
Abstract
Background
Patients with type 1 diabetes are more at risk of coronary artery disease than the general population. Although evidence points to a genetic risk there have been no study investigating genetic risk factors of coronary artery disease specific to individuals with type 1 diabetes. To identify low frequency and common genetic variations associated with coronary artery disease in populations of individuals with type 1 diabetes.
Methods
A two-stage genome wide association study was conducted. The discovery phase involved the meta-analysis of three genome-wide association cohorts totaling 434 patients with type 1 diabetes and coronary artery disease (cases) and 3123 T1D individuals with no evidence of coronary artery disease (controls). Replication of the top association signals (p < 10−5) was performed in five additional independent cohorts totaling 585 cases and 2612 controls.
Results
One locus (rs115829748, located upstream of the MAP1B gene) reached the statistical threshold of 5 × 10−8 for genome-wide significance but did not replicate. Nevertheless, three single nucleotide polymorphisms provided suggestive evidence for association with coronary artery disease in the combined studies: CDK18 rs138760780 (OR = 2.60 95% confidence interval [1.75–3.85], p = 2.02 × 10−6), FAM189A2 rs12344245 (OR = 1.85 [1.41–2.43], p = 8.52 × 10−6) and PKD1 rs116092985 (OR = 1.53 [1.27–1.85], p = 1.01 × 10−5). In addition, our analyses suggested that genetic variations at the ANKS1A, COL4A2 and APOE loci previously found associated with coronary artery disease in the general population could have stronger effects in patients with type 1 diabetes.
Conclusions
This study suggests three novel candidate genes for coronary artery disease in the subgroup of patients affected with type 1 diabetes. The detected associations deserve to be definitively validated in additional epidemiological studies.
Similar content being viewed by others


Background
Type 1 diabetes (T1D) is a chronic disease characterized by an increase in blood glucose due to a lack of insulin production. Diabetes is a major health concern globally with a prevalence ranging between 4 and 7.8% in industrialized countries [1, 2]; among persons with diabetes, it is estimated that 5–10% are affected with T1D [3]. Recent large-scale epidemiological studies suggest that T1D is associated with a reduced lifespan of approximately 12 years [4] due to at least two-fold increased risk for death due to cardiovascular (CV) events.
Of note, in a nation-based registry of type 1 diabetes, coronary heart disease was 4 times more prevalent in patients with type 1 diabetes compared to sex- and age-matched controls [5].
However, studies exploring cardiovascular disease (CVD) risk variants in T1D patients are lacking. Traditional risk factors for CVD including age, circulating lipid levels, and smoking are well established in the general population [6] and in patients with type 1 diabetes [7]. Coronary artery disease (CAD) could be strongly influenced by genetic factors [8]. However, to the best of our knowledge, the relationship between genetic factors and CAD in type 1 diabetes was largely examined with a candidate gene approach [9,10,11,12]. Another important question is whether genetic markers of CAD established in the general population, known to roughly explain 10% of the heritability [13, 14], also play a role in individuals with T1D.
In an attempt to unravel the genetic determinants of CAD among T1D patients, we examined the association of genome-wide genotype array data with CAD in multiple T1D cohorts of European descent (The British Isles, Denmark and France).
Materials and methods
General workflow
The present work reports the results of a two-stage research strategy for common genetic variations associated with CAD risk in T1D patients. The first (discovery) stage was based on the meta-analysis of three GWAS cohorts totaling 434 T1D patients with CAD (cases) and 3123 T1D patients with no evidence of CAD (controls). The second stage consisted of a replication of the top discovery signals with association p < 10−5 in five additional T1D studies totaling 585 cases and 2612 controls.
Participating cohorts for the discovery and replication stages
All participants were patients with T1D diagnosed using ADA criteria [15].
Controls were patients with T1D without history of CAD while cases were patients with T1D and a personal history of myocardial infarction or coronary artery revascularization (coronary artery angioplasty or by-pass grafting).
The discovery phase was composed of European-ancestry adults from (1) France (pooled cohorts of T1D from Corbeil Essonnes, Poitiers, Nantes, Paris, Toulouse [16] and two large scale multicenter cohorts i.e. GENESIS/GENEDIAB [16]), (2) Denmark (After-EU cohort [17]) and (3) British Isles (UK-ROI study [18]). The replication phase included five independent T1D cohorts recruited in North America (Additional file 1: Table S1).
All participating studies were approved by their respective institutional review board/ethics committee and an informed consent was obtained from all participating individuals.
Genotype determinations and imputation
For each participating study, DNA samples were genotyped with high-density SNP arrays and further imputed for SNPs available in the 1000 Genomes reference dataset. Summary descriptions of genotyping technologies, quality control procedures, and used imputation methods (MACH/Impute2) are shown in Additional file 1: Table S1.
Discovery phase: meta-analysis of discovery GWAS
Association analyses of imputed SNPs with CAD risk were performed separately in each study. Analyses were performed using either of the MACH [19], Quicktest (http://toby.freeshell.org/software/quicktest.shtml), or Plink [20] analyses tools implementing a logistic regression model where the allele dosage representing the expected number of a given reference allele at the imputed SNP was used as covariate to estimate SNP effect. Analyses were adjusted for sex, age, DN status and potential population sub-structure as defined by SNPs derived principal components.
Only SNPs with acceptable imputation quality (r2 > 0.3) in the three discovery cohorts and with estimated minor allele frequency (MAF) ≥ 1% were kept for meta-analysis. This was performed by use of a fixed-effects model based on the inverse-variance weighting method as implemented in the METAL software [21]. The statistical threshold (p < 5 × 10−8) was used for declaring genome-wide statistical significance while controlling for the number of independent tests across the genome. The Cochran’s Q statistic was used to assess heterogeneity of the SNP associations across studies whose magnitude was expressed by the I2 index [22]. Power calculations were performed using the CaTS power calculator (http://csg.sph.umich.edu/abecasis/cats/) [23].
Replication phase
Similar logistic regression models as those employed in the discovery were used for assessing the association of tested SNPs with CAD risk (Additional file 1: Table S1). Results obtained in the independent replication cohorts were then meta-analyzed using the same methodology as in the discovery step. The Bonferroni threshold corresponding to 0.05 divided by the number of tested SNPs was used to declare statistical replication. Unilateral hypothesis testing was adopted at the replication stage. For SNPs that replicated, a meta-analysis of the combined discovery and replications cohorts was performed to produce a more robust estimate of the effect size.
Results
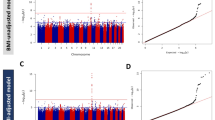
A total of 6,728,637 imputed SNPs were tested for association with CAD in 3557 T1D patients made of 434 with CAD cases and 3123 controls in the discovery dataset. The meta-analysis results of the discovery GWAS have been summarized in the Manhattan and QQ plots shown in Additional file 2: Figure S1, Additional file 3: Figure S2, respectively.
One locus at chromosome 5q13.2 reached genome-wide significance (p < 5 × 10−8) with the lead SNP, rs115829748, located upstream of the MAP1B gene. The T allele of this low frequency SNP (MAF ~ 0.04), was associated with an Odds Ratio (OR) of 3.16 [95% confidence interval (CI) 2.18–4.59] (p = 1.36 × 10−9). No other SNP demonstrated suggestive association with CAD at this locus (Additional file 4: Figure S3).
At the p < ~ 1.0 × 10−5 threshold, 20 additional loci demonstrated evidence for suggestive association with CAD with little heterogeneity across cohorts (Table 1). Imputation metrics of the top SNPs are provided in the Additional file 5: Table S2. Therefore, we sought to replicate the top 21 signals in five independent T1D cohorts totaling 585 CAD cases and 2612 controls. Replication was feasible for 17 SNPs while four SNPs (rs34319244, rs373009901, rs143723948, rs571622299) were not properly imputed in the replication stage (Table 1). While none of the 17 SNPs reached the pre-specified Bonferroni threshold of 3.0 × 10−3 for positive statistical replication, three were however nominally (p < 0.05) associated with CAD in the replication stage, with genetic effects consistent between the discovery and replication studies (Table 1). Of note, no trend for association was observed (p = 0.279) with the MAP1B rs115829748 that came out first in the discovery GWAS and that showed similar allele frequencies in the discovery and replication studies.
The strongest association was observed at the CDK18 locus where, in the replication stage, the rs138760780-T allele, with frequency 0.02, was associated with an increased odds ratio (OR) for disease of 1.88 [1.07–3.31] (p = 0.014). This value has to compare with 3.48 [2.00–6.04] observed in the discovery cohorts. In the combined discovery and replication cohorts, the meta-analyzed OR associated with the rs138760780 T allele was 2.60 [1.75–3.85] (p = 2.02 × 10−6) with no heterogeneity across the discovery and replication stage (p = 0.545).
The second suggestive association holds at the FAM189A2 locus. The rs12344245-G allele found associated with an increased OR of 2.52 [1.68–3.81] in the discovery cohorts also demonstrated a trend for association with the disease in the replication stage, OR = 1.45 [1.01–2.08] (p = 0.022). Combining the discovery and replication study led to a meta-analyzed OR for disease of 1.85 [1.41–2.43] (p = 8.52 × 10−6) with no significant evidence for heterogeneity across stage (p = 0.426).
The third suggestive association was observed for the PKD1 locus. The rs116092985 minor G associated with a 1.85 [1.408–2.44] and a 1.29 [1.00–1.67] increased risk of CAD in the discovery and replication cohorts, respectively. Altogether, the combined statistical evidence for association of the rs116092985-G allele reached p = 1.01 × 10−5 (OR = 1.53 [1.27–1.85]) (p = 0.220 for heterogeneity across studies).
Candidate CAD SNPs
About 90 loci have been found, through GWAS studies, to harbor common susceptibility alleles associated with CAD in the general population. We sought to investigate how these loci associate with CAD in T1D patients. Results of this investigation are summarized in Table 2. From CAD SNPs identified in previous GWAS [13, 14, 24,25,26], 95 were well imputed in our discovery phase and showed genetic effects with directionality in our T1D populations that were consistent with those previously reported (Table 2). Imputation quality for these 95 SNPs is given in Additional file 6: Table S3. For three SNPs ANKS1A_rs17609940, COL4A2_rs11838776 and TOMM40_rs2075650 (near the APOE locus), the statistical evidence for association with the disease was rather strong with p < 5 × 10−3 even though it did not achieve multiple testing correction for the number of tested SNPs (~ 5 × 10−4 = 0.05/95). For these three SNPs, the amplitude of the genetic association even tended to be stronger in our T1D patients than that previously reported (Table 2). As an illustration, in our discovery T1D population, the COL4A1 rs11838776-A allele was associated with an OR of 1.33 [1.11–1.61] while the OR reported in the literature was slightly lower (OR = 1.07). Conversely, the association of the polymorphism at the non-coding ANRIL loci on 9p21, that is known to associate the most with CAD among common polymorphisms, showed a very similar association in our T1D patients (OR = 1.16 [0.993–1.362], p = 0.03) compared to that previously reported (OR ~ 1.21).
Discussion
The present work was aimed at identifying susceptibility alleles for CAD risk in patient population of T1D using a GWAS approach with a two-step framework (discovery + replication). Albeit we identified one locus (MAP1B) reaching genome-wide significance in the discovery stage, it did not replicate with similar effects. Nevertheless, in the combined T1D dataset of 1019 cases and 5735 controls, we observed strong statistical evidence for association with CAD at three biological candidate genes, CDK18, PKD1 and FAM189A2.
We consider our study as very original as it is the first one, to the best of our knowledge, to use a GWAS approach for CAD, in patients with type 1 diabetes. We have thus pooled the largest collection of type 1 diabetes patients with available genetic data.
The low frequency CDK18 rs138760780-T allele (frequency ~ 0.02) was found associated with ~ 2.5 fold increased risk of CAD. According to public database (e.g. Haploreg [27]), this SNP does not show strong linkage disequilibrium (LD) (pairwise r2 > 0.80) with other SNPs at this locus, consistent with the regional association plot that does not suggest any evidence of disease associated SNPs (Additional file 7: Figure S4). Interrogating the functional status of this SNP through HaploReg tool [27] suggested that this SNP may be involved in some epigenetic regulatory mechanisms. CDK18 encodes for a cyclin-dependent kinase, suggesting a role in cell cycle. This predicted protein is also related to CDK1, which is involved in the G2/M transition in eukaryotic cells [28]. Although cell cycle is a very broad pathway, CDK1 has also been associated with T1D [29], but at this point little is known about a potential involvement of CDK18 in the pathophysiology of T1D or its complications.
We also observed some evidence that the low frequency FAM189A2 rs12344245 G allele, (frequency ~ 0.04), associated with a ~ 1.8 fold-increased risk of CAD. We did not find any evidence suggesting that this intronic SNP, or any other SNPs in LD (Additional file 8: Figure S5: regional association plot) with it, could be functional. Nevertheless, even though not much is known about the role of the encoded protein, this locus is a good candidate. Indeed, genetic variations at this locus have been found associated with albumin to creatinine ratio [30]. More interestingly, two FAM189A2 SNPs (rs10780297 and rs10120442) have been reported to moderately associate (p = 9.3 × 10−4) in a large GWAS for CAD in ~ 63,000 non-diabetic populations [14], suggesting that this locus could be a CAD locus in some specific at-risk groups of diabetic patients. The latter two SNPs are in moderate LD (D’ = 1 but r2 = 0.05) with our lead rs12344245 SNP, indicating that a fine mapping analysis of this locus would warrant further investigations. Of interest, it was not identified as a common gene in both type 2 diabetes and CAD. It can thus be speculated this gene is an important gene in high-glucose environment rather than a gene leading to high-glucose.
Finally, we observed an association of the non-synonymous PKD1 rs116092985 (Trp1399Arg) with CAD among T1D patients where the Arg1399 minor allele (frequency ~ 0.10), was associated with an increased CAD risk (OR ~ 1.5). The regional plot (Additional file 9: Figure S6) shows that there are several SNPs in LD with this PKD1 top SNP that associate with CAD. PKD1 encodes for the Polycystin 1, Transient Receptor Potential Channel Interacting protein, a member of the polycystin protein family. Recent reports have suggested a role of PKD1 not only in renal tubular function and structure [31] but rare mutations in this gene as the main also cause underlying polycystic kidney disease [32], highlighting its importance in kidney complications. One important question is whether PKD1 risk allele is involved in a common genetic background linking DN and CAD. This question was not duly analyzed due to power issue. However, no clear association was established with DN in previous GWAS focusing on this question [18].
Our study also enabled us to assess in patients with T1D the impact of common SNPs that have been found associated with CAD in large GWAS performed in unselected individuals. Beyond the observation that most of the previously reported SNPs showed consistent association with CAD in our T1D population, this look-up identified a few CAD loci (ANKS1A, COL4A2, TOMM40/APOE) where the reported CAD associated SNP could have a stronger effect in T1D patients. However, this hypothesis would require further investigation.
Some limitations must be acknowledged. We did not consider differently T1D patients with and without DN, and all of the analyses were not stratified on DN status in order to keep all CAD patients in the analysis. However, this should have limited impact on our main results as none of the SNPs identified here were positive considering previously reported GWAS with regard to DN as primary endpoint [18]. Another limitation pertains to limited power of our sample size required, particular to overcome the harsh genome-wide statistical significance threshold. Indeed, our discovery GWAS was not well powered to identify common SNPs associated with moderate genetic effects as those frequently encountered in a GWAS context. For instance, our discovery study had no power to detect at the genome-wide statistical threshold the genetic effect of a variant with an associated allelic OR less than 1.40. It was only well powered (> 80%) to detect OR greater than 1.6 as soon as the allele frequency of the disease allele is greater than 0.28 and well powered to detect OR greater than ~ 2 for allele frequency greater than 0.05. In particular, we had no power to detect the well-established association of the 9p21 locus at the 5 × 10−8 threshold while we had a chance of 60% to detect it would the liberal threshold of 0.05 had been used. Similarly, we acknowledge the low power of our replication studies where none of the tested associations achieved the Bonferroni threshold of 3 × 10−3. We only had a power of 53, 38 and 26% to detect at this threshold a significant association at the CDK18 rs13876070, PKD1 rs116092985 and the FAM189A2 loci, respectively.
Despite these limitations, we have assembled the largest cohort available and conducted novel analyses to discover novel candidate loci for CAD in T1D patients that need to be further studied with additional epidemiological data and functional work to confirm our findings. Of interest, our negative study could suggest a role of epigenetics beyond genomics. In this regard, insulin promotes the expression of DNA methyltransferases leading to methylation resulting in atherosclerosis [33], broadening the research field of CAD in type 1 diabetes.
Conclusions
We identified three new candidate loci for CAD in individuals with T1D, these loci weren’t previously found associated with CAD in the general population. Three other loci previously reported in the general population were found associated with CAD in our setting, namely ANKS1A, COL4A2 and TOMM40/APOE. Although this work needs further investigation, studying the function of these loci could lead to a better understanding of the physiological pathways involved in the development of CAD as a complication of T1D.
Abbreviations
- CAD:
-
coronary artery disease
- T1D:
-
type 1 diabetes
- GWAS:
-
genome wide association study
- CVD:
-
cardiovascular disease
- DN:
-
diabetic nephropathy
- SNP:
-
single nucleotide polymorphism
- MAF:
-
minor allele frequency
- OR:
-
odds ratio
- LD:
-
linkage disequilibrium
- CI:
-
confidence interval
References
Menke A, Casagrande S, Geiss L, Cowie CC. Prevalence of and trends in diabetes among adults in the United States, 1988–2012. JAMA. 2015;314:1021–9.
Kusnik-Joinville O, Weill A, Ricordeau P, Allemand H. Diabète traité en France en 2007: un taux de prévalence proche de 4% et des disparités géographiques croissantes. BEH. 2008;43:409–13.
Daneman D. Type 1 diabetes. Lancet. 2006;367:847–58.
Livingstone SJ, Levin D, Looker HC, Lindsay RS, Wild SH, Joss N, et al. Estimated life expectancy in a Scottish cohort with type 1 diabetes, 2008–2010. JAMA. 2015;313:37–44.
Lind M, Svensson AM, Kosiborod M, Gudbjörnsdottir S, Pivodic A, Wedel H, et al. Glycemic control and excess mortality in type 1 diabetes. N Engl J Med. 2014;371:1972–82.
Yusuf S, Hawken S, Ounpuu S, Dans T, Avezum A, Lanas F, et al. Effect of potentially modifiable risk factors associated with myocardial infarction in 52 countries (the INTERHEART study): case-control study. Lancet. 2004;364:937–52.
Nathan DM, Cleary PA, Backlund J-YC, Genuth SM, Lachin JM, Orchard TJ, et al. Intensive diabetes treatment and cardiovascular disease in patients with type 1 diabetes. N Engl J Med. 2005;353:2643–53.
Earle K, Walker J, Hill C, Viberti G. Familial clustering of cardiovascular disease in patients with insulin-dependent diabetes and nephropathy. N Engl J Med. 1992;326:673–7.
Hokanson JE, Cheng S, Snell-Bergeon JK, Fijal BA, Grow MA, Hung C, et al. A common promoter polymorphism in the hepatic lipase gene (LIPC-480C > T) is associated with an increase in coronary calcification in type 1 diabetes. Diabetes. 2002;51:1208–13.
Pettersson-Fernholm K, Karvonen MK, Kallio J, Forsblom CM, Koulu M, Pesonen U, et al. Leucine 7 to proline 7 polymorphism in the preproneuropeptide Y is associated with proteinuria, coronary heart disease, and glycemic control in type 1 diabetic patients. Diabetes Care. 2004;27:503–9.
Kretowski A, Hokanson JE, McFann K, Kinney GL, Snell-Bergeon JK, Maahs DM, et al. The apolipoprotein A-IV Gln360His polymorphism predicts progression of coronary artery calcification in patients with type 1 diabetes. Diabetologia. 2006;49:1946–54.
Pettersson-Fernholm K, Forsblom C, Hudson BI, Perola M, Grant PJ, Groop P-H, et al. The functional -374 T/A RAGE gene polymorphism is associated with proteinuria and cardiovascular disease in type 1 diabetic patients. Diabetes. 2003;52:891–4.
CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33.
Nikpay M, Goel A, Won H-H, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–30.
Association AD. Diagnosis and classification of diabetes mellitus. Diabetes Care. 2010;33:S62–9.
Hadjadj S, Cariou B, Fumeron F, Gand E, Charpentier G, Roussel R, et al. Death, end-stage renal disease and renal function decline in patients with diabetic nephropathy in French cohorts of type 1 and type 2 diabetes. Diabetologia. 2016;59:208–16.
Lajer M, Jorsal A, Tarnow L, Parving H-H, Rossing P. Plasma growth differentiation factor-15 independently predicts all-cause and cardiovascular mortality as well as deterioration of kidney function in type 1 diabetic patients with nephropathy. Diabetes Care. 2010;33:1567–72.
Sandholm N, Salem RM, McKnight AJ, Brennan EP, Forsblom C, Isakova T, et al. New susceptibility loci associated with kidney disease in type 1 diabetes. PLoS Genet. 2012;8:e1002921.
Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol. 2010;34:816–34.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Higgins JPT, Thompson SG. Quantifying heterogeneity in a meta-analysis. Stat Med. 2002;21:1539–58.
Hsieh FY, Bloch DA, Larsen MD. A simple method of sample size calculation for linear and logistic regression. Stat Med. 1998;17:1623–34.
Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. 2017;49:1385–91.
Howson JMM, Zhao W, Barnes DR, Ho W-K, Young R, Paul DS, et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. 2017;49:1113–9.
Webb TR, Erdmann J, Stirrups KE, Stitziel NO, Masca NGD, Jansen H, et al. Systematic evaluation of pleiotropy identifies 6 further loci associated with coronary artery disease. J Am Coll Cardiol. 2017;69:823–36.
Ward LD, Kellis M. HaploReg: a resource for exploring chromatin states, conservation, and regulatory motif alterations within sets of genetically linked variants. Nucleic Acids Res. 2012;40:D930–4.
Okuda T, Cleveland JL, Downing JR. PCTAIRE-1 and PCTAIRE-3, two members of a novel cdc2/CDC28-related protein kinase gene family. Oncogene. 1992;7:2249–58.
Bradfield JP, Qu H-Q, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7:e1002293.
Böger CA, Chen M-H, Tin A, Olden M, Köttgen A, de Boer IH, et al. CUBN is a gene locus for albuminuria. J Am Soc Nephrol. 2011;22:555–70.
Song X, Di Giovanni V, He N, Wang K, Ingram A, Rosenblum ND, et al. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet. 2009;18:2328–43.
The International Polycystic Kidney Disease Consortium. Polycystic kidney disease: the complete structure of the PKD1 gene and its protein. Cell. 1995;81:289–98.
Min J, Weitian Z, Peng C, Yan P, Bo Z, Yan W, Yun B, Xukai W. Correlation between insulin-induced estrogen receptor methylation and atherosclerosis. Cardiovasc Diabetol. 2016;15:156.
Authors’ contributions
RC performed the GWAS for the French cohort as well as the meta-analyses for the three discovery cohorts and the five replication cohorts. SD performed the GWAS for the Ireland-UK cohort. SK performed the GWASes for the five replication cohorts. TSA performed the GWAS for the Danish cohort. DAT and SH interpreted the results of the analysis and contributed in writing the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
Summary statistics of the discovery GWAS are available in the GRASP resources database (https://grasp.nhlbi.nih.gov/FullResults.aspx). The French cohorts details are available in additional files.
Ethics approval and consent to participate
All participating studies were approved by their respective institutional review board/ethics committee and an informed consent was obtained from all participating individuals.
Funding
R.C was supported by a Ph.D. grant from the Region Ile de France (CORDDIM program).
The UK-ROI component was funded by Diabetes UK, the JDRF, the Health Research Board, Science Foundation Ireland, and Department for the Economy NI 15/IA/3152; this includes the Warren 3/UK GoKinD Study Group, which includes, Belfast: A. P. Maxwell, A. J. McKnight, D. A. Savage; Edinburgh: J. Walker; London: S. Thomas, G. C. Viberti; Manchester: A. J. M. Boulton; Newcastle: S. Marshall; Plymouth: A. G. Demaine, B. A. Millward; Swansea: S. C. Bain. SD was supported by a Ph.D. studentship from the Northern Ireland Department of Education and Learning.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding authors
Additional files
Additional file 1: Table S1.
Main design and sample characteristics of the discovery and replication studies.
Additional file 2: Figure S1.
Quantile-Quantile plot representation of the discovery meta-GWAS results.
Additional file 3: Figure S2.
Manhattan plot representation of the discovery meta-GWAS results.
Additional file 4: Figure S3.
Regional association plot at the MAP1B locus.
Additional file 5: Table S2.
Imputation quality of SNPs with association p-values < 1.0 × 10-5 in the discovery cohorts.
Additional file 6: Table S3.
Imputation quality of the established CAD associated SNPs in the discovery cohorts.
Additional file 7: Figure S4.
Regional association plot at the CKD18 locus.
Additional file 8: Figure S5.
Regional association plot at the FAM189A2 locus.
Additional file 9: Figure S6.
Regional association plot at the PKD1 locus.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
Cite this article
Charmet, R., Duffy, S., Keshavarzi, S. et al. Novel risk genes identified in a genome-wide association study for coronary artery disease in patients with type 1 diabetes. Cardiovasc Diabetol 17, 61 (2018). https://doi.org/10.1186/s12933-018-0705-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12933-018-0705-0