
Abstract
Background
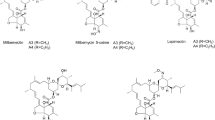
Milbemycins, a group of 16-membered macrolides with potent anthelminthic and insecticidal activity, are produced by several Streptomyces and used widely in agricultural, medical and veterinary fields. Milbemycin A3 and A4, the main components produced by Streptomyces bingchenggensis, have been developed as an acaricide to control mites. The subsequent structural modification of milbemycin A3/A4 led to other commercial products, such as milbemycin oxime, lepimectin and latidectin. Despite its importance, little is known about the regulation of milbemycin biosynthesis, which has hampered efforts to enhance milbemycin production via engineering regulatory genes.
Results
milR, a regulatory gene in the milbemycin (mil) biosynthetic gene cluster of S. bingchenggensis, encodes a large ATP-binding regulator of the LuxR family (LAL family), which contains an ATPase domain at its N-terminus and a LuxR-like DNA-binding domain at the C-terminus. Gene disruption and genetic complementation revealed that milR plays an important role in the biosynthesis of milbemycin. β-glucuronidase assays and transcriptional analysis showed that MilR activates the expression of the milA4-E operon and milF directly, and activates the other mil genes indirectly. Site-directed mutagenesis confirmed that the ATPase domain is indispensable for MilR’s function, and particularly mutation of the conserved amino acids K37A, D122A and D123A, led to the loss of MilR function for milbemycin biosynthesis. Overexpression of an extra copy of milR under the control of its native promoter significantly increased production of milbemycin A3/A4 in a high-producing industrial strain S. bingchenggensis BC04.
Conclusions
A LAL regulator, MilR, was characterized in the mil gene cluster of S. bingchenggensis BC04. MilR could activate milbemycin biosynthesis through direct interaction with the promoter of the milA4-E operon and that of milF. Overexpression of milR increased milbemycin A3/A4 production by 38 % compared with the parental strain BC04, suggesting that genetic manipulation of this activator gene could enhance the yield of antibiotics.
Similar content being viewed by others
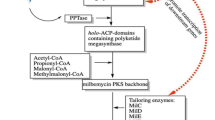

Background
Milbemycins are a group of 16-membered macrolides that share a similar lactone ring with avermectin, the discovery of which won a Nobel Prize in 2015. Similar to avermectins, milbemycins have attracted considerable attention and have been developed as acaricides, insecticides and anthelmintics because of their excellent activities against a variety of nematodes, parasitic insects and arthropod parasites, and their low toxicities to the host organisms [1–4]. Milbemycins have been used to control mites, liriomyza, aphidoidea and aleyrodidae that have developed resistance to avermectin and organophosphorus pesticides on 24 plant species, including apple, citrus, strawberry and tea. Milbemycin A3/A4 show high aricidal activity and have been used widely to control mites since 1990 in developed countries [5]. Milbemycin oxime, a semi-synthetic analog of milbemycin A3/A4, has been used commercially against worms, insects and mites of pet animals in China, Europe, Japan and the USA [6, 7]. In addition, lepimectin, another milbemycin A3/A4 derivative, has also been developed and used widely in the agricultural field.
Milbemycins were first isolated from Streptomyces hygroscopicus subsp. aureolacrimosus in 1967 in Japan [8]. Thereafter, another milbemycin-producing strain, Streptomyces bingchenggensis, was isolated by our laboratory and used as an important industrial producer of milbemycins [9–11]. Streptomyces bingchenggensis produces milbemycin A3, A4, and four α-class and β-class C5-O-methylmilbemycins (B2, B3, β1 and β2) as the main components, together with the polyether nanchangmycin and trace amounts of new milbemycin analogs [9–12]. In 2010, the genome information of S. bingchenggensis was published (GenBank Accession No. CP002047) and the milbemycin biosynthetic gene cluster (mil gene cluster) was identified [13]. The mil gene cluster (SBI00726–SBI00790) contains 10 genes, which are homologs to those of avermectin gene cluster, including one putative regulatory gene (SBI_00734, milR), four multifunctional modular type I polyketide synthase components (PKS; milA1, milA2, milA3 and milA4), four tailoring enzyme genes (milC, milD, milE and milF), a putative orf1 with unknown function and a large (62 kb) insertion fragment between milR and milA1. However, the overall gene organization of the milbemycin gene cluster is different from that of avermectin, e.g. the mil cluster PKS genes (milA1, milA2, milA3 and milA4) are organized into four operons and are transcribed independently, while the avermectin PKS genes are organized as two groups (aveA1–aveA2 and aveA3–aveA4). Initially, the biosynthetic pathway of milbemycin was proposed based on physicochemical characterization, bioconversion data and some studies on the biosynthesis of avermectin and meilingmycin [14–16]. Seven malonyl-CoA units and five methylmalonyl-CoA units are condensed to the starting unit derived from acetate or propionate, catalyzed by the PKSs to form the polyketide backbone, in a stepwise process. The tailoring enzyme genes milD (encoding C5-O-methyltransferase) and milF (encoding C5-ketoreductase) from the cluster were also characterized. MilD is responsible for the methylation of the hydroxyl group on C-5 of milbemycin A3/A4 [17], while MilF catalyzes the reduction of the C-5 keto group in the biosynthesis of milbemycin [18]. However, no report has been published so far on the regulation of milbemycin biosynthesis. A better understanding of the regulation mechanisms will be useful to construct high-producing industrial strains [19, 20].
In this study, to dissect the underlying regulation mechanism of milbemycin biosynthesis in S. bingchenggensis, the only candidate regulatory gene in the mil gene cluster, milR, was characterized. The deduced product of this gene is a LAL family transcriptional regulator. milR is the homolog of aveR, which is the regulatory gene for avermectin biosynthesis. We demonstrated that MilR shows a different regulatory pattern compared with that of aveR. MilR controls the type I polyketide chain termination and modification steps of milbemycin biosynthesis by regulating milA4-E operon and milF promoters directly. Overexpression of milR by one extra copy significantly increased the production of milbemycin A3/A4.
Results
milR encodes a putative LAL transcriptional regulator
milR encodes a putative polypeptide of 964 aa with a predicted molecular mass of 102.4 kDa. Bioinformatics analysis indicated that MilR is a LAL family (large ATP-binding regulator of the LuxR family) regulatory protein (Fig. 1a). The N-terminus of MilR contains an AAA (ATPases associated with diverse cellular activities) domain (amino acids 1–150) (Pfam No. PF00004), and the C-terminal portion contains an HTH-LuxR-like DNA-binding domain (amino acids 890–947) (SMART No. SM00421). MilR shows strong similarity to several regulatory proteins, such as AveR (49 % identity) and OlmRI (39 % identity) from Streptomyces avermitilis, PikD (37 % identity) from Streptomyces venezuelae, and AstG1 (35 % identity) from Streptomyces sp. XZQH13 [21–25].

Domain structure and amino acid alignment of parts of the MilR. a Predicted domain structure of MilR. AAA_16: A domain abbreviation for ATPases associated with diverse cellular activities; HTH-LUXR: A DNA-binding, helix-turn-helix (HTH) domain of about 65 amino acids, present in transcription regulators of the LuxR family. b Alignment of the AAA domain of MilR with related proteins. Numbers indicate amino acid residues from the N-terminus of the protein. Identical amino acid residues are black, and similar residues are shaded. AveR, a cluster-situated regulator (CSR) of avermectin biosynthesis in Streptomyces avermitilis; OlmRI, a CSR of oligomycin biosynthesis in Streptomyces avermitilis; PikD, a CSR of pikromycin biosynthesis in Streptomyces venezuelae; AstG1, a CSR of ansatrienins biosynthesis in Streptomyces sp. XZQH13. The conserved Walker A and B motifs are indicated by red and blue overlines respectively. c Comparison of the HTH-LUXR domain of MilR with those of other proteins
The AAA domain of MilR belongs to the P-loop NTPases (nucleoside triphosphatases) superfamily, which is defined by the presence of a conserved nucleotide phosphate-binding motif, also known as the Walker A motif (GX4GK[S/T], where X is any amino acid), a glycine-rich sequence followed by a conserved lysine and a serine or threonine, and the second Walker B motif (ΨΨΨΨ[D/E], where Ψ is a hydrophobic residue) (Fig. 1b) [26, 27]. Proteins of this family exert their activity through assembly and disassembly that are driven by the ATP binding and hydrolysis cycle of the AAA domain [28].
Interestingly, one leucine in the C-terminal portion (amino acid 834) of MilR is encoded by the rare TTA codon. This indicated that the translation of milR is controlled by bldA, which encodes the tRNA responsible for translating TTA into leucine [29].
milR is essential for milbemycin biosynthesis
To determine the role of milR in milbemycin biosynthesis, a milR disruption mutant (ΔmilR) was constructed via homologous recombination (Fig. 2a). In ΔmilR, a 1753-bp fragment internal to milR was replaced by the kanamycin resistance gene, neo. The resulting ΔmilR was further confirmed by PCR (Additional file 1: Figure S1). ΔmilR was cultured in fermentation medium for 9 days and the production of milbemycin was tested. The result showed that no milbemycin was produced by ΔmilR in comparison with BC04 and BC04/pSET152 controls (Fig. 2b). To verify that the phenotype was the result of milR disruption, a complementation experiment was carried out, in which an integrating plasmid, pSET152::milR, was used to complement ΔmilR. In pSET152::milR, milR was driven by its own promoter. Milbemycin production was restored in the complemented strain (ΔmilR/pSET152::milR) (Fig. 2b). These results demonstrated that MilR is indispensable for milbemycin production in S. bingchenggensis.

Effect of milR disruption on Milbemycin A3/A4 production. a Genetic organization of mil gene cluster in Streptomyces bingchenggensis BC04 and diagram of milR disruption and complementation constructions. Each arrow indicates a separate open reading frame (ORF) and orientation of transcription. b HPLC analysis of milbemycin A3/A4 production in S. bingchenggensis BC04, BC04/pSET152, ΔmilR, ΔmilR/pSET152::milR. Absorbance at 242 nm was monitored
MilR regulates the promoter regions of milA4-E operon, orf1 and milF directly
milR is situated in the middle of the mil gene cluster; therefore, it is possible that the transcription of some mil genes might be regulated by MilR. To determine the potential targets of MilR, first, co-transcription analysis was performed to confirm the putative operons. Total RNAs were extracted from S. bingchenggensis BC04 after the onset of milbemycin production (3 days of cultivation in fermentation medium) and used as templates for cDNA synthesis and reverse transcription polymerase chain reaction (RT-PCR) analysis. Primers flanking four intergenic regions (regions 1–4) within the mil gene cluster were used: generation of PCR-amplified products by these primers would indicate transcription across the intergenic region (Additional file 2: Figure S2). The results showed that the mil gene cluster contains four transcriptional units (milA2-C, milA4-E, milR-A3 and milA1-D) and two individually transcribed genes (milF and orf1). Based on the above results, the promoters of the six transcriptional units were then cloned separately upstream of gusA [encoding β-glucuronidase (GUS)]. The resulting plasmids were integrated into the ФC31 attB site of Streptomyces coelicolor M1146. At the same time, the coding region of milR was cloned downstream of P hrdB in pIJ10500 to generate pIJ10500::P hrdB milR. pIJ10500::P hrdB milR was subsequently integrated into the ФBT1 attB site of the S. coelicolor M1146 derivatives containing the six different reporter constructs. Then gusA transcriptional fusions were assessed in agar-based chromogenic assays using 5-bromo-4-chloro-3-indolyl-β-d-glucuronide as the substrate (Fig. 3). None of the strains containing mil promoter::gusA plasmids that lacked constitutively expressed MilR gave GUS activity. When MilR was constitutively expressed, transcription of gusA from P milA4 , P orf1 and P milF was readily detected; however, transcription of gusA from P milA2 , P milR and P milA1 was not detected. These results indicated that the promoters of milA4-E operon, orf1 and milF, are probably the direct targets of MilR.

MilR directly activates promoters of milA4-E, orf1 and milF. a MilR-regulating regions are indicated by two vertical green arrows in the mil gene cluster. b Chromogenic assays on AS-1 agar plates containing the substrate 5-bromo-4-chloro-3-indolyl-β-d-glucoronide. Data are representative of three independent experiments
MilR activates the transcription of the mil gene cluster
To determine the effect of milR on its target genes, quantitative real-time RT-PCR (qRT-PCR) analysis was performed to assess the transcriptional levels of milA4, milE, and milF. RNAs were prepared from cultures of BC04 and ΔmilR grown at different time points (1, 2, 3, 5, 7 and 9 days). These RNA samples were then subjected to qRT-PCR analysis. The results showed that transcripts of milA4, milE and milF were almost undetectable in ΔmilR compared with BC04, indicating that MilR activates the transcription of milA4, milE and milF directly (Fig. 4). In addition, we detected the expression of milA2, milC, milR and milA1. Transcription of these four genes decreased in ΔmilR (Fig. 4). However, GUS assays did not detect any interaction of MilR with the milA2, milR or milA1 promoters (Fig. 3), indicating an indirect activation on these genes exerted by MilR. The decreased transcriptional levels of milA2, milC, milR and milA1 were probably due to some unknown signals repressing the mil gene expression to avoid unnecessary consumption of nucleoside triphosphates (NTPs), by perceiving the inability of milbemycin production. Note that transcription of orf1, another putative target of MilR, was also undetectable in ΔmilR (data not shown). orf1 encodes a enoylreductase, but is supposed to be unnecessary for milbemycin A3/A4 production. Therefore, further research on the biological significance of orf1 as a target of MilR will not be mentioned in this work.

Quantitative real-time RT-PCR transcription profile analysis of milA4, milE, milF, milA2, milC, milR and milA1. All RNA samples were isolated from 1, 2, 3, 5, 7 and 9 days cultures. Data are presented as the averages of three independent experiments conducted in triplicate. 16S rRNA transcription was monitored and used as the internal control. Error bars show standard deviations, *P < 0.05
Importance of the conserved AAA domain for the transcriptional regulation of MilR
In MilR, the Walker A motif is GGPGCGKS and the Walker B motif is LVIAVDD. To define the relative contributions of Walker A and B to the function of MilR, site-directed mutations were introduced into certain conserved and less conserved residues in these two motifs (Additional file 3: Figure S3). We constructed pIJ10500::milR, pIJ10500::G31A, pIJ10500::G32A, pIJ10500::G34A, pIJ10500::G36A, pIJ10500::K37A, pIJ10500::K37R, pIJ10500::S38A, pIJ10500::D122A and pIJ10500::D123A. These integrative plasmids containing wild-type and mutant milR-coding nucleotide sequences were introduced into ΔmilR to obtain the complementary strains ΔmilR/MilR, ΔmilR/G31A, ΔmilR/G32A, ΔmilR/G34A, ΔmilR/G36A, ΔmilR/K37A, ΔmilR/K37R, ΔmilR/S38A, ΔmilR/D122A and ΔmilR/D123A, respectively. The strains above were verified by PCR (data not shown). Milbemycin production decreased in all strains containing the mutant MilR compared with ΔmilR/MilR (Fig. 5a), implying that the mutation of MilR seriously affected milbemycin production. Among them, ΔmilR/G37A, ΔmilR/G37R, ΔmilR/D122A and ΔmilR/D123A exhibited decreases of 98.5–99.5 %. ΔmilR/S38A decreased by 87 %, ΔmilR/G34A decreased by 78 %, ΔmilR/G36A decreased by 65 %, and ΔmilR/G31A and ΔmilR/G32A decreased by 15 %. These results showed that the functional importance of these amino acid residues is as follows: Lys37 ≈ Asp122 ≈ Asp123 > Ser38 > Gly34 > Gly36 > Gly31 ≈ Gly32.

Effect of site-directed mutation in the AAA domain of MilR on its function. a Milbemycin production in the complementation strains containing the WT and mutant MilRs. MilR:ΔmilR/milR, G31A:ΔmilR/G31A, G32A:ΔmilR/G32A, G34A:ΔmilR/G34A, G36A:ΔmilR/G36A, K37A:ΔmilR/K37A, K37R:ΔmilR/K37R, S38A:ΔmilR/S38A, D122A:ΔmilR/D122A, D123A:ΔmilR/D123A. b Western blotting analysis of the WT and several selected mutant MilRs. The proteins of MilR and mutant MilRs (G31A, K37A, S38A, D122A and D123A) showed similar expression levels. c Transcriptional analysis of milR, milA4, milE and milF by semiquantitative RT-PCR. hrdB transcript was used as an internal control. MilR:ΔmilR/milR, 31A:ΔmilR/G31A, 32A:ΔmilR/G32A, 34A:ΔmilR/G34A, 36A:ΔmilR/G36A, 37A:ΔmilR/K37A, 37R:ΔmilR/K37R, 38A:ΔmilR/S38A, 122A:ΔmilR/D122A, 123A:ΔmilR/D123A; *P < 0.05
To determine the effect of these mutant milRs on milbemycin biosynthetic genes, western blotting and RT-PCR analysis were carried out to assess the expression levels of milR and several structural genes. The wild-type and mutant milRs were transcribed and translated into proteins, as demonstrated by RT-PCR and western blotting (Fig. 5b, c), indicating that the site-directed mutagenesis did not influence the expression of MilR. In the mutants, except for ΔmilR/G31A and ΔmilR/G32A, the transcripts of milA4, milE and milF decreased substantially, whereas the transcript levels of milA2, milC, milA3 and milA1 were not affected significantly compared with that of ΔmilR/MilR (Additional file 4: Figure S4).These results strongly suggest that the Walker A and B sequences are essential for the transcriptional function of MilR.
Enhancement of milbemycin A3/A4 production by overexpression of milR
Overexpression of transcriptional activators is an efficient approach to increase production of antibiotics, especially engineered activators under the control of strong constitutive promoters. Using this strategy, nikkomycin production in Streptomyces ansochromogenes and oxytetracycline biosynthesis in Streptomyces rimosus were enhanced significantly by introducing an extra copy of activator genes sanG (driven by constitutive hrdB promoter) and otcR (controlled by the constitutive SF14 promoter), respectively [30, 31]. In this work, milR was confirmed to be a direct activator of milbemycin biosynthesis (Fig. 2); thus, engineering the expression levels of milR might be a rational strategy to improve milbemycin production in S. bingchenggensis BC04, a high-producing industrial strain for milbemycin A3/A4 production. First, milR was placed under the control of a strong constitutive hrdB promoter in an integrative plasmid (pSET152) and in a multicopy plasmid (pKC1139). These two recombinant plasmids were transformed into BC04 to create BC04::hrdBmilR and BC04::hrdBmilRs, respectively. Unexpectedly, milbemycin A3/A4 production in BC04::hrdBmilR or BC04::hrdBmilRs was lower compared with that of BC04 (Additional file 5: Figure S5). Meanwhile, cell growth rate of these two strains was reduced in seed medium. BC04::hrdBmilRs exhibited a slower growth rate than that of BC04::hrdBmilR. These results indicated that overexpression of milR under a strong constitutive promoter did not increase milbemycin production. A single copy of milR with its own promoter was then cloned into pSET152 to generate pSET152::milR. This plasmid was transformed into BC04, resulting in BC04::milR. Notably, in BC04::milR, the final production of milbemycin A3/A4 increased by 38 %, reaching 4069 mg/l, compared with 2947 mg/l in BC04 (Fig. 6a). We also determined the biomass of BC04 and BC04::milR. The results showed that they had comparable growth rates and final biomass, and that the increase in milbemycin A3/A4 production was attributed to the overexpression of milR (Fig. 6b).

Effect of overexpression of milR on antibiotics production and cell dry weight. a Milbemycin production in S. bingchenggensis BC04 and BC04::milR. b Cell dry weight of S. bingchenggensis BC04 and BC04::milR. Data are presented as the averages of the results of three independent experiments. Error bars show standard deviations, *P < 0.05
To verify whether the enhancement of milbemycin A3/A4 production was caused by the increased expression of mil genes, we measured the transcript levels of several mil genes in BC04 and BC04::milR. Transcription of milR was increased two–fold in BC04::milR over BC04 at 5 d, and remained higher than that in BC04 for several days thereafter (Fig. 7). MilR exerts its effect by activating the transcription of milA4-E and milF; therefore, the expression of milA4 and milF was also detected (Fig. 7). As expected, the expressions of both milA4 and milF were higher in BC04::milR than in BC04. Finally, based on the indirect activation by MilR on the milA2-C, milR-A3 and milA1-D operons, transcript levels of milA2, milA3 and milA1 were assessed and shown to be increased two-fold at 5 days. These results showed that the overexpressed milR and the subsequent direct activation of milA4-E, milF and indirect activation of milA2-C, milA3 and milA1-D accounted for the enhanced production of milbemycin A3/A4 in the engineered strain.

Quantitative real-time RT-PCR analysis of genes involved in milbemycin biosynthesis. The transcriptional levels of mil genes (milR, milA4, milF, milA2, milA3 and milA1) are presented relative to that of BC04 sample collected after fermentation for 5 days, which was assigned a value of 1. Data are presented as the averages of the results of three independent experiments conducted in triplicate. 16S rRNA transcription was monitored and used as the internal control. Error bars show standard deviations, *P < 0.05
To further increase milbemycin A3/A4 production, a plasmid containing two copies of milR driven by its native promoter was constructed and introduced into BC04 to create BC04::2milR. However, the milbemycin A3/A4 production was less than that of BC04::milR (Additional file 5: Figure S5), accompanied by a decrease in cell growth in seed medium, indicating that excessive expression of milR does not coordinate the physiological conditions of the cell and lead to growth retardation.
Discussion
Improving the titer of commercially valuable antibiotics is important for pharmaceutical industries to maintain sustainable development. Previously, random mutagenesis such as UV irradiation, N-methyl-N′-nitroso-N-nitrosoguanidine (NTG), atmospheric and room temperature plasma (ARTP) mutation, and rational metabolic engineering, have been carried out to improve milbemycin production in S. bingchenggensis [17, 18, 32]. However, a lack of knowledge about the regulation of milbemycin biosynthesis has meant that titer improvement of milbemycin production by manipulating regulatory genes has not been reported. In this study, a LAL activator gene, milR, the only regulator embedded within the milbemycin gene cluster, was identified, and its regulatory mechanism in milbemycin biosynthesis was characterized.
LAL regulators are widespread in Streptomyces with a varying number (from several to dozens), and mainly situated in clusters encoding type I polyketide synthases. To date, about 19 LALs have been reported to be cluster-situated activators in the regulation of antibiotic biosynthesis [19]. Among them, only the regulatory mechanisms of AveR in avermectin biosynthesis and PikD in pikomycin biosynthesis have been studied in any depth. PikD controls pikomycin biosynthesis by regulating two operons: the pikAI–AIII operon (comprising genes encoding polyketide chain initiation and elongation but not chain termination), and the desI–V operon (five co-transcribed genes encoding proteins responsible for desosamine biosynthesis) [24]. AveR controls the whole process of avermectin biosynthesis (including polyketide chain initiation, elongation and termination, polyketide modification, oleandrose biosynthesis and transglycosylation) by direct interaction with all ave promoters [21]. In the present study, we showed that MilR is a cluster-situated activator of milbemycin production, similar to the major LALs characterized in Streptomyces. The MilR sequence is highly similar to those of AveR and PikD, but exhibits a different method of regulation of milbemycin biosynthesis. In milbemycin biosynthesis, MilR controls the polyketide chain termination and modification specifically by directly activating the transcription of milA4 (encoding the fourth PKS responsible for the final elongation and chain termination), milE and milF. In addition, MilR in S. bingchenggensis BC04 only affects milbemycin production and has no influence on polyether nanchangmycin production (another compound produced by S. bingchenggensis), while AveR in S. avermitilis not only activates avermectin production, but also cross-regulates oligomycin biosynthesis by directly repressing certain oli genes (e.g. olmA1 and olmA4) [21].
In this study, we also observed a decrease in milA2, milC, milR and milA1 transcripts in ΔmilR; however these genes might not be the direct targets of MilR, as shown by GUS assays (Fig. 3). The onset and production levels of each antibiotic are controlled by a large and diverse set of regulatory proteins, some of which can respond to (bind) physiological signals (e.g., antibiotics produced by the host) [19]. There is growing evidence that endogenous antibiotics can act as this type of physiological signal. In this scenario, they control their own biosynthesis by modulating the DNA-binding activity of cluster-situated or global regulatory proteins in a feed-forward or feedback mechanism [33]. For example, in Streptomyces coelicolor, the “pseudo” GBL receptor, ScbR2, can respond to (bind) endogenous Act and Red, thereby causing indirect activation of Act and Red production [34]. In Streptomyces venezuelae, JadR2, another homolog of ScbR2, binds the endogenous antibiotic chloramphenicol, thus activating the transcription of the cml structural gene cmlJ [34]. Recently, in Streptomyces globisporus, AtrA, a highly conserved TetR-family global regulator, was observed to activate lidamycin production by binding to the promoter region of sgcR1R2 (via two CSRs within the lidamycin biosynthetic gene cluster). AtrA could bind the lidamycin intermediate heptaene, leading to its dissociation from the sgcR1R2 promoter [35]. Based on these examples, we speculate that some unknown signals (i.e., certain regulatory proteins), could repress the milbemycin structural genes directly, or indirectly via regulatory cascades. We hypothesized that in the process of milbemycin biosynthesis, milbemycins accumulate to a threshold, which is sensed by the repressors. The repressors then dissociate from their target promoters and release the expression of milbemycin structural genes for milbemycin biosynthesis. Meanwhile in ΔmilR, cells could not produce milbemycins; therefore, these repressors could repress the expression of milbemycin structural genes continuously, leading to the observed decreases in milA2, milC, milR and milA1 transcripts. We do not know the identity of these repressors; perhaps some of them are homologs of the “pseudo” GBL receptor or AtrA, which will require further confirmation. The search for upstream regulatory genes of milA2-C, milR and milA1 is an important subject that we are currently working on.
Deep research on the Escherichia coli LAL protein, MalT, indicated that the Walker A and B motifs (AAA domain) in LALs are thought to be responsible for ATP binding and hydrolysis cycles. These cycles drive the protein to cycle between an ADP-bound, resting form and an ATP-bound, active form, which oligomerizes and initiates gene expression [36, 37]. However, data on the significance of Walker A and B sequences in Streptomyces LAL functions are limited: only two amino acid residues (K38 and D138 in AAA domain) have been verified as essential for PikD activity. To further understand the correlation between Walker A and B sequences and LAL function, we took MilR as a representative, and performed nine point mutations to measure the relative contribution of Walker A and B sequences in detail (Additional file 3: Figure S3). The putative ATP-binding site K37 (positively charged) was changed to Ala or a similar charged Arg (positively charged), Mg2+ binding site S38, and four Glycines in Walker A sequence were mutated to Ala, separately. In addition, the two Asps (aspartic acids D122 and D123) in Walker B were also changed to Ala, separately. Then MilR and the nine mutants mentioned above were checked for milbemycin A3/A4 production and target gene transcription. The results showed that changing K37 to Ala or to a similar charged Arg destroyed both milbemycin A3/A4 production and the efficiencies of MilR transcriptional activation (Fig. 5a, c), which is consistent with the previous conclusion that the K37 (Lys37) residue in the Walker A sequence is crucial for transcriptional regulation of AAA-containing regulatory proteins [38, 39]. Likewise, mutation of D122 to Ala and D123 to Ala also led to the loss of MilR function, while in PikD, the second Asp of Walker B (corresponding to D123 in MilR) has no influence on PikD activity [24], indicating a slight functional difference in the same Walker B site between MilR and PikD. S38 also has a large contribution in MilR function. Besides K37, D122, D123 and S38, G31 and G36 are two other highly conserved amino acid residues in the AAA domain of LAL regulators: our results indicated that G36 is more important than G31 in MilR function, but less important than K37 (Fig. 5a, c). Finally, G32 and G34 are two less conserved residues in Walker A. Interestingly, G32 and G34 exert an obvious influence in MilR activity. Expression of intact MilR in E. coli failed despite much effort, and we were unable to assess whether each mutation influenced ATP-binding affinity, ATPase activity or the aggregation state of MilR using in vitro experiments. However, these mutation data confirmed the functional importance of Walker A and B in MilR, and further indicated that certain amino acid residues in the walker A and B sequences might be the optimal combination for MilR function and its association with the cellular physiological environment. In addition, these results extended our understanding of the importance of the AAA domain in LALs that are widespread in Streptomyces.
Overexpression of transcriptional activators is often associated with an increase in production of the corresponding antibiotics [30]. The optimal product formation phenotype is obtained when activators are engineered to express at the optimum level and time (an expression profile that correlates well with cell physiological conditions) [40, 41]. In this work, milR was overexpressed using both native and strong constitutive hrdB promoters. Our results showed that the native promoter had a beneficial effect on milbemycin production, but the constitutive hrdB promoter had no effect. Overexpression of milR by introducing an extra copy driven by its native promoter improved milbemycin A3/A4 production significantly by 38 % in BC04 (Fig. 6). However, introduction of milR under the control of a strong constitutive hrdB promoter in an integrative plasmid pSET152 or in a multicopy plasmid pKC1139 did not result in a further increase but rather a lower level of milbemycin A3/A4 production and a decrease in cell growth compared with BC04. An additional two copies of milR driven by its own promoter also led to a decrease in milbemycin A3/A4 production and cell growth compared with that of BC04::milR (Additional file 5: Figure S5). These results suggested that the expression increase gained by integrating one copy of milR may be enough to improve milbemycin A3/A4 production in BC04, and that excessive expression of milR or expressing milR at a much earlier stage of growth has adverse effects on milbemycin A3/A4 biosynthesis and cell growth. This was partially confirmed by research on OTC, where two copies of overexpressed otcR could confer the highest OTC production but three additional copies caused a burden to the cell and led to decreased antibiotic production [31]. As for milbemycin biosynthesis, one additional copy of milR might be a better strategy to improve antibiotic production.
Conclusions
In this study, we provided the first report of the regulatory mechanism in milbemycin biosynthesis in S. bingchenggensis BC04. MilR was confirmed to be a pathway-specific activator of milbemycin production. Moreover, our investigations demonstrated that overexpression of one copy of milR is an effective strategy to increase milbemycin production in an industrial strain. This is of practical importance for future industrial applications to improve milbemycin production in Streptomyces.
Methods
Bacterial strains and growth conditions
Streptomyces bingchenggensis BC04 is a derivative of S. bingchenggensis CGMCC 1734 deposited at China General Microbiological Culture Collection (CGMCC). Streptomyces bingchenggensis BC04 is a high-level producer of milbemycin A3/A4 generated from S. bingchenggensis CGMCC 1734 after random mutagenesis. S. coelicolor 1146 was used for GUS assays. Escherichia coli TOP 10 was used as a general host strain for propagating plasmids. Escherichia coli ET12567 (pUZ8002) was used for transferring DNA from E. coli to Streptomyces by conjugation [42]. For conjugation from E. coli to Streptomyces, Streptomyces strains were grown on mannitol/soya (MS) agar at 28 °C [42]. For genomic DNA extraction and spore collection, S. bingchenggensis BC04 and its derivatives were grown in SSPY (1 % sucrose, 0.1 % skimmed milk powder, 0.35 % peptone, 0.5 % yeast extract, and 0.05 % K2HPO4·3H2O, pH 7.2) liquid medium or on SSPY agar at 28 °C [18]. For milbemycin production, spore suspensions were inoculated in liquid SSPY and incubated at 28 °C on a rotary shaker (250 rpm) for 46 h as the seed culture, and then 6 ml of seed cultures was transferred into 250-ml Erlenmeyer flasks containing 50 ml fermentation medium (8 % sucrose, 2 % soybean powder, 0.1 % skimmed milk powder, 0.3 % CaCO3, 0.1 % K2HPO4, and 0.01 % FeSO4·7H2O, pH 7.0) for milbemycin production. For GUS assays, S. coelicolor 1146 derivatives were grown on AS-1 (0.1 % yeast extract, 0.02 % l-alanine, 0.02 % l-arginine, 0.02 % l-asparagine, 0.5 % soluble starch, 0.25 % NaCl, and 1 % Na2SO4) agar.
Plasmids and DNA manipulation
pBluescript KS (+) was used for routine DNA cloning. The kanamycin resistance gene (neo) was obtained from a recombinant plasmid pUC119::neo. The E. coli-Streptomyces shuttle plasmid pKC1139, which contains a temperature-sensitive origin of replication from pSG5 [43], was used to construct recombinant plasmids for gene disruption and overexpression. pSET152 and pIJ10500, which can integrate into the Streptomyces chromosome by site-specific recombination at the phage ФC31 or ФBT1 attachment site (attB) respectively [43, 44], were used to create recombinant plasmids for introducing milR into Streptomyces. pIJ10500 also contains 3× FLAG tag, and was used in western blotting experiment. Plasmids and genomic DNA were isolated according to standard techniques [42]. Conjugal transfer from E. coli ET12567 (pUZ8002) into S. bingchenggensis was carried out as described previously [42].
Sequence analysis
Protein sequence alignment and domain architectures were analyzed by using the BLAST (http://www.blast.ncbi.nlm.nih.gov/Blast.cgi), SMART (http://www.smart.embl-heidelberg.de/), Pfam (http://www.pfam.xfam.org/) and CDD (http://www.ncbi.nlm.nih.gov/cdd/) databases and software tool (Gene Doc).
Construction of milR disruption mutant and its complementation
Disruption of milR was performed by gene replacement via homologous recombination. For construction of milR disruption mutant, two 2.2-kb fragments corresponding to the upstream and downstream sequences of milR were amplified from the S. bingchenggensis BC04 genomic DNA by polymerase chain reaction (PCR) using RD-LF/R and RD-RF/R as primer pairs (Additional file 6: Table S1). The upstream fragment was digested with EcoRI and KpnI, and the downstream fragment was digested with BamHI and XbaI. The kanamycin-resistance gene (neo) was obtained from pUC119::neo after digestion with KpnI and BamHI. These three resulting DNA fragments were ligated between the EcoRI and XbaI sites of pKC1139 to give pKC1139::milR::neo, in which a 1753-bp fragment of milR was replaced by neo. pKC1139::milR::neo was subsequently introduced into S. bingchenggensis BC04 via conjugation. Spores were harvested and spread on MS agar containing kanamycin. After incubation at 37 °C for 8 days, kanamycin resistant (KanR) and apramycin sensitive (AprS) colonies were identified, and further confirmed as milR disruption mutant (ΔmilR) by PCR.
For the complementation of milR in ΔmilR, a 3278-bp DNA fragment containing the coding region of milR and its upstream region was amplified using RCOMF and RCOMR (Additional file 6: Table S1) as primers, and then inserted between the EcoRV and XbaI sites of pSET152 to obtain pSET152::milR. Introduction of pSET152::milR into ΔmilR and the empty vector pSET152 into S. bingchenggensis BC04 by conjugation resulted in the complemented strain ΔmilR/pSET152::milR and the control strain BC04/pSET152, respectively.
HPLC analysis of milbemycin A3/A4
The HPLC conditions for the detection of milbemycins were the same as previous report [18]. HPLC was performed with a Shimadzu LC-2010 CHT system (Shimadzu, Koyoto, Japan) by using a NOVA-PAKR C18 column (3.9 × 150 mm, 5 μm, Waters) at a flow rate of 1.0 ml/min with a linear gradient from 0 to 100 % of solvent B in 15 min (Solvent A: CH3CN–H2O–CH3OH (350:50:100, v/v/v); Solvent B: CH3OH) and detected at 242 nm.
Construction of the gusA reporter systems and GUS assays
A 1818-bp DNA fragment containing the complete gusA coding region was amplified from pIJ10500::gusA by PCR using gusA-F and gusA-R as primers (Additional file 1: Table S1). The six promoters of milA2, milA4, milR, milA1, orf1 and milF were obtained from the genomic DNA of S. bingchenggensis BC04 by PCR with primer pair milA2-pF/pR, milA4-pF/pR, milF-pF/pR, orf1-pF/pR, milR-pF/pR and milA1-pF/pR, respectively (Additional file 6: Table S1). Prior to PCR amplification, primers gusA-F, milA2-pR, milA4-pR, milF-pR, orf1-pR, milR-pR and milA1-pR were phosphorylated with T4 polynucleotide kinase to facilitate subsequent ligation reactions. P milA2 , P milA4 , P milF , P orf1 , P milR and P milA1 were cut with XbaI and the gusA coding region was cut with EcoRI. The promoters and the gusA coding region were ligated together with XbaI/EcoRI double digested pSET152 in a three-piece ligation reaction to generate pSET152::P milA2 gusA, pSET152::P milA4 gusA, pSET152::P orf1 gusA, pSET152::P milF gusA, pSET152::P milR gusA and pSET152::P milA1 gusA. The authenticity of all PCR amplicons was verified by sequencing. After restriction digestion analysis, these pSET152-derived plasmids were integrated into the ФC31 attB site of S. coelicolor M1146 after conjugation via the donor strain E. coli ET12567/pUZ8002. Exconjugants were selected using apramycin and confirmed by PCR amplifications. These strains were used as negative controls in β-glucuronidase (GUS) assays and as conjugation recipients for construct in which MilR was expressed from the constitutive promoter P hrdB . For the construction of the latter construct, the hrdB promoter (P hrdB ) was amplified with primer pair hrdB-pF and hrdB-pR, the milR coding region was amplified with primer pair milR-F and milR-R (Additional file 6: Table S1). Prior to PCR amplification, primers hrdB-pR and milR-F were phosphorylated with T4 polynucleotide kinase to facilitate subsequent ligation reactions. The milR coding region and P hrdB digested with SpeI were ligated together with SpeI/StuI double digested pIJ10500 in a three-piece ligation reaction to generate pIJ10500::P hrdB milR. Exconjugants were selected using hygromycin and further confirmed by PCR amplifications. For GUS assays, spores of S. coelicolor 1146 derived strains were harvested, resuspended in double-distilled water (ddH2O), and optical density at 450 nm was measured. The spore suspension was normalized to the same level, series diluted, and spotted on 25-ml plates of AS-1 agar containing 100 μl of 40 mg/ml 5-bromo-4-chloro-3-indolyl-β-D-glucoronide and photographed after 3 days at 28 °C [45, 46]. For GUS assay of milA4-E promoter, the plate of AS-1 agar containing chromogenic substrate was photographed after 7 days cultivation at 28 °C.
RNA isolation, RT-PCR and quantitative real-time RT-PCR
RNAs were isolated from S. bingchenggensis BC04 grown at 28 °C at different time points (1, 2, 3, 5, 7 and 9 days). The detailed steps for RNA extraction were described previously [47]. To exclude the possibility of genomic DNA contamination, each RNA sample was treated with RQ1 RNase-free DNase I (Promega). The quality and quantity of RNAs were examined by UV spectroscopy and agarose gel electrophoresis. For RT-PCR and quantitative real time RT-PCR, first-strand cDNA synthesis was carried out with the superscript III first-strand synthesis system (Invitrogen, California, USA) using 1 μg total RNAs following the manufacturer’s instructions. All cDNA synthesis reactions included a replicate reaction without reverse transcriptase to ensure the complete removal of contaminating DNA from the RNA samples. For RT-PCR, after 31 cycles of amplification, the products were examined by the 1.5 % agarose gel electrophoresis and then visualized by staining with ethidium bromide. For quantitative real time RT-PCR, oligonucleotides were designed to amplify fragments of 100-200 bp (Additional file 6: Table S1). The PCR procedures were as follows: reactions were performed in 96-well plates using a ROCHE LightCycler®-96. Each 20 μl reaction contained 10 μl of 2× SuperReal PreMix (SYBR Green I included), 6 pmol of each primer and 1 μl five-fold diluted cDNA. The reaction parameters were as follows: 95 °C for 10 min, followed by 40 three-step amplification cycles consisting of denaturation at 95 °C for 15 s, annealing at 60 °C for 60 s and extension at 72 °C for 30 s. A final dissociation stage was run to generate a melting curve and consequently verify the specificity of the amplification products. After the PCR amplifications, data were analyzed by LightCycler®-96 Series Software. All samples were run in triplicate. The transcriptional level of target genes was normalized internally to the level to 16S rRNA transcription according to the Livak’s method [48].
Western blotting of MilR and its derivatives in S. bignchenggensis
For western blotting analysis, cell extracts from ΔmilR/MilR, ΔmilR/G31A, ΔmilR/G37A, ΔmilR/G38A, ΔmilR/D122A and ΔmilR/D123A grown in the fermentation medium at 3 days, were sonicated on ice. The concentration of total protein was determined by BCA protein assay using BSA as the standard sample. Proteins with equal concentrations (80 μg) samples were loaded onto 12 % polyacrylamide/SDS gel electrophoresis. Proteins in the gels were transferred to PVDF western blotting membranes (Roche, Germany) and probed with monoclonal ANTI-FLAGM2 antibody (Sigma-Aldrich, USA) as recommended by the manufacturer. The antibodies on the membranes were hybridized with the Horse Anti-Mouse IgG (H + L)-AP as secondary antibody (ZSGB-BIO, China) and the position of tagged-FLAG was visualized through BCIP/NBT method.
Site-directed mutagenesis of milR
For construction of wild-type milR complement plasmid, a 3278-bp DNA fragment containing the complete milR coding region and its promoter region was amplified using RCOMF2 and RCOMR2 (Additional file 6: Table S1) as primers, and then cloned between the SpeI and StuI sites of pIJ10500 to obtain pIJ10500::milR. Meanwhile the 3278-bp fragment was also inserted into the EcoRV site of pBluescriptKS (+) to generate pBluescriptKS (+)::milR, which was used as a template plasmid. For the construction of mutant milR, the primer pairs (Additional file 6: Table S1) were phosphorylated at the 5′ end with T4 polynucleotide kinase, and were further used for amplication of mutant milR fragment by PCR from the template plasmid pBluescriptKS (+):: milR. The PCR products were purified by gel extraction and ligated by self-ligation using T4 DNA ligase to generate mutagenized milR plasmids. The mutants of milR are as follows: (1) G31A (GGC to GCG), (2) G32A (GGG to GCG), (3) G34A (GGG to GCG), (4) G36A (GGC to GCG), (5) K37A (AAG to GCG), (6) S38A (AGC to GCG), (7) D122A (GAC to GCG), (8) D123A (GAT to GCG) and (9) K37R (AAG to CGC). These mutated plasmids were then digested with SpeI and AhdI respectively, giving a 721-bp restriction fragment that contains the mutated site inside milR. The SpeI–AhdI fragments were then cloned between the SpeI and AhdI sites of the pIJ10500::milR to generate pIJ10500::G31A, pIJ10500::G32A, pIJ10500::G34A, pIJ10500::G36A, pIJ10500::K37A, pIJ10500::K37R, pIJ10500::S38A, pIJ10500::D122A and pIJ10500::D123A. The authenticity of the whole sequence of milR was verified by sequencing. Subsequently, pIJ10500::milR and these pIJ10500::milR-derived plasmids were then integrated into the chromosomal ФBT1 attB site of milR disruption mutant (ΔmilR) by conjugation.
Construction of milR overexpression strains in S. bingchenggensis BC04
For the construction of pSET152::P hrdB milR and pKC1139::P hrdB milR, the hrdB promoter (P hrdB ) was amplified with primer pair hrdB-pF and hrdB-pR, and the milR coding region was amplified with primer pair milR-F and milR-R (Additional file 6: Table S1). Prior to PCR amplification, primers hrdB-pR and milR-F were phosphorylated with T4 polynucleotide kinase to facilitate subsequent ligation reactions. P hrdB digested with XbaI and the milR coding region were ligated together with XbaI/EcoRV double digested pSET152 and pKC1139 in a three-piece ligation reaction to generate pSET152::P hrdB milR and pKC1139::P hrdB milR. For the construction of pSET152::2milR, two 3278-bp DNA fragments containing the complete milR coding region and its promoter region were amplified with primer pairs ROE-F/ROE-R1 and ROE-F1/ROE-R (Additional file 6: Table S1). The fragment amplified with ROE-F/ROE-R1 was digested with XbaI and BamHI, another fragment amplified with ROE-F1/ROE-R was digested with BamHI. Then the two digested fragments were ligated together with XbaI/EcoRV double digested pSET152 in a three-piece ligation reaction to generate pSET152::2milR. These three plasmids (pSET152::P hrdB milR, pKC1139::P hrdB milR and pSET152::2milR) together with pSET152::milR were introduced into S. bingchenggensis BC04 by conjugation and resulted in the overexpressed strain BC04::hrdBmilR, BC04::hrdBmilRs, BC04::milR and BC04::2milR.
Determination of cell dry weight
Five-milliliter cell cultures were collected by vacuum filtration and dried at 60 °C to a constant weight.
Statistical analysis
All experiments were carried out independently at least three times, and the mean values ±SD were presented. The data were analyzed by Student’s t test. P < 0.05 is used as a standard criterion of statistical significance.
Abbreviations
- AAA:
-
ATPases associated with diverse cellular activities
- Ala (A):
-
alanine
- ARTP:
-
atmospheric and room temperature plasma
- CDD:
-
conserved domain database
- CSR:
-
cluster-situated regulator
- D:
-
Asp, aspartic acid
- G:
-
Gly, glycine
- GBL:
-
γ-butyrolactones
- GUS:
-
β-glucuronidase
- HPLC:
-
high performance liquid chromatography
- HTH-LuxR:
-
helix_turn_helix, luminescence regulator
- K:
-
Lys, lysine
- LAL:
-
large ATP-binding regulator of the LuxR family
- MS:
-
mannitol/soya
- NTG:
-
N-methyl-N′-nitroso-N-nitrosoguanidine
- NTPases:
-
nucleoside triphosphatases
- NTPs:
-
nucleoside triphosphates
- ORF:
-
open reading frame
- OTC:
-
oxytetracycline
- PCR:
-
polymerase chain reaction
- PKS:
-
polyketide synthase
- qRT-PCR:
-
quantitative real-time RT-PCR
- R:
-
Arg, arginine
- RT-PCR:
-
reverse transcription polymerase chain reaction
- S:
-
Ser, serine
- SMART:
-
simple modular architecture research tool
References
Shoop WL, Mrozik H, Fisher MH. Structure and activity of avermectins and milbemycins in animal health. Vet Parasitol. 1995;59:139–56.
Danaher M, Radeck W, Kolar L, Keegan J, Cerkvenik-Flajs V. Recent developments in the analysis of avermectin and milbemycin residues in food safety and the environment. Curr Pharm Biotechnol. 2012;13:936–51.
Jacobs CT, Scholtz CH. A review on the effect of macrocyclic lactones on dung-dwelling insects: toxicity of macrocyclic lactones to dung beetles. Onderstepoort J Vet Res. 2015;82:858.
Nicastro RL, Sato ME, da Silva MZ. Fitness costs associated with milbemectin resistance in the two-spotted spider mite Tetranychus urticae. Int J Pest Manag. 2011;57:223–8.
Pluschkell U, Horowitz AR, Ishaaya I. Effect of milbemectin on the sweetpotato whitefly, Bemisia tabad. Phytoparasitica. 1999;27:183–91.
Rinaldi L, Pennacchio S, Musella V, Maurelli MP, La Torre F, Cringoli G. Helminth control in kennels: is the combination of milbemycin oxime and praziquantel a right choice? Parasit Vectors. 2015;8:30.
McCall JW. The safety-net story about macrocyclic lactone heartworm preventives: a review, an update, and recommendations. Vet Parasitol. 2005;133:197–206.
Takiguchi Y, Mishima H, Okuda M, Terao M, Aoki A, Fukuda R. Milbemycins, a new family of macrolide antibiotics: fermentation, isolation and physico-chemical properties. J Antibiot. 1980;33:1120–7.
Xiang W, Wang J, Wang X, Zhang J, Wang Z. Further new milbemycin antibiotics from Streptomyces bingchenggensis. Fermentation, isolation, structural elucidation and biological activities. J Antibiot. 2007;60:608–13.
Xiang W, Wang J, Wang X, Zhang J. Two new beta-class milbemycins from Streptomyces bingchenggensis: fermentation, isolation, structure elucidation and biological properties. J Antibiot. 2007;60:351–6.
Xiang W, Wang J, Fan H, Wang X, Zhang J. New seco-milbemycins from Streptomyces bingchenggensis: fermentation, isolation and structure elucidation. J Antibiot. 2008;61:27–32.
Wang X, Wang J, Xiang W, Zhang J. Three new milbemycin derivatives from Streptomyces bingchenggensis. J Asian Nat Prod Res. 2009;11:597–603.
Wang X, Yan Y, Zhang B, An J, Wang J, Tian J, Jiang L, Chen Y, Huang S, Yin M, et al. Genome sequence of the milbemycin-producing bacterium Streptomyces bingchenggensis. J Bacteriol. 2010;192:4526–7.
He Y, Sun Y, Liu T, Zhou X, Bai L, Deng Z. Cloning of separate meilingmycin biosynthesis gene clusters by use of acyltransferase–ketoreductase didomain PCR amplification. Appl Environ Microbiol. 2010;76:3283–92.
Nonaka K, Kumasaka C, Okamoto Y, Maruyama F, Yoshikawa H. Bioconversion of milbemycin-related compounds: biosynthetic pathway of milbemycins. J Antibiot. 1999;52:109–16.
Nonaka K, Tsukiyama T, Sato K, Kumasaka C, Maruyama F, Yoshikawa H. Bioconversion of milbemycin-related compounds: isolation and utilization of non-producer, strain RNBC-5-51. J Antibiot. 1999;52:620–7.
Zhang J, An J, Wang J, Yan Y, He H, Wang X, Xiang W. Genetic engineering of Streptomyces bingchenggensis to produce milbemycins A3/A4 as main components and eliminate the biosynthesis of nanchangmycin. Appl Microbiol Biotechnol. 2013;97:10091–101.
Wang H, Zhang J, Zhang Y, Zhang B, Liu C, He H, Wang X, Xiang W. Combined application of plasma mutagenesis and gene engineering leads to 5-oxomilbemycins A3/A4 as main components from Streptomyces bingchenggensis. Appl Microbiol Biotechnol. 2014;98:9703–12.
Liu G, Chater KF, Chandra G, Niu G, Tan H. Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol Mol Biol Rev. 2013;77:112–43.
Liu G, Tian Y, Yang H, Tan H. A pathway-specific transcriptional regulatory gene for nikkomycin biosynthesis in Streptomyces ansochromogenes that also influences colony development. Mol Microbiol. 2005;55:1855–66.
Guo J, Zhao J, Li L, Chen Z, Wen Y, Li J. The pathway-specific regulator AveR from Streptomyces avermitilis positively regulates avermectin production while it negatively affects oligomycin biosynthesis. Mol Genet Genomics. 2010;283:123–33.
Kitani S, Ikeda H, Sakamoto T, Noguchi S, Nihira T. Characterization of a regulatory gene, aveR, for the biosynthesis of avermectin in Streptomyces avermitilis. Appl Microbiol Biotechnol. 2009;82:1089–96.
Luo S, Sun D, Zhu J, Chen Z, Wen Y, Li J. An extracytoplasmic function sigma factor, sigma, differentially regulates avermectin and oligomycin biosynthesis in Streptomyces avermitilis. Appl Microbiol Biotechnol. 2014;98:7097–112.
Wilson DJ, Xue Y, Reynolds KA, Sherman DH. Characterization and analysis of the PikD regulatory factor in the pikromycin biosynthetic pathway of Streptomyces venezuelae. J Bacteriol. 2001;183:3468–75.
Xie C, Deng J, Wang H. Identification of AstG1, A LAL family regulator that positively controls ansatrienins production in Streptomyces sp. XZQH13. Curr Microbiol. 2015;70:859–64.
Saraste M, Sibbald PR, Wittinghofer A. The P-loop—a common motif in ATP- and GTP-binding proteins. Trends Biochem Sci. 1990;15:430–4.
Snider J, Houry WA. AAA+ proteins: diversity in function, similarity in structure. Biochem Soc Trans. 2008;36:72–7.
Leipe DD, Koonin EV, Aravind L. STAND, a class of P-loop NTPases including animal and plant regulators of programmed cell death: multiple, complex domain architectures, unusual phyletic patterns, and evolution by horizontal gene transfer. J Mol Biol. 2004;343:1–28.
Leskiw BK, Lawlor EJ, Fernandez-Abalos JM, Chater KF. TTA codons in some genes prevent their expression in a class of developmental, antibiotic-negative, Streptomyces mutants. Proc Natl Acad Sci USA. 1991;88:2461–5.
Du D, Zhu Y, Wei J, Tian Y, Niu G, Tan H. Improvement of gougerotin and nikkomycin production by engineering their biosynthetic gene clusters. Appl Microbiol Biotechnol. 2013;97:6383–96.
Yin S, Wang W, Wang X, Zhu Y, Jia X, Li S, Yuan F, Zhang Y, Yang K. Identification of a cluster-situated activator of oxytetracycline biosynthesis and manipulation of its expression for improved oxytetracycline production in Streptomyces rimosus. Microb Cell Fact. 2015;14:46.
Wang X, Wang X, Xiang W. Improvement of milbemycin-producing Streptomyces bingchenggensis by rational screening of ultraviolet- and chemically induced mutants. World J Microbiol Biotechnol. 2009;25:1051–6.
Niu G, Chater KF, Tian Y, Zhang J, Tan H. Specialised metabolites regulating antibiotic biosynthesis in Streptomyces spp. FEMS Microbiol Rev. 2016;40:554–73.
Xu G, Wang J, Wang L, Tian X, Yang H, Fan K, Yang K, Tan H. “Pseudo” gamma-butyrolactone receptors respond to antibiotic signals to coordinate antibiotic biosynthesis. J Biol Chem. 2010;285:27440–8.
Li X, Yu T, He Q, McDowall KJ, Jiang B, Jiang Z, Wu L, Li G, Li Q, Wang S, et al. Binding of a biosynthetic intermediate to AtrA modulates the production of lidamycin by Streptomyces globisporus. Mol Microbiol. 2015;96:1257–71.
Hanson PI, Whiteheart SW. AAA+ proteins: have engine, will work. Nat Rev Mol Cell Biol. 2005;6:519–29.
Marquenet E, Richet E. How integration of positive and negative regulatory signals by a STAND signaling protein depends on ATP hydrolysis. Mol Cell. 2007;28:187–99.
Li R, Liu G, Xie Z, He X, Chen W, Deng Z, Tan H. PolY, a transcriptional regulator with ATPase activity, directly activates transcription of polR in polyoxin biosynthesis in Streptomyces cacaoi. Mol Microbiol. 2010;75:349–64.
Hsu JL, Peng HL, Chang HY. The ATP-binding motif in AcoK is required for regulation of acetoin catabolism in Klebsiella pneumoniae CG43. Biochem Biophys Res Commun. 2008;376:121–7.
Sohoni SV, Fazio A, Workman CT, Mijakovic I, Lantz AE. Synthetic promoter library for modulation of actinorhodin production in Streptomyces coelicolor A3(2). PLoS ONE. 2014;9:e99701.
Alper H, Fischer C, Nevoigt E, Stephanopoulos G. Tuning genetic control through promoter engineering. Proc Natl Acad Sci USA. 2005;102:12678–83.
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA. Practical Streptomyces genetics. Norwich: The John Innes Foundation; 2000.
Bierman M, Logan R, O’Brien K, Seno ET, Rao RN, Schoner BE. Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene. 1992;116:43–9.
Kuhstoss S, Richardson MA, Rao RN. Plasmid cloning vectors that integrate site-specifically in Streptomyces spp. Gene. 1991;97:143–6.
Sherwood EJ, Bibb MJ. The antibiotic planosporicin coordinates its own production in the actinomycete Planomonospora alba. Proc Natl Acad Sci USA. 2013;110:2500–9.
Myronovskyi M, Welle E, Fedorenko V, Luzhetskyy A. Beta-glucuronidase as a sensitive and versatile reporter in actinomycetes. Appl Environ Microbiol. 2011;77:5370–83.
Zhang Y, Pan G, Zou Z, Fan K, Yang K, Tan H. JadR*-mediated feed-forward regulation of cofactor supply in jadomycin biosynthesis. Mol Microbiol. 2013;90:884–97.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–8.
Authors’ contributions
YZ performed the experiments, analyzed the primary data and drafted the manuscript. HH carried out the GUS assays. HL participated in the fermentation analysis. HW assisted with experiments. XW revised the manuscript. WX supervised the whole work and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
We would like to thank Professor Mervyn Bibb (John Innes Centre, Norwich, UK) for providing S. coelicolor M1146, and Professor Mark Buttner (John Innes Centre, Norwich, UK) for providing pIJ10500.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its Additional files.
Funding
This work was supported by grants from the National Outstanding Youth Foundation (No. 31225024), the National Natural Science Foundation of China (No. 31401814), the Outstanding Youth Foundation of Heilongjiang Province (JC201201) and The Agricultural Science and Technology Innovation Program. The funding bodies had no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional files
12934_2016_552_MOESM1_ESM.pdf
Additional file 1: Figure S1. Confirmation of milR disruption by PCR amplification. A. The schematic diagram showing the position of primers in the chromosome of double-crossover strains. Among the two primer pairs used for PCR, one primer (conR1F or conR2R) in each pair is designed outside of the upstream (L) or downstream (R) sequence we used for construction of milR disruption mutant. B. Agarose gel electrophoresis showing PCR amplified fragments. PCR templates were genomic DNAs from the Streptomyces bingchenggensis BC04 (lane 1) and the three independent mutants as indicated (lanes 2–4). The primer pairs used are also shown. Then theoretical size of DNA fragments (4.5-kb and 3.5-kb) for ΔmilR were obtained by PCR, and further confirmed by DNA sequencing (data not shown).
12934_2016_552_MOESM2_ESM.pdf
Additional file 2: Figure S2. Co-transcriptional analysis of the biosynthetic genes in mil cluster. A. Intergenic regions numbered 1-4 were subjected to RT-PCR analysis and the occurrence of translational coupling between milA1 and milD were marked by “▲”, this location was not subjected to RT-PCT analysis. B. RT-PCR analysis of co-transcribed genes. Primers were designed to amplify 4 intergenic regions (numbered 1-4, Table S1). cDNA was synthesized from RNA samples isolated from S. bingchenggensis BC04 cultures (3 d cultivation in fermentation medium). Positive controls: PCR reactions using genomic DNA of BC04 as template which gave products of the same molecular sizes as those amplified from cDNA template obtained by reverse transcription. Negative controls: PCR amplification of hrdB using “cDNA” template synthesized without reverse transcriptase, which did not give products, thus excluding the possible contamination of genomic DNA. c: cDNA template; g: genomic DNA template; NRT: “cDNA” template synthesized in the absence of reverse transcriptase. These results show that milA2 and milC, milA4 and milE, milR and milA3, milA1 and milD form an operon respectively, whereas the other two genes milF and orf1 are transcribed individually.
12934_2016_552_MOESM3_ESM.pdf
Additional file 3: Figure S3. Diagrams of site-directed mutation of Walker A and Walker B motifs in MilR. A: Mutation in Walker A motif. The first line shows the wild-type Walker A sequence. From the second to the eighth line, red words indicate the Ala or Arg substitution was performed in the corresponding position. B: Mutation in Walker B motif. The first line shows the wild-type Walker B sequence, from the second to the third line, blue words indicate the Ala substitution was carried out to replace Asp in the corresponding position.
12934_2016_552_MOESM4_ESM.pdf
Additional file 4: Figure S4. Effect of site-directed mutation in the AAA domain of MilR on the transcription of milA2, milC, milA3 and milA1. Transcriptional analysis of milA2, milC, milA3 and milA1 were performed by semiquantitative RT-PCR. MilR:ΔmilR/milR, 31A:ΔmilR/G31A, 32A:ΔmilR/G32A, 34A:ΔmilR/G34A, 36A:ΔmilR/G36A, 37A:ΔmilR/K37A, 37R:ΔmilR/K37R, 38A:ΔmilR/S38A, 122A:ΔmilR/D122A, 123A:ΔmilR/D123A.
12934_2016_552_MOESM5_ESM.pdf
Additional file 5: Figure S5. Effect of milR engineering on antibiotic production. Data are presented as the averages of the results of three independent experiments. Error bars show standard deviations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y., He, H., Liu, H. et al. Characterization of a pathway-specific activator of milbemycin biosynthesis and improved milbemycin production by its overexpression in Streptomyces bingchenggensis . Microb Cell Fact 15, 152 (2016). https://doi.org/10.1186/s12934-016-0552-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12934-016-0552-1