds synthesized by nonribosomal peptide synthetases and type-I polyketide synthases derived from marine microbiomes. Mar Drugs. 2016;14:E80.
Weissman KJ. Genetic engineering of modular PKSs: from combinatorial biosynthesis to synthetic biology. Nat Prod Rep. 2016;33:203–30.
Dhakal D, Sohng JK. Commentary: toward a new focus in antibiotic and drug discovery from the Streptomyces arsenal. Front Microbiol. 2015;6:727.
Wohlleben W, Mast Y, Muth G, Röttgen M, Stegmann E, Weber T. Synthetic biology of secondary metabolite biosynthesis in actinomycetes: engineering precursor supply as a way to optimize antibiotic production. FEBS Lett. 2012;586:2171–6.
Dutta S, Whicher JR, Hansen DA, Hale WA, Chemler JA, Congdon GR, Narayan AR, Håkansson K, Sherman DH, Smith JL, Skiniotis G. Structure of a modular polyketide synthase. Nature. 2014;510:512.
Whicher JR, Dutta S, Hansen DA, Hale WA, Chemler JA, Dosey AM, Narayan AR, Håkansson K, Sherman DH, Smith JL, Skiniotis G. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature. 2014;510:560–4.
Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416.
Chen AY, Schnarr NA, Kim CY, Cane DE, Khosla C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide b synthase. J Am Chem Soc. 2006;128:3067–74.
Fischbach MA, Walsh CT. Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev. 2006;106:3468–96.
Keatinge-Clay AT. The structures of type I polyketide synthases. Nat Prod Rep. 2012;29:1050–73.
Xu W, Qiao K, Tang Y. Structural analysis of protein-protein interactions in type I polyketide synthases. Crit Rev Biochem Mol Biol. 2013;48:98–122.
Baltz RH, Sen ET. Properties of Streptomyces fradieae mutants blocked in biosynthesis of the macrolide antibiotic tylosin. Antimicrob Agents Chemother. 1981;20:214–25.
Cundliffe E, Bate N, Butler A, Fish S, Gandecha A, Merson-Davies L. The tylosin-biosynthetic genes of Streptomyces fradiae. Antonie Van Leeuwenhoek. 2001;79:229–34.
Fiers WD, Dodge GJ, Li Y, Smith JL, Fecik RA, Aldrich CC. Tylosin polyketide synthase module 3: stereospecificity, stereoselectivity and steady-state kinetic analysis of β-processing domains via diffusible, synthetic substrates. Chem Sci. 2015;6:5027–33.
Baltz RH, Seno ET, Stonesifer J, Wild GM. Biosynthesis of the macrolide antibiotic tylosin. A preferred pathway from tylactone to tylosin. J Antibiot (Tokyo). 1983;36:131–41.
Chen H, Du L. Iterative polyketide biosynthesis by modular polyketide synthases in bacteria. Appl Microbiol Biotechnol. 2016;100:541–57.
Beck BJ, Yoon YJ, Reynolds KA, Sherman DH. The hidden steps of domain skipping: macrolactone ring size determination in the pikromycin modular polyketide synthase. Chem Biol. 2002;9:575–83.
El-Sayed AK, Hothersall J, Cooper SM, Stephens E, Simpson TJ, Thomas CM. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem Biol. 2003;10:419–30.
Helfrich EJ, Piel J. Biosynthesis of polyketides by trans-AT polyketide synthases. Nat Prod Rep. 2016;33:231–316.
Ames BD, Nguyen C, Bruegger J, Smith P, Xu W, Ma S, Wong E, Wong S, Xie X, Li JWH, Vederas JC. Crystal structure and biochemical studies of the trans-acting polyketide enoyl reductase LovC from lovastatin biosynthesis. Proc Natl Acad Sci USA. 2012;109:11144–9.
Xu W, Zhai G, Liu Y, Li Y, Shi Y, Hong K, Hong H, Leadlay PF, Deng Z, Sun Y. An iterative module in the azalomycin F polyketide synthase contains a switchable enoylreductase domain. Angew Chem Int Ed Engl. 2017;2017(56):5503–6.
Kotowska M, Pawlik K. Roles of type II thioesterases and their application for secondary metabolite yield improvement. Appl Microbiol Biotechnol. 2014;98:7735–46.
Li Y, Zhang W, Zhang H, Tian W, Wu L, Wang S, Zheng M, Zhang J, Sun C, Deng Z, Sun Y. Structural basis of a broadly selective acyltransferase from the polyketide synthase of splenocin. Angew Chem Int Ed Engl. 2018;130:5925–9.
Musiol-Kroll EM, Wohlleben W. Acyltransferases as tools for polyketide synthase engineering. Antibiotics (Basel). 2018;7:E62.
Carpenter SM, Williams GJ. Extender unit promiscuity and orthogonal protein interactions of an aminomalonyl-ACP utilizing trans-acyltransferase from zwittermicin biosynthesis. ACS Chem Biol. 2018. https://doi.org/10.1021/acschembio.8b00867.
Cummings M, Breitling R, Takano E. Steps towards the synthetic biology of polyketide biosynthesis. FEMS Microbiol Lett. 2014;351:116–25.
Olano C, Méndez C, Salas JA. Post-PKS tailoring steps in natural product-producing actinomycetes from the perspective of combinatorial biosynthesis. Nat Prod Rep. 2010;27:571–616.
Nepal KK, Wang G. Streptomycetes: surrogate hosts for the genetic manipulation of biosynthetic gene clusters and production of natural products. Biotechnol Adv. 2018;37(1):1–20.
Wohlleben W, Pelzer S. New compounds by combining “modern” genomics and “old-fashioned” mutasynthesis. Chem Biol. 2002;9:1163–4.
Demain AL, Elander RP. The β-lactam antibiotics: past, present, and future. Antonie Van Leeuwenhoek. 1999;75:5–19.
Kinoshita K, Pfeifer BA, Khosla C, Cane DE. Precursor-directed polyketide biosynthesis in Escherichia coli. Bioorg Med Chem Lett. 2003;13:3701–4.
Jacobsen JR, Hutchinson CR, Cane DE, Khosla C. Precursor-directed biosynthesis of erythromycin analogs by an engineered polyketide synthase. Science. 1997;277:367–9.
Jacobsen JR, Keatinge-Clay AT, Cane DE, Khosla C. Precursor-directed biosynthesis of 12-ethyl erythromycin. Bioorg Med Chem. 1998;6:1171–7.
Weissman KJ, Bycroft M, Cutter AL, Hanefeld U, Frost EJ, Timoney MC, Harris R, Handa S, Roddis M, Staunton J, Leadlay PF. Evaluating precursor-directed biosynthesis towards novel erythromycins through in vitro studies on a bimodular polyketide synthase. Chem Biol. 1998;5:743–54.
Leaf T, Cadapan L, Carreras C, Regentin R, Ou S, Woo E, Ashley G, Licari P. Precursor-directed biosynthesis of 6-deoxyerythronolide B analogs in Streptomyces coelicolor: understanding precursor effects. Biotechnol Progress. 2000;16:553–6.
Lee HY, Harvey CJ, Cane DE, Khosla C. Improved precursor-directed biosynthesis in E. coli via directed evolution. J Antibiot (Tokyo). 2011;64:59–64.
Kinoshita K, Williard PG, Khosla C, Cane DE. Precursor-directed biosynthesis of 16-membered macrolides by the erythromycin polyketide synthase. J Am Chem Soc. 2001;123:2495–502.
Mehlhorn H, Jones HL, Weatherley AJ, Schumacher B. Doramectin, a new avermectin highly efficacious against gastrointestinal nematodes and lungworms of cattle and pigs: two studies carried out under field conditions in Germany. Parasitol Res. 1993;79:603e7.
Eustaquio AS, Moore BS. Mutasynthesis of fluorosalinosporamide, a potent and reversible inhibitor of the proteasome. Angew Chem Int Ed Engl. 2008;47:3936e8.
Menzella HG, Reeves CD. Combinatorial biosynthesis for drug development. Curr Opin Microbiol. 2007;10:238–45.
Zhang W, Tang Y. Combinatorial biosynthesis of natural products. J Med Chem. 2008;51:2629–33.
Sánchez C, Salas AP, Braña AF, Palomino M, Pineda-Lucena A, Carbajo RJ, Méndez C, Moris F, Salas JA. Generation of potent and selective kinase inhibitors by combinatorial biosynthesis of glycosylated indolocarbazoles. Chem Commun (Camb). 2009;27:4118–20.
Miao V, Coëffet-Le Gal MF, Nguyen K, Brian P, Penn J, Whiting A, Steele J, Kau D, Martin S, Ford R, Gibson T, Bouchard M, Wrigley SK, Baltz RH. Genetic engineering in Streptomyces roseosporus to produce hybrid lipopeptide antibiotics. Chem Biol. 2006;13:269–76.
Heide L, Gust B, Anderle C, Li SM. Combinatorial biosynthesis, metabolic engineering and mutasynthesis for the generation of new aminocoumarin antibiotics. Curr Top Med Chem. 2008;8:667–79.
Weissman KJ, Leadlay PF. Combinatorial biosynthesis of reduced polyketides. Nat Rev Microbiol. 2005;3:925–36.
Cropp TA, Wilson DJ, Reynolds KA. Identification of a cyclohexylcarbonyl CoA biosynthetic gene cluster and application in the production of doramectin. Nat Biotechnol. 2000;18:980e3.
Sheehan LS, Lill RE, Wilkinson B, Sheridan RM, Vousden WA, Kaja AL, et al. Engineering of the spinosyn PKS: directing starter unit incorporation. J Nat Prod. 2006;69:1702e10.
Yoon YJ, Beck BJ, Kim BS, Kang HY, Reynolds KA, Sherman DH. Generation of multiple bioactive macrolides by hybrid modular polyketide synthases in Streptomyces venezuelae. Chem Biol. 2002;9:203–14.
Hong JSJ, Park SH, Choi CY, Sohng JK, Yoon YJ. New olivosyl derivatives of methymycin/pikromycin from an engineered strain of Streptomyces venezuelae. FEMS Microbiol Lett. 2004;238:391–9.
Jung WS, Han AR, Hong JS, Park SR, Choi CY, Park JW, Yoon YJ. Bioconversion of 12-, 14-, and 16-membered ring aglycones to glycosylated macrolides in an engineered strain of Streptomyces venezuelae. Appl Microbiol Biotechnol. 2007;76:1373–81.
Han AR, Park SR, Park JW, Lee EY, Kim DM, Kim BG, Yoon YJ. Biosynthesis of glycosylated derivatives of tylosin in Streptomyces venezuelae. J Microbiol Biotechnol. 2011;21:613–6.
Han AR, Shinde PB, Park JW, Cho J, Lee SR, Ban YH, Yoo YJ, Kim EJ, Kim E, Park SR, Kim BG, Lee DG, Yoon YJ. Engineered biosynthesis of glycosylated derivatives of narbomycin and evaluation of their antibacterial activities. Appl Microbiol Biotechnol. 2012;93:1147–56.
Shinde PB, Han AR, Cho J, Lee SR, Ban YH, Yoo YJ, Kim EJ, Kim E, Song MC, Park JW, Lee DG, Yoon YJ. Combinatorial biosynthesis and antibacterial evaluation of glycosylated derivatives of 12-membered macrolide antibiotic YC-17. J Biotechnol. 2013;168:142–8.
Katz L. Manipulation of modular polyketide synthases. Chem Rev. 1997;97:2557–76.
Zhang X, Li S. Expansion of chemical space for natural products by uncommon P450 reactions. Nat Prod Rep. 2017;34:1061–89.
Lee SK, Basnet DB, Hong JSJ, Jung WS, Choi CY, Lee HC, Sohng JK, Ryu KG, Kim DJ, Ahn JS, Kim BS. Structural diversification of macrolactones by substrate-flexible cytochrome P450 monooxygenases. Adv Synth Catal. 2005;347:1369–78.
Luo Y, Li BZ, Liu D, Zhang L, Chen Y, Jia B, Zeng BX, Zhao H, Yuan YJ. Engineered biosynthesis of natural products in heterologous hosts. Chem Soc Rev. 2015;44:5265–90.
Zhang MM, Wang Y, Ang EL, Zhao H. Engineering microbial hosts for production of bacterial natural products. Nat Prod Rep. 2016;33:963–87.
Rokem JS, Lantz AE, Nielsen J. Systems biology of antibiotic production by microorganisms. Nat Prod Rep. 2007;24:1262e87.
Kim E, Moore BS, Yoon YJ. Reinvigorating natural product combinatorial biosynthesis with synthetic biology. Nat Chem Biol. 2015;11:649–59.
Breitling R, Takano E. Synthetic biology of natural products. Cold Spring Harb Perspect Biol. 2016;8:a023994.
Olano C, Lombó F, Méndez C, Salas JA. Improving production of bioactive secondary metabolites in actinomycetes by metabolic engineering. Metab Eng. 2008;10:281–92.
Liu R, Deng Z, Liu T. Streptomyces species: ideal chassis for natural product discovery and overproduction. Metab Eng. 2018;50:74–84.
Lechner A, Brunk E, Keasling JD. The need for integrated approaches in metabolic engineering. Cold Spring Harb Perspect Biol. 2016;8:a023903.
Huh JH, Kim DJ, Zhao XQ, Li M, Jo YY, Yoon TM, Shin SK, Yong JH, Ryu YW, Yang YY, Suh JW. Widespread activation of antibiotic biosynthesis by S-adenosylmethionine in streptomycetes. FEMS Microbiol Lett. 2004;238:439–47.
Chaudhary AK, Dhakal D, Sohng JK. An insight into the “-omics” based engineering of streptomycetes for secondary metabolite overproduction. Biomed Res Int. 2013;2013:968518.
Reeves AR, Brikun IA, Cernota WH, Leach BI, Gonzalez MC, Weber JM. Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab Eng. 2007;9:293–303.
Weber JM, Cernota WH, Gonzalez MC, Leach BI, Reeves AR, Wesley RK. An erythromycin process improvement using the diethyl methylmalonate-responsive (Dmr) phenotype of the Saccharopolyspora erythraea mutB strain. Appl Microbiol Biotechnol. 2012;93:1575–83.
Dhakal D, Chaudhary AK, Yi JS, Pokhrel AR, Shrestha B, Parajuli P, Shrestha A, Yamaguchi T, Jung HJ, Kim SY, Kim BG, Sohng JK. Enhanced production of nargenicin A1 and creation of a novel derivative using a synthetic biology platform. Appl Microbiol Biotechnol. 2016;100:9917–31.
Jung WS, Kim E, Yoo YJ, Ban YH, Kim EJ, Yoon YJ. Characterization and engineering of the ethylmalonyl-CoA pathway towards the improved heterologous production of polyketides in Streptomyces venezuelae. Appl Microbiol Biotechnol. 2014;98:3701–13.
Dhakal D, Le TT, Pandey RP, Jha AK, Gurung R, Parajuli P, Pokhrel AR, Yoo JC, Sohng JK. Enhanced production of nargenicin A1 and generation of novel glycosylated derivatives. Appl Biochem Biotechnol. 2015;175:2934–49.
Yi JS, Kim M, Kim EJ, Kim BG. Production of pikromycin using branched chain amino acid catabolism in Streptomyces venezuelae ATCC 15439. J Ind Microbiol Biotechnol. 2018;45:293–303.
Shao Z, Rao G, Li C, Abil Z, Luo Y, Zhao H. Refactoring the silent spectinabilin gene cluster using a plug-and-play scaffold. ACS Synth Biol. 2013;2:662e9.
Luo Y, Huang H, Liang J, Wang M, Lu L, Shao Z, Cobb RE, Zhao H. Activation and characterization of a cryptic polycyclic tetramate macrolactam biosynthetic gene cluster. Nat Commun. 2013;4:2894.
Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng. 2013;19:98–106.
Myronovskyi M, Luzhetskyy A. Native and engineered promoters in natural product discovery. Nat Prod Rep. 2016;33:1006–19.
Liu Q, Xiao L, Zhou Y, Deng K, Tan G, Han Y, Liu X, Deng Z, Liu T. Development of Streptomyces sp. FR-008 as an emerging chassis. Synth Syst Biotechnol. 2016;1:207–14.
Chi H, Wang X, Shao Y, Qin Y, Deng Z, Wang L, Chen S. Engineering and modification of microbial chassis for systems and synthetic biology. Synth Syst Biotechnol. 2019;4:25–33.
Tang Y, Kim CY, Mathews AA, Cane DE, Khosla C. The 2.7-Å crystal structure of a 194-kDa homodimeric fragment of the 6-deoxyerythronolide B synthase. Proc Natl Acad Sci USA. 2006;103:11124–9.
Miyanaga A, Iwasawa S, Shinohara Y, Kudo F, Eguchi T. Structure-based analysis of the molecular interactions between acyltransferase and acyl carrier protein in vicenistatin biosynthesis. Proc Natl Acad Sci USA. 2016;113:1802–7.
Keatinge-Clay A. Crystal structure of the erythromycin polyketide synthase dehydratase. J Mol Biol. 2008;2008(384):941–53.
Tsai SC, Miercke LJ, Krucinski J, Gokhale R, Chen JC, Foster PG, Cane DE, Khosla C, Stroud RM. Crystal structure of the macrocycle-forming thioesterase domain of the erythromycin polyketide synthase: versatility from a unique substrate channel. Proc Natl Acad Sci USA. 2001;98:14808–13.
Akey DL, Kittendorf JD, Giraldes JW, Fecik RA, Sherman DH, Smith JL. Structural basis for macrolactonization by the pikromycin thioesterase. Nat Chem Biol. 2006;2(10):537–42.
Sundermann U, Bravo-Rodriguez K, Klopries S, Kushnir S, Gomez H, Sanchez-Garcia E, Schulz F. Enzyme-directed mutasynthesis: a combined experimental and theoretical approach to substrate recognition of a polyketide synthase. ACS Chem Biol. 2013;8:443–50.
Yuzawa S, Deng K, Wang G, Baidoo EE, Northen TR, Adams PD, Katz L, Keasling JD. Comprehensive in vitro analysis of acyltransferase domain exchanges in modular polyketide synthases and its application for short-chain ketone production. ACS Synth Biol. 2017;6:139–47.
Park JW, Yoon YJ. Recent advances in the discovery and combinatorial biosynthesis of microbial 14-membered macrolides and macrolactones. J Ind Microbiol Biotechnol. 2018. https://doi.org/10.1007/s10295-018-2095-4.
Bravo-Rodriguez K, Ismail-Ali AF, Klopries S, Kushnir S, Ismail S, Fansa EK, Wittinghofer A, Schulz F, Sanchez-Garcia E. Predicted incorporation of non-native substrates by a polyketide synthase yields bioactivenatural product derivatives. ChemBioChem. 2014;15(13):1991–7.
Koryakina I, Kasey C, McArthur JB, Lowell AN, Chemler JA, Li S, Hansen DA, Sherman DH, Williams GJ. Inversion of extender unit selectivity in the erythromycin polyketide synthase by acyltransferase domain engineering. ACS Chem Biol. 2017;12:114–23.
Kim HJ, Han CY, Park JS, Oh SH, Kang SH, Choi SS, Kim JM, Kwak JH, Kim ES. Nystatin-like Pseudonocardia polyene B1, a novel disaccharide-containing antifungal heptaene antibiotic. Sci Rep. 2018;8:13584.
Hans M, Hornung A, Dziarnowski A, Cane DE, Khosla C. Mechanistic analysis of acyl transferase domain exchange in polyketide synthase modules. J Am Chem Soc. 2003;125:5366–74.
Jiang M, Pfeifer BA. Metabolic and pathway engineering to influence native and altered erythromycin production through E. coli. Metab Eng. 2013;19:42–9.
Tripathi A, Choi SS, Sherman DH, Kim ES. Thioesterase domain swapping of a linear polyketide tautomycetin with a macrocyclic polyketide pikromycin in Streptomyces sp. CK4412. J Ind Microbiol Biotechnol. 2016;43:1189–93.
Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science. 2001;291:1790–2.
Jiang M, Fang L, Pfeifer BA. Improved heterologous erythromycin A production through expression plasmid re-design. Biotechnol Prog. 2013;29:862–9.
Fang L, Guell M, Church GM, Pfeifer BA. Heterologous erythromycin production across strain and plasmid construction. Biotechnol Prog. 2018;34:271–6.
Zhang G, Li Y, Fang L, Pfeifer BA. Tailoring pathway modularity in the biosynthesis of erythromycin analogs heterologously engineered in E. coli. Sci Adv. 2015;1:e1500077.
Luo Y, Enghiad B, Zhao H. New tools for reconstruction and heterologous expression of natural product biosynthetic gene clusters. Nat Prod Rep. 2016;33:174–82.
Jung WS, Lee SK, Hong JS, Park SR, Jeong SJ, Han AR, Sohng JK, Kim BG, Choi CY, Sherman DH, Yoon YJ. Heterologous expression of tylosin polyketide synthase and production of a hybrid bioactive macrolide in Streptomyces venezuelae. Appl Microbiol Biotechnol. 2006;2006(72):763–9.
Komatsu M, Komatsu K, Koiwai H, Yamada Y, Kozone I, Izumikawa M, Hashimoto J, Takagi M, Omura S, Shin-ya K, Cane DE, Ikeda H. Engineered Streptomyces avermitilis host for heterologous expression of biosynthetic gene cluster for secondary metabolites. ACS Synth Biol. 2013;2:384–96.
Myronovskyi M, Rosenkränzer B, Nadmid S, Pujic P, Normand P, Luzhetskyy A. Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab Eng. 2018;49:316–24.
Rodriguez E, Ward S, Fu H, Revill WP, McDaniel R, Katz L. Engineered biosynthesis of 16-membered macrolides that require methoxymalonyl-ACP precursors in Streptomyces fradiae. Appl Microbiol Biotechnol. 2004;66:85–91.
Hansen DA, Rath CM, Eisman EB, Narayan AR, Kittendorf JD, Mortison JD, Yoon YJ, Sherman DH. Biocatalytic synthesis of pikromycin, methymycin, neomethymycin, novamethymycin, and ketomethymycin. J Am Chem Soc. 2013;135:11232–8.
Lowell AN, DeMars MD 2nd, Slocum ST, Yu F, Anand K, Chemler JA, Korakavi N, Priessnitz JK, Park SR, Koch AA, Schultz PJ, Sherman DH. Chemoenzymatic total synthesis and structural diversification of tylactone-based macrolide antibiotics through late-stage polyketide assembly, tailoring, and C-H functionalization. J Am Chem Soc. 2017;139:7913–20.
Pandey RP, Sohng JK. In vitro type I modular polyketide synthase catalysis for new antibiotics. Bull Korean Chem Soc. 2018;39:421–2.
Ji CH, Kim JP, Kang HS. Library of synthetic Streptomyces regulatory sequences for use in promoter engineering of natural product biosynthetic gene clusters. ACS Synth Biol. 2018;7:1946–55.
Osswald C, Zipf G, Schmidt G, Maier J, Bernauer HS, Müller R, Wenzel SC. Modular construction of a functional artificial epothilone polyketide pathway. ACS Synth Biol. 2014;3:759–72.
Tan GY, Deng K, Liu X, Tao H, Chang Y, Chen J, Chen K, Sheng Z, Deng Z, Liu T. Heterologous biosynthesis of spinosad: an omics-guided large polyketide synthase gene cluster reconstitution in Streptomyces. ACS Synth Biol. 2017;6:995–1005.
Song MC, Kim E, Ban YH, Yoo YJ, Kim EJ, Park SR, Pandey RP, Sohng JK, Yoon YJ. Achievements and impacts of glycosylation reactions involved in natural product biosynthesis in prokaryotes. Appl Microbiol Biotechnol. 2013;97:5691–704.
Cao H, Chen X, Jassbi AR, Xiao J. Microbial biotransformation of bioactive flavonoids. Biotechnol Adv. 2015;33:214–23.
Pandey RP, Parajuli P, Sohng JK. Metabolic engineering of glycosylated polyketide biosynthesis. Emerg Top Life Sci. 2018;2:389–403.
Muffler K, Leipold D, Scheller MC, Haas C, Steingroewer J, Bley T, Neuhaus HE, Mirata MA, Schrader J, Ulber R. Biotransformation of triterpenes. Process Biochem. 2010;46:1–15.
Park JW, Park SR, Han AR, Ban YH, Yoo YJ, Kim EJ, Kim E, Yoon YJ. Microbial transformation of trichostatin A to 2,3-dihydrotrichostatin A. J Nat Prod. 2011;74:1272–4.
Lee WH, Kim MD, Jin YS, Seo JH. Engineering of NADPH regenerators in Escherichia coli for enhanced biotransformation. Appl Microbiol Biotechnol. 2013;97:2761–72.
Ringle M, Khatri Y, Zapp J, Hannemann F, Bernhardt R. Application of a new versatile electron transfer system for cytochrome P450-based Escherichia coli whole cell bioconversions. Appl Microbiol Biotechnol. 2013;97:7741–54.
Lee WH, Kim JW, Park EH, Han NS, Kim MD, Seo JH. Effects of NADH kinase on NADPH-dependent biotransformation processes in Escherichia coli. Appl Microbiol Biotechnol. 2013;97:1561–9.
Pandey RP, Parajuli P, Koffas MAG, Sohng JK. Microbial production of natural and non-natural flavonoids: pathway engineering, directed evolution and systems/synthetic biology. Biotechnol Adv. 2016;34:634–62.
Peirú S, Rodríguez E, Menzella HG, Carney JR, Gramajo H. Metabolically engineered Escherichia coli for efficient production of glycosylated natural products. Microb Biotechnol. 2008;1:476–86.
Useglio M, Peirú S, Rodríguez E, Labadie GR, Carney JR, Gramajo H. TDP-L-megosamine biosynthesis pathway elucidation and megalomicin a production in Escherichia coli. Appl Environ Microbiol. 2010;76:3869–77.
Goodman CD, Useglio M, Peirú S, Labadie GR, McFadden GI, Rodríguez E, Gramajo H. Chemobiosynthesis of new antimalarial macrolides. Antimicrob Agents Chemother. 2013;57:907–13.
Thibodeaux CJ, Melançon CE 3rd, Liu HW. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew Chem Int Ed Engl. 2008;47:9814–59.
Huong NL, Hoang NH, Shrestha A, Sohng JK, Yoon YJ, Park JW. Biotransformation of rosamicin antibiotic into 10,11-dihydrorosamicin with enhanced in vitro antibacterial activity against MRSA. J Microbiol Biotechnol. 2014;24:44–7.
Park JW, Park SR, Han AR, Ban YH, Yoo YJ, Kim EJ, Kim E, Yoon YJ. Generation of reduced macrolide analogs by regio-specific biotransformation. J Antibiot (Tokyo). 2011;64:155–7.
Hoang NH, Huong NL, Shrestha A, Sohng JK, Yoon YJ, Park JW. Regio-selectively reduced streptogramin A analogue, 5,6-dihydrovirginiamycin M1 exhibits improved potency against MRSA. Lett Appl Microbiol. 2013;57:393–8.
Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, et al. Minimum information about a biosynthetic gene cluster. Nat Chem Biol. 2015;11:625–31.
Blin K, Wolf T, Chevrette MG, Lu X, Schwalen CJ, Kautsar SA, et al. antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 2017;45:W36–45.
Machado H, Tuttle RN, Jensen PR. Omics-based natural product discovery and the lexicon of genome mining. Curr Opin Microbiol. 2017;39:136–42.
Genilloud O. Mining actinomycetes for novel antibiotics in the omics era: are we ready to exploit this new paradigm? Antibiotics (Basel). 2018;7(4):E85.
Du C, van Wezel GP. Mining for microbial gems: integrating proteomics in the postgenomic natural product discovery pipeline. Proteomics. 2018;18(18):e1700332.
Nothias LF, Nothias-Esposito M, da Silva R, Wang M, Protsyuk I, Zhang Z, Sarvepalli A, Leyssen P, Touboul D, Costa J, Paolini J, Alexandrov T, Litaudon M, Dorrestein PC. Bioactivity-based molecular networking for the discovery of drug leads in natural product bioassay-guided fractionation. J Nat Prod. 2018;81:758–67.
Robertsen HL, Weber T, Kim HU, Lee SY. Toward systems metabolic engineering of streptomycetes for secondary metabolites production Biotechnol J. 2018. https://doi.org/10.1002/biot.201700465.
Wang HH, Kim H, Cong L, Jeong J, Bang D, Church GM. Genome-scale promoter engineering by coselection MAGE. Nat Methods. 2012;9:591–3.
Li L, Wei K, Liu X, Wu Y, Zheng G, Chen S, Jiang W, Lu Y. Multiplexed site-specific genome engineering for overproducing bioactive secondary metabolites in actinomycetes. Metab Eng. 2017;40:80–92.
Doudna JA, Charpentier E. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096.
Jakočiūnas T, Pedersen LE, Lis AV, Jensen MK, Keasling JD. CasPER, a method for directed evolution in genomic contexts using mutagenesis and CRISPR/Cas9. Metab Eng. 2018;48:288–96.
Xu M, Wright GD. Heterologous expression-facilitated natural products’ discovery in actinomycetes. J Ind Microbiol Biotechnol. 2019;46:415–31.
Huo L, Hug JJ, Fu C, Bian X, Zhang Y, Müller R. Heterologous expression of bacterial natural product biosynthetic pathways. Nat Prod Rep. 2019. https://doi.org/10.1039/C8NP00091C.
Han SJ, Park SW, Park BW, Sim SJ. Selective production of epothilone B by heterologous expression of propionyl-CoA synthetase in Sorangium cellulosum. J Microbiol Biotechnol. 2008;18:135–7.
Jiang H, Wang Y-Y, Ran X-X, Fan W-M, Jiang X-H, Guan W-J, et al. Improvement of natamycin production by engineering of phosphopantetheinyl transferases in Streptomyces chattanoogensis L10. Appl Environ Microbiol. 2013;79:3346e54.
Stratigopoulos G, Bate N, Cundliffe E. Positive control of tylosin biosynthesis: pivotal role of TylR. Mol Microbiol. 2004;54:1326e34.
Stratigopoulos G, Cundliffe E. Expression analysis of the tylosin-biosynthetic gene cluster: pivotal regulatory role of the tylQ product. Chem Biol. 2002;9:71e8.
Stratigopoulos G, Gandecha AR, Cundliffe E. Regulation of tylosin production and morphological differentiation in Streptomyces fradiae by TylP, a deduced gamma-butyrolactone receptor. Mol Microbiol. 2002;45:735e44.
Karray F, Darbon E, Nguyen HC, Gagnat J, Pernodet JL. Regulation of the biosynthesis of the macrolide antibiotic spiramycin in Streptomyces ambofaciens. J Bacteriol. 2010;192:5813–21.
Zhang Y, He H, Liu H, Wang H, Wang X, Xiang W. Characterization of a pathway-specific activator of milbemycin biosynthesis and improved milbemycin production by its overexpression in Streptomyces bingchenggensis. Microb Cell Fact. 2016;15:152.
Laureti L, Song L, Huang S, Corre C, Leblond P, Challis GL, Aigle B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc Natl Acad Sci USA. 2011;108:6258–63.
Mo SJ, Ban YH, Park JW, Yoo YJ, Yoon YJ. Enhanced FK506 production in Streptomyces clavuligerus CKD1119 by engineering the supply of methylmalonyl-CoA precursor. J Ind Microbiol Biotechnol. 2009;36:1473–82.
Maharjan S, Oh TJ, Lee HC, Sohng JK. Heterologous expression of metK1-sp and afsR-sp in Streptomyces venezuelae for the production of pikromycin. Biotechnol Lett. 2008;30:1621–6.
Jung WS, Jeong SJ, Park SR, Choi CY, Park BC, Park JW, Yoon YJ. Enhanced heterologous production of desosaminyl macrolides and their hydroxylated derivatives by overexpression of the pikD regulatory gene in Streptomyces venezuelae. Appl Environ Microbiol. 2008;74:1972–9.
Deng Q, Zhou L, Luo M, Deng Z, Zhao C. Heterologous expression of Avermectins biosynthetic gene cluster by construction of a Bacterial Artificial Chromosome library of the producers. Synth Syst Biotechnol. 2017;2:59–64.
Ward SL, Hu Z, Schirmer A, Reid R, Revill WP, Reeves CD, Petrakovsky OV, Dong SD, Katz L. Chalcomycin biosynthesis gene cluster from Streptomyces bikiniensis: novelfeatures of an unusual ketolide produced through expression of the chm polyketide synthase in Streptomyces fradiae. Antimicrob Agents Chemother. 2004;48:4703–12.
Tang L, Shah S, Chung L, Carney J, Katz L, Khosla C, Julien B. Cloning and heterologous expression of the epothilone gene cluster. Science. 2000;287:640–2.
Julien B, Shah S. Heterologous expression of epothilone biosynthetic genes in Myxococcus xanthus. Antimicrob Agents Chemother. 2002;46:2772–8.
Mutka SC, Carney JR, Liu Y, Kennedy J. Heterologous production of epothilone C and D in Escherichia coli. Biochemistry. 2006;45:1321–30.
Park SR, Park JW, Jung WS, Han AR, Ban Y-H, Kim EJ, Sohng JK, Sim SJ, Yoon YJ. Heterologous production of epothilone B and D in Streptomyces venezuelae. Appl Microbiol Biotechnol. 2008;81:109–17.
Lau J, Frykman S, Regentin R, Ou S, Tsuruta H, Licari P. Optimizing the heterologous production of epothilone D in Myxococcus xanthus. Biotechnol Bioeng. 2002;78:280–8.
Bian X, Tang B, Yu Y, Tu Q, Gross F, Wang H, Li A, Fu J, Shen Y, Li YZ, Stewart AF, Zhao G, Ding X, Müller R, Zhang Y. Heterologous production and yield improvement of epothilones in Burkholderiales strain DSM 7029. ACS Chem Biol. 2017;12:1805–12.
Kao CM, Katz L, Khosla C. Engineered biosynthesis of a complete macrolactone in a heterologous host. Science. 1994;265:509–12.
Zhang H, Wang Y, Wu J, Skalina K, Pfeifer BA. Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem Biol. 2010;17:1232–40.
Volchegursky Y, Hu Z, Katz L, McDaniel R. Biosynthesis of the anti-parasitic agent megalomicin: transformation of erythromycin to megalomicin in Saccharopolyspora erythraea. Mol Microbiol. 2000;40:1045–6.
Jung WS, Lee SK, Hong JSJ, Park SR, Jeong SJ, Han AR, Sohng JK, Kim BG, Choi CY, Sherman DH, Yoon YJ. Heterologous expression of tylosin polyketide synthase and production of a production of a hybrid bioactive macrolide in Streptomyces venezuelae. Appl Microbiol Biotechnol. 2006;72:763–9.
Tang L, Fu H, McDaniel R. Formation of functional heterologous complexes using subunits from the picromycin, erythromycin and oleandomycin polyketide synthases. Chem Biol. 2000;7:77–84.
Pyeon HR, Nah HJ, Kang SH, Choi SS, Kim ES. Heterologous expression of pikromycin biosynthetic gene cluster using Streptomyces artificial chromosome system. Microb Cell Fact. 2017;16:96.
Hashimoto T, Hashimoto J, Kozone I, Amagai K, Kawahara T, Takahashi S, Ikeda H, Shin-ya K. Biosynthesis of quinolidomicin, the largest known macrolide of terrestrial origin: identification and heterologous expression of a biosynthetic gene cluster over 200 kb. Org Lett. 2018. https://doi.org/10.1021/acs.orglett.8b03570.
Liu C, Zhang J, Lu C, Shen Y. Heterologous expression of galbonolide biosynthetic genes in Streptomyces coelicolor. Antonie Van Leeuwenhoek. 2015;107:1359–66.
Jones AC, Gust B, Kulik A, Heide L, Buttner MJ, Bibb MJ. Phage p1-derived artificial chromosomes facilitate heterologous expression of the FK506 gene cluster. PLoS ONE. 2013;8:e69319.
Zhang B, Wang KB, Wang W, Bi SF, Mei YN, Deng XZ, Jiao RH, Tan RX, Ge HM. Discovery, biosynthesis, and heterologous production of streptoseomycin, an anti-microaerophilic bacteria macrodilactone. Org Lett. 2018;20:2967–71.
Funding
This research was supported by grant from National Research Foundation of Korea to RPP (NRF-2017R1C1B5018056), JKS (NRF-2017R1A2A2A05000939) and DD (NRF-2017R1D1A1B03036273).
Author information
Authors and Affiliations
Contributions
DD and RPP wrote the manuscript. JKS gave valuable suggestions. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Dhakal, D., Sohng, J.K. & Pandey, R.P. Engineering actinomycetes for biosynthesis of macrolactone polyketides. Microb Cell Fact 18, 137 (2019). https://doi.org/10.1186/s12934-019-1184-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-019-1184-z