
Abstract
Oxygen is essentially required by most eukaryotic organisms as a scavenger to remove harmful electron and hydrogen ions or as a critical substrate to ensure the proper execution of enzymatic reactions. All nucleated cells can sense oxygen concentration and respond to reduced oxygen availability (hypoxia). When oxygen delivery is disrupted or reduced, the organisms will develop numerous adaptive mechanisms to facilitate cells survived in the hypoxic condition. Normally, such hypoxic response will cease when oxygen level is restored. However, the situation becomes complicated if hypoxic stress persists (chronic hypoxia) or cyclic normoxia-hypoxia phenomenon occurs (intermittent hypoxia). A series of chain reaction-like gene expression cascade, termed hypoxia-mediated gene regulatory network, will be initiated under such prolonged or intermittent hypoxic conditions and subsequently leads to alteration of cellular function and/or behaviors. As a result, irreversible processes occur that may cause physiological disorder or even pathological consequences. A growing body of evidence implicates that hypoxia plays critical roles in the pathogenesis of major causes of mortality including cancer, myocardial ischemia, metabolic diseases, and chronic heart and kidney diseases, and in reproductive diseases such as preeclampsia and endometriosis. This review article will summarize current understandings regarding the molecular mechanism of hypoxia in these common and important diseases.
Similar content being viewed by others
Introduction
In metazoan organisms, the oxygen delivery and cellular adaptation to oxygen deprivation are accelerated through the hypoxic signaling pathway in order to sustain oxygen homeostasis [130]. Lack of oxygen supply or an excessive oxygen consumption could result in insufficient oxygen levels for maintaining normal cellular function, a condition defined as hypoxia. Hypoxia may not be considered as the inequivalent to ambient oxygen concentration (21% oxygen) as many tissues physiologically function at levels equal to 5% oxygen or even as low as 1% oxygen [61]. Mostly, hypoxia is referred to the relatively low (generally < 2%) oxygen content compared to normal status in a given organ, tissue, or cell type. Hypoxia is a state of continuously lack of oxygen for a short (acute hypoxia, e.g., ischemia) or long (chronic hypoxia, e.g., chronic kidney disease, cancer) period of time. Therefore, a wider range of oxygen concentrations and feedback to acute stresses from seconds to days, even weeks to months, shall be put into consideration when referring the mechanisms of pathophysiological relevancies.
Hypoxia is usually considered to have pathological effects; however, it also involves in maintaining normal physiological functions. Taking human as an example, the central and peripheral chemoreceptors sense the reduction of oxygen tension and send signals to respiratory center in medulla and pons, where a series of processes are initiated to increase pulmonary ventilation and cardiac output to maintain normal functions of human body. Oxygen exchange occurs in the alveoli of lung with more than 95% of oxygen diffuses into the capillary vessels via the alveolar-capillary exchange system and then binds to hemoglobin. The oxygenated blood returns to left atrium through pulmonary vein. The heart then pumps out the oxygenated blood from left ventricle to its periphery to maintain the proper function of every single cell. In this minireview, we will not discuss the physiological effects of hypoxia but focus on pathological impacts of hypoxia in several key human diseases including cancer, cardiovascular diseases, chronic kidney diseases, metabolic diseases, preeclampsia, and endometriosis. Understanding the disease pathological process shall help us dissecting the molecular mechanisms of causing the disorders and designing better therapeutic regimens against them.
Molecular basis of hypoxia in regulating gene expression
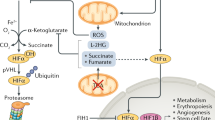
Oxygen is essentially required by most eukaryotic organisms as a scavenger to remove harmful electron and hydrogen ions generated as by-products of mitochondrial oxidative phosphorylation. At the cellular level, adaptation involves a switch of energy metabolism from oxidative phosphorylation to anaerobic glycolysis, which increases glucose uptake, and expression of stress proteins related to cell survival or death [22]. All nucleated cells can sense oxygen concentration and respond to reduced oxygen availability in one of two distinctive ways. Alterations of preexisting proteins (such as phosphorylation or changing redox state) primarily occur in response to acute hypoxia (within minutes) while alterations in gene expression principally occur in response to chronic hypoxia (lasting from minutes to hours or longer). The expression of hypoxia responsive genes is mainly regulated by hypoxia inducible factor (HIF)- or nuclear factor-κB (NF-κB)- dependent manners. There are three HIF-αs (HIF-1α, −2α, and -3α) identified thus far [128]. All three HIFs dimerize with constitutively expressed HIF-1β (also known as aryl hydrocarbon nuclear translocator, ARNT) to form a heterodimeric functional unit. HIF-1α is expressed in most, if not all, human tissues [165] while HIF-2α and HIF-3α are expressed in more restricted tissues and developing stages such as fetal lung or developing vascular endothelium [39, 43, 146]. In reflecting to their specific tissue expression patterns, HIF-1α appears to play a general role in transcriptional regulation of all cells in response to hypoxia whereas HIF-2α and HIF-3α play more limited or specialized roles in oxygen homeostasis. Under normoxia, HIF-αs protein are hydroxylated by prolyl-hydroxylases (PHDs) and factor inhibiting HIF (FIH). These two oxygen-dependent enzymes are activated under normoxia and suppress HIF-αs activity via distinct mechanisms. PHDs catalyze the proline hydroxylation of HIF-αs so the E3 ubiquitin ligase, von Hippel–Lindau (VHL) can bind to HIF-αs protein and promote the degradation via the ubiquitin proteasome degradation pathway (Fig. 1). FIH hydroxylates the asparagine residue in the C-terminal transactivation domain of HIF-αs prevents the recruitment of transcriptional coactivator CREB-binding protein (CBP) and its homolog, p300, and thus inhibits HIF-αs transcription activity. In addition, PHDs and FIH can inactivate NF-κB by direct hydroxylation of inhibitor of κB kinase (IKK) complex [159]. Under hypoxia, insufficient oxygen inhibits the activity of PHDs, thus prevents HIF-αs from VHL-dependent protein degradation. In addition, mitogen-activated protein kinases (MAPKs) phosphorylate HIF-αs, which also increases the stability of α subunit. The phosphorylated HIF-αs protein translocate to the nucleus and associate with HIF-1β, which forms a HIF-1α/β heteroduplex and binds to the hypoxia responsive element (HRE) of target genes. Suppression of FIH activity under hypoxia results in increasing CBP/p300 recruitment to enhance the transcription of HIF target genes [60, 130]. Besides, lack of oxygen molecules prevents hydroxylation of IKK. Nonhydroxylated IKK complex promotes the phosphorylation, ubiquitination, and degradation of inhibitor of NF-κB (IκB). Therefore, NF-κB is released and translocates to the nucleus to regulate the transcription of target genes [32].

Schematic diagram illustrates the regulation of HIF-1α and NF-κB under normoxic and hypoxic conditions
Hypoxia and human diseases
Cancer
Impaired oxygen delivery and consumption are typical features of hypoxia in tumor microenvironment. The rise of hypoxia first comes from the restriction of oxygen diffusion in avascular primary tumors along with the higher oxygen consumption due to hyperproliferation of cancer cells [19]. Cellular responses to hypoxia are essential for tumor progression in many aspects, such as cancer cell survival, proliferation, epithelial-to-mesenchymal transition (EMT), invasion, angiogenesis, drug resistance, and metastasis [127]. According to findings involved in cancer biology from the initial tumor formation to advanced cancer dissemination, it is clear that tumor hypoxia is not only a hallmark of tumor microenvironment but also plays crucial roles in molecular and cellular responses to drive cancer progression. At the molecular level, hypoxia stabilizes HIFs to help cells adapting to hypoxic stress via transactivating downstream genes [130]. HIF-1α and HIF-2α are clinically correlated with advanced stages and poor survival of cancer patients [127]. In addition, the HIF signaling is essential for promoting the metastatic ability of cancer cells [117], suggesting that HIF pathway plays a vital role in cancer biology [127]. Furthermore, other pathways such as the mammalian target of rapamycin (mTOR) and the unfolded protein response (UPR) act cooperatively with HIF to regulate cellular functions [168]. Hypoxia suppresses mTORC1 activity through multiple pathways. Prolonged hypoxia causes energy stress that activates AMP-activated protein kinase (AMPK), which induces the transcription of regulated in development and DNA damage responses 1 (REDD1). REDD1 suppresses mTOR activity through tuberous sclerosis complex (TSC)1/TSC2-mediated mTOR inactivation [20]. Hypoxia can also inhibit mTORC1 activity through BCL-2 interacting protein 3 (BNIP3) and the promyelocytic leukemia (PML) tumor suppressor [168]. The suppressed mTORC1 activity results in decreased EIF4E-binding protein 1 (4E-BP1) dephosphorylation and thus sequesters eukaryotic translation initiation factor 4E (EIF4E) from cap-dependent translation initiation. The hypoxic inhibition of translation initiation is also reported to act through enhanced association with EIF4E-transporter (4E-T). Hypoxia induces endoplasmic reticulum (ER) stress sensors PKR-like ER kinase (PERK), inositol-requiring protein 1 (IRE1), and activating transcription factor 6 (ATF6) to induce the UPR pathway. The PERK-mediated phosphorylation of EIF2α results in globally suppressed translation initiation, while the other two ER stress sensors induce transcription through ATF6 and IRE1-activated X-box binding protein 1 (XBP1). These pathways have been long known to orchestrate a network with HIF to regulate gene expression at different molecular levels [168]. The widespread regulation by hypoxia/HIF signaling explains the molecular basis of hypoxia biology in cancer, from the stress (hypoxia), regulators (HIFs), targets (functional proteins), to phenotypes. Herein, we summarize hypoxia-regulated pathophysiological processes that play critical roles in cancer development and progression (Fig. 2).

Hypoxia-regulated cancer progression. Hypoxia is a typical feature of tumor microenvironment, which contributes to initial tumorigenesis, induced angiogenesis, drug resistance, and cancer metastasis. The major upstream regulators (gray), functional downstream genes (blue), and resulting cellular consequences (yellow) under the control of hypoxia signaling are indicated
Angiogenesis
The physiological diffusion of oxygen in microenvironment soon becomes a limiting factor during tumor growth, which stimulates new blood vessel formation (angiogenesis) to provide essential oxygen and nutrients for further tumor outgrowth. Tumor angiogenesis is tightly regulated by multiple pro- and anti-angiogenic factors. HIFs have been identified as master enhancers for vascular endothelial cell migration and proliferation. As a transcription factor, stabilized HIF-1α/β binds to HRE of target genes and transactivates their expression [48]. Among these genes, vascular endothelial growth factor (VEGF), platelet-derived growth factor B (PDGF-B), fibroblast growth factor (FGF), plasminogen activator inhibitor-1 (PAI-1), matrix metalloproteinases (MMP-2 and MMP-9), interleukin 8 (IL-8), and angiopoietins (ANG-1 and ANG − 2) are known as pro-angiogenic factors playing crucial roles during tumor angiogenesis [82, 94]. Moreover, VEGF and PAI-1 has also been found to enhance tumor angiogenesis under the control of HIF-2α [119, 138]. Among these secretory factors, VEGF, PDGF-B, FGF, ANG-1, and ANG-2 bind to their specific receptors VEGFR2, PDGFRβ, FGFR, TIE-1 and TIE-2, respectively, on vascular endothelial cells to activate cell proliferation, migration, tube formation, and vascularization [57, 113, 114], while other factors (e.g., MMPs, PAI-1) participate in remodeling extracellular matrix (ECM) and are also involved in local invasion of cancer cells [111].
Survival advantages from adaptive cellular responses
Malignant tumors tend to exhibit enhanced anaerobic glycolysis as their energy source. This metabolic shift enables cancer cells adapting to microenvironmental stresses including hypoxia [98]. Under hypoxia, cancer cell adapt to the reduced available oxygen and nutrient through upregulation of glucose transporters (GLUT1 and GLUT3), carbonic anhydrases (CA9 and CA12), pyruvate dehydrogenase kinases (PDK1 and PDK3), lactate dehydrogenase A (LDHA), phosphoglycerate kinase 1 (PGK-1), and hexokinases (HK1 and HK2) to cooperatively modulate a metabolic shift from oxidative phosphorylation to anaerobic glycolysis [92, 129]. Epigenetically, histone lysine demethylase 3A (KDM3A) removes demethylated histone 3 lysine 9 (H3K9me2) from PGK1 promoter to enhance HIF-1α-dependent PGK1 transcription thus facilitate glycolysis under hypoxia. Under hypoxia, the HIF-1α-dependent histone demethylase KDM4B induction removes H3K9me3 from the promoters of hypoxia-inducible genes involved in cell survival [124]. While the nonadaptive precancer/cancer cells undergo cell death, the hypoxia-induced anti-apoptosis pathways help other cancer cells survive. These evolutionally survived cells tend to express reduced pro-apoptotic factors such as Bcl-2-associated X protein (Bax), Bcl-2 associated agonist of cell death (Bad), and BH3 interacting domain death agonist (Bid), and enhanced anti-apoptotic factors such as B-cell lymphoma 2 (Bcl-2) and B-cell lymphoma-extra large (Bcl-xL) [50, 131]. Consequently, both of the poly (ADP-ribose) polymerase (PARP) cleavage and caspase activity are suppressed under hypoxia [40, 65]. These adaptive responses, including the metabolic and pro-survival shifts, not only cooperatively maintain the survival of cancer cell, but may also continually facilitate tumorigenesis from initial tumor formation to secondary tumorigenesis after/under treatment.
Epithelial-mesenchymal transition
Hypoxia/HIF signaling also regulates cellular behaviors, including migration/invasion, intra−/extra-vasation, colonization and tumorigenesis at distant organs, to drive cancer metastasis. EMT is a common process which enables noninvasive (epithelial-like) cancer cells to invade and metastasize (mesenchymal-like). It is now clear that hypoxia enhances the metastatic ability through promoting EMT of cancer cells. Hypoxia/HIF signaling directly facilitates the EMT gene profile to induce the invasiveness of cancer cells [117] through recognition of HREs of EMT transcription factors zinc finger E-box-binding homeobox 1 (ZEB1), snail family transcriptional repressor 1 (SNAI1), and twist family bHLH transcription factor 1 (TWIST1) genes [154]. Alternatively, HIF modulates Notch [123], transforming growth factor beta (TGF-β) [29], integrin-linked kinase (ILK) [26], tyrosine kinase receptors (TKRs) [34], Hedgehog [75, 140], AXL receptor tyrosine kinase [116], lysyl oxidase (LOX) [162], and protein 3-phosphoinositide-dependent protein kinase 1 [110] to indirectly facilitate EMT. In addition, histone deacetylase 3 (HDAC3) interacts with WD repeat domain 5 (WDR5) to enhance deacetylation of H3K4Ac in the promoters of EMT regulators under hypoxia [174]. These genetic regulations drive the phenotypic shift from epithelial-like (noninvasive) to mesenchymal-like (invasive), which help cancer cell motility during the multistep metastasis process, such as migration/invasion and intra−/extra-vasation.
Cancer stemness and drug resistance
Cancer stem cells (CSCs) expressing stem cell markers (e.g., CD133, CD44) and transcription factors (e.g., OCT3/4, SOX2, KLF4, c-MYC) have been identified to exhibit undifferentiated (stem cell-like) and tumorigenic properties [12]. Hypoxic microenvironment also facilitates the maintenance of CSCs [53]. It is known that both HIF-1α and HIF-2α induction activate OCT4, c-MYCc, SOX2 and enrich the expression of CD133- and CD44-positive cancer cells, along with the enhanced self-renewal functions and tumorigenic potential [4], while several studies proposed that cancer stemness is predominately controlled by HIF-2α [30, 62]. In addition to maintaining the unlimited primary tumor growth, CSCs have slow growing rate (quiescent) thus are also relatively insensitive to chemotherapy targeting proliferative cancer cells [25]. Thus, it is also suggested that CSCs within tumors determine the therapeutic efficacy and cancer prognosis, as the reservoir CSCs result in resistant subpopulation after chemotherapy and become the cellular sources for continued cancer propagation and recurrence. Irrespective of the involvement of CSCs in cancer recurrence, hypoxia signaling also activates several drug resistant pathways to protect cancer cells [151]. The nature of hypoxic region with poor vascularization may partly limit the diffusion of circulating drugs. Furthermore, HIF-1α induces the expression of multidrug resistance (MDR) genes under hypoxia. Expression of MDR gene products, the drug efflux pump protein ABC transporters, enables cancer cells to pump out intracellular chemotherapeutic drugs, thus reduces the therapeutic effects and enhances drug resistance [151].
Exosome secretion and priming
Exosomes released to microenvironment have been known to participate in intercellular communication since these extracellular microvesicles containing functional nucleic acids and proteins [100] . Recent studies demonstrated the clinical significance of hypoxic exosome in cancer. First, the contents of exosome, including proteins and nucleic acids, have significant changes under hypoxia [132] . Many of the proteins and RNAs enriched in hypoxic exosomes are canonical downstream gene products of hypoxia, such as HIF-1α, MMPs, and LOX [2, 69]. It is also known that the exosome secretion is enhanced in hypoxic cells to regulate cancer metastasis through the upregulation of small guanosine triphosphatase RAB22A [161]. Exosomes secreted by hypoxic cancer cells containing high levels of oncogenic proteins to enhance EMT, stemness and invasiveness through degradation of E-cadherin and activation of β-catenin pathway [115]. The currently identified extracellular secretion and functions of hypoxic exosomes make them as a possible hypoxic biomarker and therapeutic target.
Hypoxia regulates miRNA biogenesis
Current knowledge based on the large-scale genomic sequencing projects illustrated that the protein-coding transcriptional output is less than 2% in human genome. For recent 20 years, the roles of non-coding RNAs, such as miRNAs, have been extensively studied and their roles in cancer progression have been well-documented [134]. Here, we will discuss the regulation of hypoxia on miRNA biogenesis. The maturation of miRNA is a multistep process involving several protein factors. It is now established that both the global level of miRNAs and the expression of miRNA biogenesis factors (Dicer, Drosha, TARPB2, and DCGR8) are downregulated under hypoxia [10] . The cytoplasmic biogenesis factor Dicer is suppressed by HIF-1α-mediated proteasomal degradation or H3K27me3 demethylases KDM6A/B-dependent epigenetic modification [70, 156]. Several miRNAs including miR-103/107, let-7, and miR-630 were also reported to target and suppress Dicer expression [96, 122, 147]. Moreover, activation of EGFR pathway under hypoxia phosphorylates AGO2 and abolishes its interaction with Dicer, thus represses miRNA maturation and activity [135]. These pathways consequently result in either global or specific miRNA downregulation to promote cancer progression. Notably, many of the genes regulated by miRNAs are also canonical hypoxia/HIF signaling downstream genes, such as ZEB1, GLUT1, and VEGF, which further suggests that the hypoxia-regulated miRNAs act post-transcriptionally and synergistically with canonical hypoxia pathway of transcriptional regulation.
Cardiovascular diseases
Cardiovascular diseases are the leading cause of mortality worldwide, representing 1 of every 3 deaths in 2018. Hypoxia is one of the common features in the pathophysiology of a variety of cardiovascular disorders [1]. Heart failure with reduced left ventricular systolic function after myocardial infarction causes the insufficient oxygen in the body. Moreover, heart failure with preserved ejection fraction in patients eventually leads to systemic and pulmonary hypertension. Pulmonary hypertension also associates with other hypoxic pulmonary diseases, including chronic obstructive pulmonary disease and obstructive sleep apnea syndrome, and promotes inflammation and atherosclerosis [95]. Atherosclerosis is a chronic inflammatory disease that can increase the risk of myocardial infarction and stroke. The thickness of arterial wall causes hypoxia in the intima, reduces the perfusion of the tissue, and further stimulates proatherosclerotic processes, like inflammation, lipid synthesis, and angiogenesis [139].
Roles of NF-κB
As mentioned above, hypoxia simultaneously activates HIF and NF-κB signaling pathways to regulate numerous biological processes (Fig. 1). It should be noted that the crosstalk of HIF-1α and NF-κB plays an important role in ischemic cardiovascular disease. The hypoxia-induced HIF-1 upregulates NF-κB, which reciprocally activates the transcription of HIF-1α [38]. This forms a positive feedback loop to worsen the disease. In addition, NF-κB also induces many other target genes such as inflammatory cytokines. The inflammatory response leads to smooth muscle cell activation, resulting in neointima formation and occlusive plaque. Importantly, elevated serum inflammatory marker, such as IL-6 and tumor necrosis factor-α (TNF-α), in patients are correlated with their prognosis in hypoxic cardiovascular disease [76, 103].
Roles of Hif-1α
The functional consequences of HIF-1α in cardiomyopathy had been investigated using transaortic constriction (TAC) murine models to mimic pressure-overload heart failure human disease. A 3-week TAC in the mice with cardiomyocyte-specific deletion of Hif1-α fails to induce Vegf expression and neoangiogenesis. As a result, acute heart failure occurs due to the lack of enough oxygen being delivered to the rapidly increasing cardiac muscle cells [125]. Another study using cardiomyocyte- and endothelial cell-specific Hif1-α knockout mice showed 1-week TAC causes severe heart failure phenotype. Moreover, the myocardial capillary density is decreased in these mice, resulting from markedly increased endothelial cell apoptosis [164]. These results implicate the proangiogenic and cardio-protective effect of Hif-1α. Conversely, Krishnan and colleagues demonstrated that, after TAC, the Hif1-α+/− mice have better cardiac function than wild-type mice. They also found Hif-1 promotes the expression of peroxisome proliferator-activated receptor γ (PPARγ), and activates lipid synthesis by engaging glycerolipid and fatty acid uptake genes, and, in turn, induces cell apoptosis [68]. These conflicting results suggest that Hif-1 mediates the complexity of adaptive responses. Hence, further research differentiating the roles of HIF-1 in cardiomyocyte is needed in order to acquire a clear picture.
Roles of miRNAs
In addition to the transcriptional regulation by HIF-1α and NF-κB, ischemic/hypoxia also modulates cardiofunction via miRNAs at the posttranscriptional level. Numerous miRNAs had been reported to be up- or down-regulated in patients with myocardial ischemia/reperfusion injury (Table 1). Among them, MiR-22, miR-134, miR-135a, miR-203, miR-144, miR-98, miR-18a, miR-210, miR-340-5p, miR-374a-5p, and miR-1192 exert protective effects in cardiovascular ischemic injury through downregulating their target genes [37, 41, 56, 77, 79, 112, 160, 163, 175, 183, 184]. On the other hand, a specific set of miRNAs has been linked to cardiac dysfunctions in varied cardiac injury models. For example, in intermittent hypoxia-induced myocardial damage, miR-146a-5p promotes cell death by targeting X-linked inhibitor of apoptosis protein [80] . MiR-327 reduces the expression of apoptosis repressor with caspase recruitment domain expression, and subsequently deteriorates myocardial ischemia/reperfusion injury [78]. MiR-429 accelerates ischemia/reperfusion injury by targeting mouse protein 25 and decreasing the protective effect of autophagy [189]. These data indicate that miRNAs do play important roles in regulating cardiovascular function after heart injury and imply they might be potential molecular targets for diagnosis or treatment of cardiovascular diseases. Indeed, miRNA-based therapies using modified oligonucleotides have been developed in treating different cardiovascular diseases. The critical role of miR-34a in cardiac ageing and function made it to be selected as a target for treating myocardial infarction [17, 144]. Targeting miR-34a by locked nucleic acid-modified anti-miR-34a attenuates adverse cardiac remodeling in myocardial infarction- or TAC-induced cardiac injury [15]. Similarly, anti-miR-92a and anti-miR-132 therapies are also effectively resistant to hypoxia-induced cardiac injury [13, 155]. Most recently, circulating extracellular vesicles-containing miRNAs attract great attention as promising biomarkers for early detection of cardiovascular diseases [3]. In lights of these recent advances, future studies focusing on elucidating key miRNA diagnostic biomarkers that can be targeted using mimetics or inhibitors to alleviate ischemic cardiovascular diseases are warranted.
Metabolic diseases
Adipocytes and metabolic diseases
Adipose tissue is one of the least metabolically dynamic structure for lipid turnover and can also communicate with other tissues through secretion of adipocyte-derived hormones, growth factors, inflammatory cytokines, leptin, adiponectin, signaling lipids, fatty acids, and miRNAs packaged in exosomes [145, 188]. These adipocyte-secreted factors (adipokines) and fatty acids regulate systemic metabolism and play important roles in the development of metabolic diseases, such as metabolic syndrome, ischemic heart disease, stroke, obesity, type 2 diabetes mellitus, and cancer [7, 148]. White adipocytes in white adipose tissue, which is the most important lipid buffering organ, can either accumulate fatty acids in lipid droplets or supply fatty acids for other tissues determined by the balance between fatty acid synthesis (lipogenesis) and lipid breakdown (lipolysis and fatty acid β-oxidation) [63]. Conversely, brown adipocytes in brown adipose tissue and beige adipocytes in beige/brite adipose tissue more frequently produce heat through fatty acid β-oxidation in mitochondria (thermogenesis) [31]. Owing to their contributions to whole-body lipid homeostasis, both white and brown adipose tissues are considered primary targets for the treatment of obesity and type 2 diabetes mellitus [23, 28]. In addition, both white and brown adipose tissues are able to elaborate adipokines to control nutritional intake, sensitivity to insulin, and inflammatory processes in other tissues [28, 188]. Therefore, lipid metabolism and adipokines secretion in adipose tissue exert an impact on whole-body metabolism and are important for the progress of metabolic diseases.
Effects of hypoxia on adipocytes
Hypoxia is one of the mechanisms responsible for the development of metabolic changes and pro-inflammatory situations of white adipose tissue [150]. In obesity, due to the enlargement of adipocytes and increased distance from the vasculature, hypoxia occurs within the expanding white adipose tissue in ob/ob and dietary obese mice. Accordingly, white adipose tissues from obese people are subjected to intra-adipose tissue hypoxia and characterized by increased HIF-1α expression [67]. In vitro experiments show that HIF-1α and HIF-2α inhibit insulin signaling in both human and murine white adipocytes [120]. Moreover, chronic hypoxia has been suggested to be part of the pathogenic pathways leading to adipose tissue dysfunction [166, 178]. Hypoxia triggers reactive oxygen species (ROS) production, ER stress, inflammatory responses, angiogenesis, and adipocyte death [66, 150, 176, 185]. HIF also regulates the expression of various adipokine genes, such as increasing leptin, visfatin, apelin, TNF-α, IL-1, IL-6, VEGF, MMP2, MMP9, angiopoietin-like protein-4, macrophage migration inhibitory factor, and PAI-1 expression, while downregulating adiponectin and PPARγ expression in adipocytes [52, 150, 176].
Hypoxia alters several key metabolic processes including glucose uptake, glycolysis, oxidative metabolism, lipolysis, and lipogenesis in adipocytes. Hypoxia stimulates glucose uptake in adipocytes through HIF-1α-upregulated GLUT expression, and increases anaerobic glycolysis and lactate production by induction of glycolytic enzymes [121, 167]. Hypoxia also induces rearrangements of lipid metabolism in adipocytes. In response to hypoxia, extracellular fatty acid uptake is reduced by inhibition of fatty acid transporters (FATP1 and CD36) and transcription factors (PPARγ and C/EBPα) while lipolysis is increased in 3 T3-L1 adipocytes [179]. Another study shows that hypoxia inhibits lipogenesis by reducing PPARγ and fatty acid synthase (FAS), and induces basal lipolysis in visceral and subcutaneous human adipocytes [109]. In addition, hypoxia inhibits adipogenesis and differentiation in 3 T3-L1 adipocytes via HIF-1α-dependent upregulation of differentiated embryo-chondrocyte expressed gene 1 (DEC1/Stra13) and subsequent repression of PPARγ2 expression [182]. HIF-1α also suppresses expression of genes involved in fatty acid β-oxidation by repression of sirtuin 2-mediated deacetylation of PPARγ coactivator 1-α (PGC-1α) in white adipocytes [67]. Besides, studies indicate that obesity induces hypoxia in brown adipose tissue and causes the loss of its thermogenic capacity [149]. Other studies show that hypoxia is a trigger for brown adipose tissue whitening with diminished β-adrenergic signaling, enlarged lipid droplets, and loss of mitochondria in the cells [137]. Increased HIF-1α and suppressed uncoupling protein 1 (UCP1) expression with lower fatty acid β-oxidation are observed in hypoxic brown adipose tissue [136]. Therefore, hypoxia alters lipid metabolism in adipocytes mainly by inhibiting lipogenesis and decreasing fatty acid β-oxidation.
Effects of hypoxia on other cell types
The effects of hypoxia on lipid metabolism are also studied in other cell types related to metabolic diseases. As fatty acid β-oxidation takes place inside mitochondria and requires oxygen, fatty acid metabolism has to be modified other than energy production under hypoxia. Furthermore, the major source of cytoplasmic acetyl-CoA from glucose converted citrate is prohibited under hypoxia due to the inhibition of the TCA cycle, so alternative sources of fatty acid precursors have to be exploited. Uptake of extracellular fatty acids for triacylglycerol synthesis is promoted by HIF-1α-induced PPARγ in cardiomyocytes under hypoxia [68]. Extracellular fatty acid influx and lipid droplet accumulation are enhanced via HIF-1α-mediated induction of fatty acid binding protein 3 and 7 (FABP3 and FABP7), while de novo lipogenesis is repressed in glioblastoma and breast cancer cells under hypoxia [14]. To maintain certain level of lipogenesis under hypoxia, production of fatty acid precursors, citrate and acetyl-CoA, are supported through reductive glutamine metabolism in several cancer cells [46, 105] and brown adipocytes [180]. HIF-induced isocitrate dehydrogenase 1 and 2 (IDH1 and IDH2) contribute to the preservation of citrate levels via conversion of α-ketoglutarate to isocitrate and its subsequent reductive carboxylation to produce citrate from glutamine under hypoxia [101]. Adequate fatty acid synthesis is further supported by HIF-1-dependent activation of sterol regulatory element-binding protein 1 (SREBP1), which in turn upregulates the expression of FAS in breast cancer cells [45]. Hypoxia also induces lipid droplet accumulation by upregulating two enzymes of the triacylglycerol biosynthesis pathway, acylglycerol-3-phosphate acyltransferase 2 (AGPAT2) and lipin-1 in different types of cancer cells [106, 152]. Furthermore, hypoxia-induced lipid droplet accumulation is accompanied by the inhibition of fatty acid β-oxidation though HIF-1 and HIF-2-dependent downregulation of PGC-1α and carnitine palmitoyltransferase 1A (CPT1A) in both hepatoma and renal cell carcinoma cells [35, 90]. Therefore, hypoxia may result in different adaptation of lipid metabolism depending on the cell types.
Effects of hypoxia on animal models of obesity
Since hypoxia is shown to inhibit fatty acid β-oxidation as a promoter of obesity and inhibit lipogenesis as a suppressor of obesity, several animal studies based on the overexpression or inhibition of HIFs in adipocytes suggest that HIF activation either promotes or inhibits metabolic diseases. As a promoter of obesity, mice overexpressing Hif-1α in adipocytes have elevated obesity and insulin resistance associated with increased inflammation and fibrosis [49, 59]. Adipocyte specific Hif-1α or Hif-1β knockout, or inhibition of Hif-1α by inhibitors decrease obesity and insulin resistance in mice fed with high-fat diet [58, 74, 142]. In agreement, adipocyte-specific Phd2 ablation enhances adiposity in mice under normal chow diet (low-fat) with lower expression of adipose triglyceride lipase (ATGL) and suppresses lipolysis in white adipocytes [102]. These obesity-promoting effects can be correlated with the capacity of Hif-1 to downregulate fatty acid β-oxidation in white and brown adipose tissue [67, 136]. On the other hand, a number of studies have shown that Hif activation decreases obesity and Hif inhibition increases obesity indicating hypoxia as a suppressor of obesity. Accordingly, transgenic mice overexpressing an adipose tissue-specific dominant negative Hif-1α mutant developed severe obesity, insulin resistance, and accumulated enlarged lipid droplets in brown adipose tissue with decreased mitochondrial biogenesis after high-fat diet [187]. Another group shows that transgenic mice with adipose tissue-specific knockout of Phd2 (for constitutive expression of Hif) are resistant to high-fat diet-induced obesity with fewer lipid droplets in white adipose tissue and increased Ucp1 expression in brown adipose tissue [97], while adipocyte-specific knockout of Phd2 induces adiposity in mice under chow diet (low-fat) [102]. Similarly, phenotypes are also observed in mice globally lacking Fih, which are resistant to high-fat diet-induced weight gain and hepatic steatosis [186]. Taken together, these studies suggest that Hif-1α may stimulate the thermogenic functions of brown adipose tissue to conquer high-fat diet-induced obesity, which is contradictory with other findings that Hif-1 suppressed expression of genes involved in fatty acid β-oxidation in white and brown adipose tissue leading to obesity [67, 136].
In summary, studies investigating the functions of HIFs in adipocytes and other metabolic diseases revealed the conflicting results due to different experimental conditions (Fig. 3). The roles of HIFs in these studies were discovered primarily through the analysis of conditional Hif-knockout mice or through some pharmacological HIF inhibitors. These inconsistent consequences might be also due to the complexity of metabolic regulation with the complicated roles of HIFs that extend further than lipid metabolism. Therefore, the actual functions of HIFs in adipocytes and related metabolic diseases must be carefully interpreted relative to different physiological conditions.

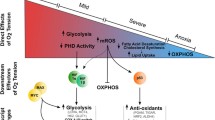
Hypoxia-regulated lipid metabolism related to obesity. Hypoxia is shown as a promoter or a suppressor of obesity by regulating lipid metabolism. The major changes (increases shown in red; decreases shown in green) involved in fatty acid β-oxidation, extracellular fatty acid influx, lipolysis, lipogenesis and lipid droplet accumulation under hypoxia for its promoting obesity or anti-obesity effects are summarized
Kidney diseases
Acute kidney injury (AKI) is notorious for its correlation with the development of chronic kidney disease (CKD). AKI is a rapid failure of kidney function, usually caused by decreased blood flow, toxic exposure, and ureteral obstruction. As the injuries are removed, renal cells undergo repair process. However, some of the AKI patients fail to fully recover. Instead, CKD starts to develop, which is characterized by the progressive loss of the kidney function [27]. Events include tubular atrophy, vascular rarefaction, and hypoxia, which lead to renal fibrosis and functional loss of kidney. Ultimately, CKD will develop into the end-stage renal disease (ESRD). Today, the only treatments for ESRD are dialysis and kidney transplantation [91]. Kidney fibrosis, including glomerulosclerosis and tubulointerstitial fibrosis, a phenomenon of excessive ECM deposit and accumulation, has been recognized as the hallmark of CKD and the major pathway leading to ESRD [107]. Fibrosis is the terminal pathway involved in the continuous progression of CKD and it is a consequence of failed recovery after kidney damage. When injuries happen, cells in kidney including fibroblasts, tubular epithelial cells, endothelial cells, pericytes, lymphocytes, and macrophages, undergo wound healing programming in an attempt to repair tissues. Sometimes, the damage is too severe for cells to overcome. Subsequently, they become victims and even act to fuel the fibrogenesis progressively [18]. As the diverse origin of cells involved in kidney fibrosis, it is complicated to figure out a comprehensive therapy for treating CKD patients. Today, there is still no effective ways to prevent CKD from getting worse. Several publications have pointed out that hinder renal fibrosis from getting worse is a potential way to prevent AKI-CKD transition and delay ESRD development [11, 16].
Hypoxia in kidney fibrosis
Tubular injury may lead to renal microvascular loss, which restricts downstream blood flow from glomerular capillaries and contributes to the development of renal hypoxia. Loss of microvasculature, reduced oxygen dispersion, and metabolic abnormality of cells in the kidney are the main causes of the hypoxic state. The initiation of hypoxia is one of the main causes of AKI, which can increase levels of HIF-1α, followed by the induction of TGF-β signaling. During the process of kidney fibrosis, hypoxia and TGF-β1 signaling are excessively upregulated. AKI can be gradually developed into CKD if TGF-β signaling remains hyperactivated for a period of time. Thus, hyperactivation of TGF-β signaling is responsible for the renal fibrosis and can also be identified as the hallmark of CKD. Hypoxia is common at the beginning of kidney fibrosis, which promotes HIF-1α expression that contributes to both a result and a cause of renal fibrosis. Hyperactivation of myofibroblasts is responsible for kidney fibrosis as they continuously produce collagens, fibronectin, and vimentin, which contribute to tubulointerstitial fibrosis and local tissue hypoxia [72, 143]. Prolonged hypoxia further promotes renal fibrosis by increasing the synthesis of type I and type IV collagen and inhibiting the expression of MMP-1 in human renal fibroblasts [108]. By using kidney-specific Vhl−/− mice, which have a stable expression of Hif-1α in kidney, researchers found that these transgenic mice exhibit more severe interstitial fibrosis after conduct 5/6 nephrectomy [64]. In contrast, intraperitoneal injection of HIF-1α inhibitor YC-1 protects unilateral ureter obstruction mice from kidney fibrosis development [126]. These data indicate that HIF-1α has a pivotal role in mediating kidney fibrosis (Fig. 4). However, the underlying mechanisms by which HIF-1 accelerates kidney fibrosis remain unclear.

Schematic diagram of the hypoxic process linked to kidney diseases. During the development of CKD, vascular endothelial cells died and causing atrophy of microvessels. This will cause local hypoxia between tissues and cause inflammatory reactions. Hypoxia induces the expression of TGF-β, which leads to fibroblast transformation into myofibroblast, increases ECM production, and causes fibrosis. Renal tubular epithelial cells may encounter cell cycle arrest, apoptosis, autophagy, and finally shrink during renal fibrosis
Preeclampsia
Preeclampsia is a severe gestational complication featured by new onset of high blood pressure after 20 weeks of gestation along with signs of proteinuria, abnormally high serum creatinine, or damaged liver function [36]. Its complications remain a major cause for morbidity and mortality in pregnant women and fetuses. Defective trophoblast invasion into the decidualized endometrium leads to poor transformation of uterine spiral arteries from high to low resistant vessels [42]. This deficient vessel remodeling causes a sustained hypoxic environment implicating the development of preeclampsia. Thus, placental ischemia and hypoxia is a major cause of preeclampsia.
Primary cilium and preeclampsia
Defective trophoblast invasion leads to placental hypoxia. The trophoblast invasion is regulated by endocrine gland-derived vascular endothelial growth factor (EG-VEGF, also known as prokineticin 1) [51]. EG-VEGF induces the expression of MMPs by triggering ERK signaling cascade for proper trophoblast invasion [157]. The receptor of EG-VEGF localizes to the primary cilium, a cellular protrusion atop from the centrosome (Fig. 5). When EG-VEGF binds to its receptor, ERK signaling is initiated from the base of the primary cilium, and then transduced throughout the cytoplasm. Inhibition of EG-VEGF signaling or disruption of primary cilia alleviates trophoblast invasion in vitro [158]. More importantly, these phenotypes are also observed in pregnant women who suffered from preeclampsia. These data suggest the important roles of EG-VEGF and primary cilia in preventing placental hypoxia.

Potential role of hypoxia in developing preeclampsia. Binding of EG-VEGF to its receptor on the primary cilium activates ERK signaling at the basal body for proper placentation. Under hypoxic condition, however, HIF-1α translocates to the base of cilia and induces cilia deacetylation, thus leading to ciliary resorption. The hypoxia-induced ciliary defects contribute to the development of preeclampsia
Primary cilium functions as a sensory hub for transducing environmental chemo- and/or mechano-signaling into the cells for proper development and differentiation. Growing body of evidence supports the important role of primary cilium in maintaining embryo development. Interestingly, hypoxia suppresses primary cilium formation [71], and elevated levels of HIF-1α in the human trophoblasts have been linked to the development of preeclampsia [5], suggesting placental hypoxia-activated HIF-1α plays a key role in the pathogenesis of preeclampsia. It has been shown that HIF-1α translocates to the base of primary cilium for the resorption of primary cilia in the nutrient deprivation model, suggesting its non-genomic function in regulating primary cilium formation (Fig. 5).
HIF-1α in ciliogenesis
Despite the role of hypoxia in preeclampsia has been demonstrated for long, the underlying molecular mechanism remains unclear. Trophoblast invasion is triggered by the binding of EG-VEGF to its congenital receptor on the primary cilium, thus inducing downstream ERK signaling for MMPs expression. Disruption of primary cilium inhibits trophoblast invasion even in the presence of EG-VEGF, supporting the importance of cilia-mediated EG-VEGF signaling [8]. Besides, fewer primary cilia are observed in the placenta tissues of pregnant women who suffered from preeclampsia, suggesting loss of primary cilia plays an important role in developing preeclampsia. Preeclamptic placenta is a hypoxic microenvironment, and HIF-1α is highly expressed in syncytiotrophoblasts of preeclamptic placenta [118]. Thus, HIF-1α might facilitate the resorption of primary cilia and disrupting the EG-VEGF signaling. Interestingly, this hypothesis is further confirmed by the effect of aspirin on preeclampsia. Aspirin has been used for treating preeclampsia for decades in clinic. Recent studies show that aspirin induces primary cilia and suppresses the expression of soluble fms-like tyrosine kinase 1 (sFLT1), a known marker of preeclampsia, thus promoting trophoblast invasion [81, 141]. A recent study analyzing a cohort of 23, 604 women who had information on placental pathology and aspirin intake during pregnancy revealed that aspirin use reduced risks of having hypoxia-related placental pathology such as thrombus, infarct, fibrin deposition, hydatid, cyst, and calcification [177]. These data provide the potential molecular mechanism by which aspirin prevents preeclampsia maybe via maintaining primary cilia. However, the precise role of HIF-1α in regulating placental hypoxic ciliopathy still needs to be further investigated.
Endometriosis
Endometriosis is one of the most common gynecological diseases that reduces life quality and fertility of patients worldwide. It is characterized by the presence of endometrial tissue outside the uterine cavity and the incidence rate of endometriosis is around 8 to 15% in women of the reproductive age [47]. The etiology of endometriosis remains unknown, but a recent report suggests that hypoxia is the driving force of endometriosis [170]. The notion is based on a series of investigations that provided crucial evidence to support the role of hypoxia during the development of endometriosis. Some key findings are summarized below (Fig. 6).

Impacts of hypoxia on endometriosis pathogenesis. Shed-off endometrial tissues will immediately suffer hypoxic stress when it retrogrades into the peritoneal cavity during menstruation. Hypoxia regulates numerous downstream target genes involved in different cellular processes including cell survival, metabolism, angiogenesis, E2 and PGE2 production, and cell adhesion to help the development of endometriosis
Hypoxia and cell adhesion
Endometriosis is initiated by retrograde menstruation. The retrograded endometrial tissues need to land on the surface of organs in the peritoneal cavity and implant in order to survive ectopically. Two challenges that the retrograded cells have to face are hypoxic stress and adhesive ability. Recently, two studies reported that hypoxia enhances cell adhesive ability of the endometrial stromal cells by inducing the expression of cell adhesion molecules such as integrin α5, αV, β3, and β5 via TGF-β1/Smad signaling and anthrax toxin receptor 2 (ANTXR2) via HIF-1α-dependent manner [84, 87]. Furthermore, hypoxia-induced ANTXR2 expression is mediated by downregulation of EZH2 causing epigenetic change in ANTXR2 locus [84]. Treatment with inhibitors of TGF β1 receptor and ANTXR2 significantly attenuates hypoxia-induced cell adhesion in normal endometrial stromal cells [84, 87]. Furthermore, ANTXR2 inhibitor can prevent and reduce endometriotic lesion formation in the mouse model of endometriosis [84], revealing its therapeutic potential for endometriosis. Taken together, it is indicated that hypoxia can promote the development of endometriosis via increasing cell adhesive ability.
Hypoxia and hormone production
Previous studies have revealed that both estrogen (E2) and prostaglandin E2 (PGE2) are crucial factors for the development of endometriosis [21, 172] and the enzymes control the rate limiting steps of E2 (aromatase and steroidogenic acute regulatory protein, StAR) and PGE2 (cyclooxygenase-2, COX-2) biogenesis are aberrantly expressed in endometriotic stromal cells [9, 55, 153, 173]. It was later revealed that hypoxia upregulates both StAR and COX-2 expression. Hypoxia represses dual-specificity phosphatase-2 (DUSP2) expression and contributes to increase COX-2 expression via activation of ERK and p38 signaling pathways [171]. Furthermore, hypoxia also inhibits chicken ovalbumin upstream promoter-transcription factor II (COUP-TFII) to de-repress COX-2 expression in endometriotic stromal cells [85]. Finally, recent study has identified that hypoxia promotes YAP1 activation, which leads to StAR and COX-2 overexpression [83]. The same study also reported that inhibition of YAP1 by its inhibitor, verteporfin, not only decreases E2 and PGE2 production but also causes the regression of endometriotic lesion in the mouse model of endometriosis [83]. These data indicate that retrograded endometrial tissues can produce E2 and PGE2 via the assistance of hypoxia to support their growth and development.
Hypoxia and angiogenesis
To sustain the growth of endometriotic lesion in the hostile peritoneal cavity, new blood vessels must be established to provide oxygen and nutrient. Heterograft animal model of endometriosis by implanting human eutopic endometrium into severe combined immunodeficiency mice has identified that hypoxia pretreatment for eutopic endometrium significantly increases the level of VEGF to promote cell proliferation and angiogenesis in vivo [93]. Critically, numerous studies have demonstrated the effects of hypoxia on angiogenesis via different mechanisms in endometriosis. Among them, leptin and VEGF-A are two well-known angiogenic factors upregulated by hypoxic treatment in normal endometrial stromal cells [133, 169]. In addition, hypoxia-induced miR-20a causes prolonged activation of ERK singling results in increasing several angiogenic factor including CYR61 and osteopontin [86]. Furthermore, hypoxia can increase angiogenin and IL-8 expression by downregulation of COUP-TFII and DUSP2 expression, respectively [44, 54]. More importantly, IL-8 receptor inhibitor, reparixin, and YAP1 inhibitor, veterporfin, inhibit angiogenesis and the growth of endometriotic lesion in the animal model of endometriosis [54, 83]. Taken together, these findings reveal the critical role of hypoxia-induced angiogenesis during the development of endometriosis.
Hypoxia and autophagy
Peritoneal cavity is an unfavorable microenvironment for the retrograded endometrial tissues due to lack of blood vessel to supply oxygen and nutrients. The study has reported that the higher level of oxidative stress-induced DNA damage is observed in endometriotic specimen compared to the normal endometrial tissues [33]. Furthermore, primary endometriotic stromal cells produce more ROS than normal endometrial stromal cells [24]. Therefore, how endometriotic cells survive under this stressful condition is an intriguing question. Previously, autophagy, a cellular mechanism to remove or recycle unnecessary or dysfunctional components to produce energy, has been thought to play crucial roles in protecting cells from oxidative stress-induced cell apoptosis [99, 104]. Indeed, autophagy is reported to be upregulated in the ovarian endometriosis [6, 89] and blocking hypoxia-induced autophagy enhances apoptosis of endometrial stromal cells [88]. Recently, it is further demonstrated that hypoxia-induced long non-coding RNA MALAT1 (lncRNA-MALAT1) is involved in autophagy to protect cells from apoptosis [88]. However, the underlying mechanism that how lncRNA-MALAT1 regulates hypoxia-induced autophagy is still unclear. Besides autophagy, the change of metabolic phenotype in endometriotic lesions may also favor the development of endometriosis. For example, higher levels of glycolysis-related genes such as pyruvate dehydrogenase kinase 1 and lactate dehydrogenase A have been reported in endometriotic specimens and stromal cells, respectively [73, 181]. Furthermore, hypoxia-upregulated pyruvate dehydrogenase kinase 1 has prevented cell death induced by H2O2 or low nutrient treatment [73]. In summary, hypoxia-induced autophagy and change of cell metabolism may help retrograded endometrial tissues to adapt to the hostile microenvironment and favor the development of endometriosis.
Conclusion and perspective
The mechanisms of cellular response to hypoxia contribute to stress-induced pathophysiological outcomes. Previous studies established the fundamental concept of hypoxia biology, while recent advances in molecular and cellular biology such as non-coding RNA and microvesicle accelerate us to demonstrate a more complete and complex view of the regulatory network under hypoxia. Notably, most of these new findings were found to be closely integrated in the canonical hypoxia pathway, suggesting a fine-tuned cellular machinery with multiple pathways cooperatively, synergistically, or mutual exclusively work together to modulate the transduction of hypoxia signaling. Identification of these new sensors, messengers, and functional modulators not only advances our knowledge of hypoxia biology but also provides insights into the development of potential diagnostic and therapeutic approaches.
Availability of data and materials
Not applicable.
Abbreviations
- 4E-BP1:
-
EIF4E-binding protein 1
- 4E-T:
-
EIF4E-transporter
- Act1:
-
Activator 1
- AGPAT2:
-
Acylglycerol-3-phosphate acyltransferase 2
- AKI:
-
Acute kidney injury
- AMPK:
-
AMP-activated protein kinase
- ANG:
-
Angiopoietin
- ANTX2:
-
Anthrax toxin receptor 2
- ARNT:
-
Aryl hydrocarbon nuclear translocator
- ATF6:
-
Activating transcription factor 6
- ATGL:
-
Adipose triglyceride lipase
- Bcl-2:
-
B-cell lymphoma 2
- Bad:
-
Bcl-2 associated agonist of cell death
- Bax:
-
Bcl-2-associated X protein
- Bcl-xL:
-
B-cell lymphoma-extra large
- BDNF:
-
Brain derived neurotrophic factor
- Bid:
-
BH3 interacting domain death agonist
- BNIP3:
-
Bcl-2 interacting protein 3
- C/EBP:
-
CCAAT/enhancer binding protein
- CA:
-
Carbonic anhydrase
- CASP3:
-
Caspase-3
- CBP:
-
CREB-binding protein
- CD36:
-
CD36 molecule
- CKD:
-
Chronic kidney disease
- COUP-TFII:
-
Chicken ovalbumin upstream promoter-transcription factor II
- COX-2:
-
Cyclooxygenase-2
- CPT1A:
-
Carnitine palmitoyltransferase 1A
- CRD:
-
Apoptosis repressor with caspase recruitment domain
- CSC:
-
Cancer stem cell
- CYR61:
-
Cysteine rich angiogenic inducer 61
- DAPK1:
-
Death-associated protein kinase 1
- DEC1:
-
Differentiated embryo-chondrocyte expressed gene 1
- DUSP2:
-
Dual-specificity phosphatase-2
- E2 :
-
Estrogen
- ECM:
-
Extracellular matrix
- EFNA3:
-
Ephrin A3
- EGFR:
-
Epidermal growth factor receptor
- EG-VEGF:
-
Endocrine gland-derived vascular endothelial growth factor
- EIF4E:
-
Eukaryotic translation initiation factor 4E
- EMT:
-
Epithelial-to-mesenchymal transition
- ER:
-
Endoplasmic reticulum
- ERK:
-
Extracellular signal-regulated kinase
- ESRD:
-
End-stage renal disease
- EZH2:
-
Enhancer of zeste 2 polycomb repressive complex 2 subunit
- FABP:
-
Fatty acid binding protein
- FAS:
-
Fatty acid synthase
- FATP1:
-
Fatty acid transport protein 1
- FGF:
-
Fibroblast growth factor
- FIH:
-
Factor inhibiting HIF
- FOXO1:
-
Forkhead box protein O1
- GLUT:
-
Glucose transporter
- H3K4Ac:
-
Histone 3 lysine 4
- HDAC3:
-
Histone deacetylase 3
- HIF:
-
Hypoxia inducible factor
- HK:
-
Hexokinase
- HRE:
-
Hypoxia responsive element
- IDH:
-
Isocitrate dehydrogenase
- IKK:
-
Inhibitor of κB kinase
- IL-8:
-
Interleukin 8
- ILK:
-
Integrin-linked kinase
- IRE1:
-
Inositol-requiring protein 1
- IκB:
-
Inhibitor of NF-κB
- KDM:
-
Histone lysine demethylase
- LDHA:
-
Lactate dehydrogenase A
- lncRNA-MALAT1:
-
Long non-coding RNA MALAT1
- LOX:
-
Lysyl oxidase
- MAP K6:
-
Mitogen-activated protein kinase 6
- MMP:
-
Matrix metalloproteinases
- MO25:
-
Mouse protein 25
- mTOR:
-
Mammalian target of rapamycin
- NF-κB:
-
Nuclear factor-κB
- NOS3:
-
Nitric oxide synthase 3
- PAI-1:
-
Plasminogen activator inhibitor-1
- PARP:
-
Poly (ADP-ribose) polymerase
- PDCD4:
-
Programmed cell death 4
- PDGF-B:
-
Platelet-derived growth factor B
- PDK:
-
Pyruvate dehydrogenase kinase
- PERK:
-
PKR-like ER kinase
- PGC-1α:
-
PPARγ coactivator 1-α
- PGE2:
-
Prostaglandin E2
- PGK-1:
-
Phosphoglycerate kinase 1
- PHDs:
-
Prolyl-hydroxylases
- PPARγ:
-
Peroxisome proliferator-activated receptor γ
- PTP1B:
-
Protein tyrosine phosphatase 1B
- REDD1:
-
Regulated in development and DNA damage responses 1
- RHEB:
-
Ras homolog enriched in brain
- ROS:
-
Reactive oxygen species
- sFLT1:
-
Soluble Fms-Like tyrosine kinase 1
- SIRT1:
-
Sirtuin 1
- SNAI1:
-
Snail family transcriptional repressor 1
- Spry1:
-
Sprouty 1
- SREBP1:
-
Sterol regulatory element-binding protein 1
- StAR:
-
Steroidogenic acute regulatory protein
- TAC:
-
Transaortic constriction
- TGF-β:
-
Transforming growth factor beta
- TKRs:
-
Tyrosine kinase receptors
- TNF-α:
-
Tumor necrosis factor-α
- TWIST1:
-
Twist family bHLH transcription factor 1
- UCP1:
-
Uncoupling protein 1
- UPR:
-
Unfolded protein response
- VEGF:
-
Vascular endothelial growth factor
- VEGF-A:
-
Vascular endothelial growth factor A
- VHL:
-
E3 ubiquitin ligase von Hippel–Lindau
- WDR5:
-
WD repeat domain 5
- XBP1:
-
X-box binding protein 1
- XIAP:
-
X-linked inhibitor of apoptosis protein
- YAP1:
-
Yes Associated Protein 1
- ZEB1:
-
Zinc finger E-box-binding homeobox 1
References
Abe H, Semba H, Takeda N. The roles of hypoxia signaling in the pathogenesis of cardiovascular diseases. J Atheroscler Thromb. 2017;24(9):884–94.
Aga M, Bentz GL, Raffa S, Torrisi MR, Kondo S, Wakisaka N, Yoshizaki T, Pagano JS, Shackelford J. Exosomal HIF1α supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene. 2014;33(37):4613–22.
Aghabozorgi AS, Ahangari N, Eftekhaari TE, Torbati PN, Bahiraee A, Ebrahimi R, Pasdar A. Circulating exosomal miRNAs in cardiovascular disease pathogenesis: new emerging hopes. J Cell Physiol. 2019;234(12):21796–809.
Al TW, Dale TP, Al-Jumaily RMK, Forsyth NR. Hypoxia-modified cancer cell metabolism. Front Cell Dev Biol. 2019;7:4.
Albers RE, Kaufman MR, Natale BV, Keoni C, Kulkarni-Datar K, Min S, Williams CR, Natale DRC, Brown TL. Trophoblast-specific expression of Hif-1α results in preeclampsia-like symptoms and fetal growth restriction. Sci Rep. 2019;9(1):2742.
Allavena G, Carrarelli P, Del Bello B, Luisi S, Petraglia F, Maellaro E. Autophagy is upregulated in ovarian endometriosis: a possible interplay with p53 and heme oxygenase-1. Fertil Steril. 2015;103(5):1244–51.
Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 2008;34(1):2–11.
Anvarian Z, Mykytyn K, Mukhopadhyay S, Pedersen LB, Christensen ST. Cellular signalling by primary cilia in development, organ function and disease. Nat Rev Nephrol. 2019;15(4):199–219.
Attar E, Bulun SE. Aromatase and other steroidogenic genes in endometriosis: translational aspects. Hum Reprod Update. 2006;12(1):49–56.
Bandara V, Michael MZ, Gleadle JM. Hypoxia represses microRNA biogenesis proteins in breast cancer cells. BMC Cancer. 2014;14:533.
Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, Kellum JA, Ronco C, Group A.X.W. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27(3):687–97.
Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. 2013;13(10):727–38.
Bellera N, Barba I, Rodriguez-Sinovas A, Ferret E, Asin MA, Gonzalez-Alujas MT, Perez-Rodon J, Esteves M, Fonseca C, Toran N, Garcia Del Blanco B, Perez A, Garcia-Dorado D. Single intracoronary injection of encapsulated antagomir-92a promotes angiogenesis and prevents adverse infarct remodeling. J Am Heart Assoc. 2014;3(5):e000946.
Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, Zhang Q, Wakelam MJO, Karpe F, Schulze A, Harris AL. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014;9(1):349–65.
Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, Gregorevic P, Obad S, Kauppinen S, Du XJ, Lin RC, McMullen JR. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A. 2012;109(43):17615–20.
Bonventre JV. Maladaptive proximal tubule repair: cell cycle arrest. Nephron Clin Pract. 2014;127(1-4):61–4.
Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, Kaluza D, Treguer K, Carmona G, Bonauer A, Horrevoets AJ, Didier N, Girmatsion Z, Biliczki P, Ehrlich JR, Katus HA, Muller OJ, Potente M, Zeiher AM, Hermeking H, Dimmeler S. MicroRNA-34a regulates cardiac ageing and function. Nature. 2013;495(7439):107–10.
Boor P, Ostendorf T, Floege J. Renal fibrosis: novel insights into mechanisms and therapeutic targets. Nat Rev Nephrol. 2010;6(11):643–56.
Brown JM, Giaccia AJ. The unique physiology of solid tumors: opportunities (and problems) for cancer therapy. Cancer Res. 1998;58(7):1408–16.
Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18(23):2893–904.
Bulun SE, Yang S, Fang Z, Gurates B, Tamura M, Sebastian S. Estrogen production and metabolism in endometriosis. Ann N Y Acad Sci. 2002;955:75–85.
Bunn HF, Poyton RO. Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev. 1996;76(3):839–85.
Carpentier AC, Blondin DP, Virtanen KA, Richard D, Haman F, Turcotte EE. Brown adipose tissue energy metabolism in humans. Front Endocrinol (Lausanne). 2018;9:447.
Chen C, Zhou Y, Hu C, Wang Y, Yan Z, Li Z, Wu R. Mitochondria and oxidative stress in ovarian endometriosis. Free Radic Biol Med. 2019;136:22–34.
Chen W, Dong J, Haiech J, Kilhoffer MC, Zeniou M. Cancer stem cell quiescence and plasticity as major challenges in cancer therapy. Stem Cells Int. 2016;2016:1740936.
Chou CC, Chuang HC, Salunke SB, Kulp SK, Chen CS. A novel HIF-1α-integrin-linked kinase regulatory loop that facilitates hypoxia-induced HIF-1α expression and epithelial-mesenchymal transition in cancer cells. Oncotarget. 2015;6(10):8271–85.
Chou YH, Huang TM, Chu TS. Novel insights into acute kidney injury-chronic kidney disease continuum and the role of renin-angiotensin system. J Formos Med Assoc. 2017;116(9):652–9.
Coelho M, Oliveira T, Fernandes R. Biochemistry of adipose tissue: an endocrine organ. Arch Med Sci. 2013;9(2):191–200.
Copple BL. Hypoxia stimulates hepatocyte epithelial to mesenchymal transition by hypoxia-inducible factor and transforming growth factor-beta-dependent mechanisms. Liver Int. 2010;30(5):669–82.
Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, Labosky PA, Simon MC, Keith B. HIF-2α regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20(5):557–70.
Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360(15):1509–17.
D'Ignazio L, Rocha S. Hypoxia induced NF-kappaB. Cells. 2016;5(1):e10.
Dai Y, Lin X, Xu W, Lin X, Huang Q, Shi L, Pan Y, Zhang Y, Zhu Y, Li C, Liu L, Zhang S. MiR-210-3p protects endometriotic cells from oxidative stress-induced cell cycle arrest by targeting BARD1. Cell Death Dis. 2019;10(2):144.
De Bock K, Mazzone M, Carmeliet P. Antiangiogenic therapy, hypoxia, and metastasis: risky liaisons, or not? Nat Rev Clin Oncol. 2011;8(7):393–404.
Du W, Zhang L, Brett-Morris A, Aguila B, Kerner J, Hoppel CL, Puchowicz M, Serra D, Herrero L, Rini BI, Campbell S, Welford SM. HIF drives lipid deposition and cancer in ccRCC via repression of fatty acid metabolism. Nat Commun. 2017;8(1):1769.
Duley L. The global impact of pre-eclampsia and eclampsia. Semin Perinatol. 2009;33(3):130–7.
Lusha E, Jiang H, Lu Z. MicroRNA-144 attenuates cardiac ischemia/reperfusion injury by targeting FOXO1. Exp Ther Med. 2019;17(3):2152–60.
Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–65.
Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94(9):4273–8.
Errami Y, Naura AS, Kim H, Ju J, Suzuki Y, El-Bahrawy AH, Ghonim MA, Hemeida RA, Mansy MS, Zhang J, Xu M, Smulson ME, Brim H, Boulares AH. Apoptotic DNA fragmentation may be a cooperative activity between caspase-activated deoxyribonuclease and the poly (ADP-ribose) polymerase-regulated DNAS1L3, an endoplasmic reticulum-localized endonuclease that translocates to the nucleus during apoptosis. J Biol Chem. 2013;288(5):3460–8.
Fasanaro P, D'Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F. MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem. 2008;283(23):15878–83.
Fisher SJ. Why is placentation abnormal in preeclampsia? Am J Obstet Gynecol. 2015;213(4 Suppl):S115–22.
Flamme I, Frolich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. 1997;173(2):206–10.
Fu JL, Hsiao KY, Lee HC, Li WN, Chang N, Wu MH, Tsai SJ. Suppression of COUP-TFII upregulates angiogenin and promotes angiogenesis in endometriosis. Hum Reprod. 2018;33(8):1517–27.
Furuta E, Pai SK, Zhan R, Bandyopadhyay S, Watabe M, Mo YY, Hirota S, Hosobe S, Tsukada T, Miura K, Kamada S, Saito K, Iiizumi M, Liu W, Ericsson J, Watabe K. Fatty acid synthase gene is up-regulated by hypoxia via activation of Akt and sterol regulatory element binding protein-1. Cancer Res. 2008;68(4):1003–11.
Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, Arreola A, Rathmell WK, Olumi A, Lopez-Larrubia P, Stephanopoulos G, Iliopoulos O. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab. 2013;17(3):372–85.
Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364(9447):1789–99.
Greer SN, Metcalf JL, Wang Y, Ohh M. The updated biology of hypoxia-inducible factor. EMBO J. 2012;31(11):2448–60.
Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, Brekken RA, Scherer PE. Hypoxia-inducible factor 1α induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol. 2009;29(16):4467–83.
Harrison LR, Micha D, Brandenburg M, Simpson KL, Morrow CJ, Denneny O, Hodgkinson C, Yunus Z, Dempsey C, Roberts D, Blackhall F, Makin G, Dive C. Hypoxic human cancer cells are sensitized to BH-3 mimetic-induced apoptosis via downregulation of the Bcl-2 protein Mcl-1. J Clin Invest. 2011;121(3):1075–87.
Hoffmann P, Feige JJ, Alfaidy N. Expression and oxygen regulation of endocrine gland-derived vascular endothelial growth factor/prokineticin-1 and its receptors in human placenta during early pregnancy. Endocrinology. 2006;147(4):1675–84.
Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, Shimomura I. Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes. 2007;56(4):901–11.
Hou PC, Li YH, Lin SC, Lin SC, Lee JC, Lin BW, Liou JP, Chang JY, Kuo CC, Liu YM, Sun HS, Tsai SJ. Hypoxia-induced downregulation of DUSP-2 phosphatase drives colon cancer stemness. Cancer Res. 2017;77(16):4305–16.
Hsiao KY, Chang N, Lin SC, Li YH, Wu MH. Inhibition of dual specificity phosphatase-2 by hypoxia promotes interleukin-8-mediated angiogenesis in endometriosis. Hum Reprod. 2014;29(12):2747–55.
Hsu CC, Lu CW, Huang BM, Wu MH, Tsai SJ. Cyclic adenosine 3′,5′-monophosphate response element-binding protein and CCAAT/enhancer-binding protein mediate prostaglandin E2-induced steroidogenic acute regulatory protein expression in endometriotic stromal cells. Am J Pathol. 2008;173(2):433–41.
Huang ZQ, Xu W, Wu JL, Lu X, Chen XM. MicroRNA-374a protects against myocardial ischemia-reperfusion injury in mice by targeting the MAPK6 pathway. Life Sci. 2019;232:116619.
Javerzat S, Auguste P, Bikfalvi A. The role of fibroblast growth factors in vascular development. Trends Mol Med. 2002;8(10):483–9.
Jiang C, Kim JH, Li F, Qu A, Gavrilova O, Shah YM, Gonzalez FJ. Hypoxia-inducible factor 1α regulates a SOCS3-STAT3-adiponectin signal transduction pathway in adipocytes. J Biol Chem. 2013;288(6):3844–57.
Jun JC, Devera R, Unnikrishnan D, Shin MK, Bevans-Fonti S, Yao Q, Rathore A, Younas H, Halberg N, Scherer PE, Polotsky VY. Adipose HIF-1α causes obesity by suppressing brown adipose tissue thermogenesis. J Mol Med (Berl). 2017;95(3):287–97.
Ke Q, Costa M. Hypoxia-inducible factor-1 (HIF-1). Mol Pharmacol. 2006;70(5):1469–80.
Keeley TP, Mann GE. Defining physiological normoxia for improved translation of cell physiology to animal models and humans. Physiol Rev. 2019;99(1):161–234.
Keith B, Johnson RS, Simon MC. HIF1α and HIF2α: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2011;12(1):9–22.
Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001;2(4):282–6.
Kimura K, Iwano M, Higgins DF, Yamaguchi Y, Nakatani K, Harada K, Kubo A, Akai Y, Rankin EB, Neilson EG, Haase VH, Saito Y. Stable expression of HIF-1α in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295(4):F1023–9.
Kitazumi I, Tsukahara M. Regulation of DNA fragmentation: the role of caspases and phosphorylation. FEBS J. 2011;278(3):427–41.
Koumenis C, Naczki C, Koritzinsky M, Rastani S, Diehl A, Sonenberg N, Koromilas A, Wouters BG. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2α. Mol Cell Biol. 2002;22(21):7405–16.
Krishnan J, Danzer C, Simka T, Ukropec J, Walter KM, Kumpf S, Mirtschink P, Ukropcova B, Gasperikova D, Pedrazzini T, Krek W. Dietary obesity-associated Hif1α activation in adipocytes restricts fatty acid oxidation and energy expenditure via suppression of the Sirt2-NAD+ system. Genes Dev. 2012;26(3):259–70.
Krishnan J, Suter M, Windak R, Krebs T, Felley A, Montessuit C, Tokarska-Schlattner M, Aasum E, Bogdanova A, Perriard E, Perriard JC, Larsen T, Pedrazzini T, Krek W. Activation of a HIF1α-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9(6):512–24.
Kucharzewska P, Christianson HC, Welch JE, Svensson KJ, Fredlund E, Ringner M, Morgelin M, Bourseau-Guilmain E, Bengzon J, Belting M. Exosomes reflect the hypoxic status of glioma cells and mediate hypoxia-dependent activation of vascular cells during tumor development. Proc Natl Acad Sci U S A. 2013;110(18):7312–7.
Lai HH, Li JN, Wang MY, Huang HY, Croce CM, Sun HL, Lyu YJ, Kang JW, Chiu CF, Hung MC, Suzuki HI, Chen PS. HIF-1α promotes autophagic proteolysis of dicer and enhances tumor metastasis. J Clin Invest. 2018;128(2):625–43.
Lavagnino M, Oslapas AN, Gardner KL, Arnoczky SP. Hypoxia inhibits primary cilia formation and reduces cell-mediated contraction in stress-deprived rat tail tendon fascicles. Muscles Ligaments Tendons J. 2016;6(2):193–7.
LeBleu VS, Taduri G, O'Connell J, Teng Y, Cooke VG, Woda C, Sugimoto H, Kalluri R. Origin and function of myofibroblasts in kidney fibrosis. Nat Med. 2013;19(8):1047–53.
Lee HC, Lin SC, Wu MH, Tsai SJ. Induction of pyruvate dehydrogenase kinase 1 by hypoxia alters cellular metabolism and inhibits apoptosis in Endometriotic stromal cells. Reprod Sci. 2019;26(6):734–44.
Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, Quehenberger O, Johnson RS, Olefsky JM. Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell. 2014;157(6):1339–52.
Lei J, Ma J, Ma Q, Li X, Liu H, Xu Q, Duan W, Sun Q, Xu J, Wu Z, Wu E. Hedgehog signaling regulates hypoxia induced epithelial to mesenchymal transition and invasion in pancreatic cancer cells via a ligand-independent manner. Mol Cancer. 2013;12:66.
Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. 1990;323(4):236–41.
Li D, Zhou J, Yang B, Yu Y. microRNA-340-5p inhibits hypoxia/reoxygenation-induced apoptosis and oxidative stress in cardiomyocytes by regulating the Act1/NF-kappaB pathway. J Cell Biochem. 2019;120(9):14618–27.
Li Q, Yang J, Zhang J, Liu XW, Yang CJ, Fan ZX, Wang HB, Yang Y, Zheng T, Yang J. Inhibition of microRNA-327 ameliorates ischemia/reperfusion injury-induced cardiomyocytes apoptosis through targeting apoptosis repressor with caspase recruitment domain. J Cell Physiol. 2020;235(4):3753–67.
Lin B, Feng D, Xu J. Cardioprotective effects of microRNA-18a on acute myocardial infarction by promoting cardiomyocyte autophagy and suppressing cellular senescence via brain derived neurotrophic factor. Cell Biosci. 2019;9:38.
Lin G, Huang J, Chen Q, Chen L, Feng D, Zhang S, Huang X, Huang Y, Lin Q. miR-146a-5p mediates intermittent hypoxia-induced injury in H9c2 cells by targeting XIAP. Oxidative Med Cell Longev. 2019;6581217:2019.
Lin L, Li G, Zhang W, Wang YL, Yang H. Low-dose aspirin reduces hypoxia-induced sFlt1 release via the JNK/AP-1 pathway in human trophoblast and endothelial cells. J Cell Physiol. 2019;234(10):18928–41.
Lin SC, Hsiao KY, Chang N, Hou PC, Tsai SJ. Loss of dual-specificity phosphatase-2 promotes angiogenesis and metastasis via up-regulation of interleukin-8 in colon cancer. J Pathol. 2017;241(5):638–48.
Lin SC, Lee HC, Hou PC, Fu JL, Wu MH, Tsai SJ. Targeting hypoxia-mediated YAP1 nuclear translocation ameliorates pathogenesis of endometriosis without compromising maternal fertility. J Pathol. 2017;242(4):476–87.
Lin SC, Lee HC, Hsu CT, Huang YH, Li WN, Hsu PL, Wu MH, Tsai SJ. Targeting anthrax toxin receptor 2 ameliorates endometriosis progression. Theranostics. 2019;9(3):620–32.
Lin SC, Li YH, Wu MH, Chang YF, Lee DK, Tsai SY, Tsai MJ, Tsai SJ. Suppression of COUP-TFII by proinflammatory cytokines contributes to the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2014;99(3):e427–37.
Lin SC, Wang CC, Wu MH, Yang SH, Li YH, Tsai SJ. Hypoxia-induced microRNA-20a expression increases ERK phosphorylation and angiogenic gene expression in endometriotic stromal cells. J Clin Endocrinol Metab. 2012;97(8):e1515–23.
Lin X, Dai Y, Xu W, Shi L, Jin X, Li C, Zhou F, Pan Y, Zhang Y, Lin X, Zhang S. Hypoxia promotes ectopic adhesion ability of endometrial stromal cells via TGF-beta1/Smad signaling in endometriosis. Endocrinology. 2018;159(4):1630–41.
Liu H, Zhang Z, Xiong W, Zhang L, Du Y, Liu Y, Xiong X. Long non-coding RNA MALAT1 mediates hypoxia-induced pro-survival autophagy of endometrial stromal cells in endometriosis. J Cell Mol Med. 2019;23(1):439–52.
Liu H, Zhang Z, Xiong W, Zhang L, Xiong Y, Li N, He H, Du Y, Liu Y. Hypoxia-inducible factor-1α promotes endometrial stromal cells migration and invasion by upregulating autophagy in endometriosis. Reproduction. 2017;153(6):809–20.
Liu Y, Ma Z, Zhao C, Wang Y, Wu G, Xiao J, McClain CJ, Li X, Feng W. HIF-1α and HIF-2α are critically involved in hypoxia-induced lipid accumulation in hepatocytes through reducing PGC-1α-mediated fatty acid beta-oxidation. Toxicol Lett. 2014;226(2):117–23.
Lovisa S, Zeisberg M, Kalluri R. Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol Metab. 2016;27(10):681–95.
Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283(42):28106–14.
Lu Z, Zhang W, Jiang S, Zou J, Li Y. Effect of oxygen tensions on the proliferation and angiogenesis of endometriosis heterograft in severe combined immunodeficiency mice. Fertil Steril. 2014;101(2):568–76.
Lv X, Li J, Zhang C, Hu T, Li S, He S, Yan H, Tan Y, Lei M, Wen M, Zuo J. The role of hypoxia-inducible factors in tumor angiogenesis and cell metabolism. Genes Dis. 2017;4(1):19–24.
Ma L, Zhang J, Liu Y. Roles and mechanisms of obstructive sleep apnea-hypopnea syndrome and chronic intermittent hypoxia in atherosclerosis: evidence and prospective. Oxidative Med Cell Longev. 2016;2016:8215082.
Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, Enzo E, Guzzardo V, Rondina M, Spruce T, Parenti AR, Daidone MG, Bicciato S, Piccolo S. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141(7):1195–207.
Matsuura H, Ichiki T, Inoue E, Nomura M, Miyazaki R, Hashimoto T, Ikeda J, Takayanagi R, Fong GH, Sunagawa K. Prolyl hydroxylase domain protein 2 plays a critical role in diet-induced obesity and glucose intolerance. Circulation. 2013;127(21):2078–87.
Maxwell PH, Pugh CW, Ratcliffe PJ. Activation of the HIF pathway in cancer. Curr Opin Genet Dev. 2001;11(3):293–9.
Mazure NM, Pouyssegur J. Hypoxia-induced autophagy: cell death or cell survival? Curr Opin Cell Biol. 2010;22(2):177–80.
Meng W, Hao Y, He C, Li L, Zhu G. Exosome-orchestrated hypoxic tumor microenvironment. Mol Cancer. 2019;18(1):57.
Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, Kelleher JK, Vander Heiden MG, Iliopoulos O, Stephanopoulos G. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481(7381):380–4.
Michailidou Z, Morton NM, Moreno Navarrete JM, West CC, Stewart KJ, Fernandez-Real JM, Schofield CJ, Seckl JR, Ratcliffe PJ. Adipocyte pseudohypoxia suppresses lipolysis and facilitates benign adipose tissue expansion. Diabetes. 2015;64(3):733–45.
Miettinen KH, Lassus J, Harjola VP, Siirila-Waris K, Melin J, Punnonen KR, Nieminen MS, Laakso M, Peuhkurinen KJ. Prognostic role of pro- and anti-inflammatory cytokines and their polymorphisms in acute decompensated heart failure. Eur J Heart Fail. 2008;10(4):396–403.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41.
Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, Yang Y, Linehan WM, Chandel NS, DeBerardinis RJ. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2011;481(7381):385–8.
Mylonis I, Sembongi H, Befani C, Liakos P, Siniossoglou S, Simos G. Hypoxia causes triglyceride accumulation by HIF-1-mediated stimulation of lipin 1 expression. J Cell Sci. 2012;125(Pt 14):3485–93.
Nath KA. Tubulointerstitial changes as a major determinant in the progression of renal damage. Am J Kidney Dis. 1992;20(1):1–17.
Norman JT, Clark IM, Garcia PL. Hypoxia promotes fibrogenesis in human renal fibroblasts. Kidney Int. 2000;58(6):2351–66.
O'Rourke RW, Meyer KA, Gaston G, White AE, Lumeng CN, Marks DL. Hexosamine biosynthesis is a possible mechanism underlying hypoxia's effects on lipid metabolism in human adipocytes. PLoS One. 2013;8(8):e71165.
Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3(3):187–97.
Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21(7):1104–17.
Planavila A, Dominguez E, Navarro M, Vinciguerra M, Iglesias R, Giralt M, Lope-Piedrafita S, Ruberte J, Villarroya F. Dilated cardiomyopathy and mitochondrial dysfunction in Sirt1-deficient mice: a role for Sirt1-Mef2 in adult heart. J Mol Cell Cardiol. 2012;53(4):521–31.
Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146(6):873–87.
Raica M, Cimpean AM. Platelet-derived growth factor (PDGF)/PDGF receptors (PDGFR) axis as target for antitumor and antiangiogenic therapy. Pharmaceuticals (Basel). 2010;3(3):572–99.
Ramteke A, Ting H, Agarwal C, Mateen S, Somasagara R, Hussain A, Graner M, Frederick B, Agarwal R, Deep G. Exosomes secreted under hypoxia enhance invasiveness and stemness of prostate cancer cells by targeting adherens junction molecules. Mol Carcinog. 2015;54(7):554–65.
Rankin EB, Fuh KC, Castellini L, Viswanathan K, Finger EC, Diep AN, LaGory EL, Kariolis MS, Chan A, Lindgren D, Axelson H, Miao YR, Krieg AJ, Giaccia AJ. Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc Natl Acad Sci U S A. 2014;111(37):13373–8.
Rankin EB, Giaccia AJ. Hypoxic control of metastasis. Science. 2016;352(6282):175–80.
Rath G, Aggarwal R, Jawanjal P, Tripathi R, Batra A. HIF-1α and placental growth factor in pregnancies complicated with preeclampsia: a qualitative and quantitative analysis. J Clin Lab Anal. 2016;30(1):75–83.
Raval RR, Lau KW, Tran MGB, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25(13):5675–86.
Regazzetti C, Peraldi P, Gremeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M, Bost F, Le Marchand-Brustel Y, Tanti JF, Giorgetti-Peraldi S. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes. 2009;58(1):95–103.
Rocha S. Gene regulation under low oxygen: holding your breath for transcription. Trends Biochem Sci. 2007;32(8):389–97.
Rupaimoole R, Ivan C, Yang D, Gharpure KM, Wu SY, Pecot CV, Previs RA, Nagaraja AS, Armaiz-Pena GN, McGuire M, Pradeep S, Mangala LS, Rodriguez-Aguayo C, Huang L, Bar-Eli M, Zhang W, Lopez-Berestein G, Calin GA, Sood AK. Hypoxia-upregulated microRNA-630 targets dicer, leading to increased tumor progression. Oncogene. 2016;35(33):4312–20.
Sahlgren C, Gustafsson MV, Jin S, Poellinger L, Lendahl U. Notch signaling mediates hypoxia-induced tumor cell migration and invasion. Proc Natl Acad Sci U S A. 2008;105(17):6392–7.
Salminen A, Kaarniranta K, Kauppinen A. Hypoxia-inducible histone lysine demethylases: impact on the aging process and age-related diseases. Aging Dis. 2016;7(2):180–200.
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, Akazawa H, Tateno K, Kayama Y, Harada M, Shimizu I, Asahara T, Hamada H, Tomita S, Molkentin JD, Zou Y, Komuro I. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444–8.
Schinner E, Schramm A, Kees F, Hofmann F, Schlossmann J. The cyclic GMP-dependent protein kinase Iα suppresses kidney fibrosis. Kidney Int. 2013;84(6):1198–206.
Schito L, Semenza GL. Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer. 2016;2(12):758–70.
Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88(4):1474–80.
Semenza GL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010;20(1):51–6.
Semenza GL. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148(3):399–408.
Sermeus A, Genin M, Maincent A, Fransolet M, Notte A, Leclere L, Riquier H, Arnould T, Michiels C. Hypoxia-induced modulation of apoptosis and BCL-2 family proteins in different cancer cell types. PLoS One. 2012;7(11):e47519.
Shao C, Yang F, Miao S, Liu W, Wang C, Shu Y, Shen H. Role of hypoxia-induced exosomes in tumor biology. Mol Cancer. 2018;17(1):120.
Sharkey AM, Day K, McPherson A, Malik S, Licence D, Smith SK, Charnock-Jones DS. Vascular endothelial growth factor expression in human endometrium is regulated by hypoxia. J Clin Endocrinol Metab. 2000;85(1):402–9.
Shen G, Li X, Jia YF, Piazza GA, Xi Y. Hypoxia-regulated microRNAs in human cancer. Acta Pharmacol Sin. 2013;34(3):336–41.
Shen J, Xia WY, Khotskaya YB, Huo LF, Nakanishi K, Lim SO, Du Y, Wang Y, Chang WC, Chen CH, Hsu JL, Wu Y, Lam YC, James BP, Liu XP, Liu CG, Patel DJ, Hung MC. EGFR modulates microRNA maturation in response to hypoxia through phosphorylation of AGO2. Nature. 2013;497(7449):383–7.
Shimizu I, Aprahamian T, Kikuchi R, Shimizu A, Papanicolaou KN, MacLauchlan S, Maruyama S, Walsh K. Vascular rarefaction mediates whitening of brown fat in obesity. J Clin Invest. 2014;124(5):2099–112.
Shimizu I, Walsh K. The whitening of brown fat and its implications for weight management in obesity. Curr Obes Rep. 2015;4(2):224–9.
Skuli N, Liu LP, Runge A, Wang T, Yuan LJ, Patel S, Iruela-Arispe L, Simon MC, Keith B. Endothelial deletion of hypoxia-inducible factor-2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood. 2009;114(2):469–77.
Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. J Pathol. 2009;218(1):7–29.
Spivak-Kroizman TR, Hostetter G, Posner R, Aziz M, Hu C, Demeure MJ, Von Hoff D, Hingorani SR, Palculict TB, Izzo J, Kiriakova GM, Abdelmelek M, Bartholomeusz G, James BP, Powis G. Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer. Cancer Res. 2013;73(11):3235–47.
Su MT, Wang CY, Tsai PY, Chen TY, Tsai HL, Kuo PL. Aspirin enhances trophoblast invasion and represses soluble fms-like tyrosine kinase 1 production: a putative mechanism for preventing preeclampsia. J Hypertens. 2019;37(12):2461–9.
Sun K, Halberg N, Khan M, Magalang UJ, Scherer PE. Selective inhibition of hypoxia-inducible factor 1α ameliorates adipose tissue dysfunction. Mol Cell Biol. 2013;33(5):904–17.
Sun YB, Qu X, Caruana G, Li J. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016;92(3):102–7.
Tabuchi T, Satoh M, Itoh T, Nakamura M. MicroRNA-34a regulates the longevity-associated protein SIRT1 in coronary artery disease: effect of statins on SIRT1 and microRNA-34a expression. Clin Sci (Lond). 2012;123(3):161–71.
Thomou T, Mori MA, Dreyfuss JM, Konishi M, Sakaguchi M, Wolfrum C, Rao TN, Winnay JN, Garcia-Martin R, Grinspoon SK, Gorden P, Kahn CR. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017;542(7642):450–5.
Tian H, McKnight SL, Russell DW. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997;11(1):72–82.
Tokumaru S, Suzuki M, Yamada H, Nagino M, Takahashi T. let-7 regulates Dicer expression and constitutes a negative feedback loop. Carcinogenesis. 2008;29(11):2073–7.
Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand. 2005;184(4):285–93.
Trayhurn P, Alomar SY. Oxygen deprivation and the cellular response to hypoxia in adipocytes - perspectives on white and brown adipose tissues in obesity. Front Endocrinol (Lausanne). 2015;6:19.
Trayhurn P, Wang B, Wood IS. Hypoxia and the endocrine and signalling role of white adipose tissue. Arch Physiol Biochem. 2008;114(4):267–76.
Tredan O, Galmarini CM, Patel K, Tannock IF. Drug resistance and the solid tumor microenvironment. J Natl Cancer Inst. 2007;99(19):1441–54.
Triantafyllou EA, Georgatsou E, Mylonis I, Simos G, Paraskeva E. Expression of AGPAT2, an enzyme involved in the glycerophospholipid/triacylglycerol biosynthesis pathway, is directly regulated by HIF-1 and promotes survival and etoposide resistance of cancer cells under hypoxia. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863(9):1142–52.
Tsai SJ, Wu MH, Lin CC, Sun HS, Chen HM. Regulation of steroidogenic acute regulatory protein expression and progesterone production in endometriotic stromal cells. J Clin Endocrinol Metab. 2001;86(12):5765–73.
Tsai YP, Wu KJ. Hypoxia-regulated target genes implicated in tumor metastasis. J Biomed Sci. 2012;19:102.
Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, Dangwal S, Kumarswamy R, Bang C, Holzmann A, Remke J, Caprio M, Jentzsch C, Engelhardt S, Geisendorf S, Glas C, Hofmann TG, Nessling M, Richter K, Schiffer M, Carrier L, Napp LC, Bauersachs J, Chowdhury K, Thum T. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078.
van den Beucken T, Koch E, Chu K, Rupaimoole R, Prickaerts P, Adriaens M, Voncken JW, Harris AL, Buffa FM, Haider S, Starmans MHW, Yao CQ, Ivan M, Ivan C, Pecot CV, Boutros PC, Sood AK, Koritzinsky M, Wouters BG. Hypoxia promotes stem cell phenotypes and poor prognosis through epigenetic regulation of DICER. Nat Commun. 2014;5:5203.
Wang CY, Tsai HL, Syu JS, Chen TY, Su MT. Primary cilium-regulated EG-VEGF signaling facilitates trophoblast invasion. J Cell Physiol. 2017;232(6):1467–77.
Wang CY, Tsai PY, Chen TY, Tsai HL, Kuo PL, Su MT. Elevated miR-200a and miR-141 inhibit endocrine gland-derived vascular endothelial growth factor expression and ciliogenesis in preeclampsia. J Physiol. 2019;597(12):3069–83.
Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92(12):5510–4.
Wang S, Cheng Z, Chen X, Xue H. microRNA-135a protects against myocardial ischemia-reperfusion injury in rats by targeting protein tyrosine phosphatase 1B. J Cell Biochem. 2019;120(6):10421–33.
Wang T, Gilkes DM, Takano N, Xiang L, Luo W, Bishop CJ, Chaturvedi P, Green JJ, Semenza GL. Hypoxia-inducible factors and RAB22A mediate formation of microvesicles that stimulate breast cancer invasion and metastasis. Proc Natl Acad Sci U S A. 2014;111(31):e3234–42.
Wang TH, Hsia SM, Shieh TM. Lysyl oxidase and the tumor microenvironment. Int J Mol Sci. 2016;18(1):62.
Wang Y, Tian MM, Mi CJ, Chen KL, Ji YC, Wang L, Zhang J, Cheng K. Exercise protects the heart against myocardial infarction through upregulation of miR-1192. Biochem Biophys Res Commun. 2019;521(4):1061–9.
Wei H, Bedja D, Koitabashi N, Xing D, Chen J, Fox-Talbot K, Rouf R, Chen S, Steenbergen C, Harmon JW, Dietz HC, Gabrielson KL, Kass DA, Semenza GL. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-beta signaling. Proc Natl Acad Sci U S A. 2012;109(14):e841–50.
Wiener CM, Booth G, Semenza GL. In vivo expression of mRNAs encoding hypoxia-inducible factor 1. Biochem Biophys Res Commun. 1996;225(2):485–8.
Wood IS, de Heredia FP, Wang B, Trayhurn P. Cellular hypoxia and adipose tissue dysfunction in obesity. Proc Nutr Soc. 2009;68(4):370–7.
Wood IS, Wang B, Lorente-Cebrian S, Trayhurn P. Hypoxia increases expression of selective facilitative glucose transporters (GLUT) and 2-deoxy-D-glucose uptake in human adipocytes. Biochem Biophys Res Commun. 2007;361(2):468–73.
Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–64.
Wu MH, Chen KF, Lin SC, Lgu CW, Tsai SJ. Aberrant expression of leptin in human endometriotic stromal cells is induced by elevated levels of hypoxia inducible factor-1α. Am J Pathol. 2007;170(2):590–8.
Wu MH, Hsiao KY, Tsai SJ. Hypoxia: the force of endometriosis. J Obstet Gynaecol Res. 2019;45(3):532–41.
Wu MH, Lin SC, Hsiao KY, Tsai SJ. Hypoxia-inhibited dual-specificity phosphatase-2 expression in endometriotic cells regulates cyclooxygenase-2 expression. J Pathol. 2011;225(3):390–400.
Wu MH, Lu CW, Chuang PC, Tsai SJ. Prostaglandin E2: the master of endometriosis? Exp Biol Med (Maywood). 2010;235(6):668–77.
Wu MH, Wang CA, Lin CC, Chen LC, Chang WC, Tsai SJ. Distinct regulation of cyclooxygenase-2 by interleukin-1beta in normal and endometriotic stromal cells. J Clin Endocrinol Metab. 2005;90(1):286–95.
Wu MZ, Tsai YP, Yang MH, Huang CH, Chang SY, Chang CC, Teng SC, Wu KJ. Interplay between HDAC3 and WDR5 is essential for hypoxia-induced epithelial-mesenchymal transition. Mol Cell. 2011;43(5):811–22.
Xiao JM, Wang JJ, Sun LL. Effect of miR-134 against myocardial hypoxia/reoxygenation injury by directly targeting NOS3 and regulating PI3K/Akt pathway. Acta Cir Bras. 2019;34(8):e201900802.
Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes. 2009;33(1):54–66.
Ye J, Chen Y, Zhu J, Chen C, Zhu X, Feng L, Ye W, Zhang J. Aspirin use during pregnancy and hypoxia-related placental pathology. Pregnancy Hypertens. 2018;14:177–88.
Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of Ob/Ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293(4):e1118–28.
Yin J, Gao Z, He Q, Zhou D, Guo Z, Ye J. Role of hypoxia in obesity-induced disorders of glucose and lipid metabolism in adipose tissue. Am J Physiol Endocrinol Metab. 2009;296(2):e333–42.
Yoo H, Antoniewicz MR, Stephanopoulos G, Kelleher JK. Quantifying reductive carboxylation flux of glutamine to lipid in a brown adipocyte cell line. J Biol Chem. 2008;283(30):20621–7.
Young VJ, Brown JK, Maybin J, Saunders PT, Duncan WC, Horne AW. Transforming growth factor-beta induced Warburg-like metabolic reprogramming may underpin the development of peritoneal endometriosis. J Clin Endocrinol Metab. 2014;99(9):3450–9.
Yun Z, Maecker HL, Johnson RS, Giaccia AJ. Inhibition of PPAR gamma 2 gene expression by the HIF-1-regulated gene DEC1/Stra13: a mechanism for regulation of adipogenesis by hypoxia. Dev Cell. 2002;2(3):331–41.
Zhai CL, Tang GM, Qian G, Hu HL, Wang SJ, Yin D, Zhang S. MicroRNA-98 attenuates cardiac ischemia-reperfusion injury through inhibiting DAPK1 expression. IUBMB Life. 2019;71(2):166–76.
Zhang J, Pan J, Yang M, Jin X, Feng J, Wang A, Chen Z. Upregulating microRNA-203 alleviates myocardial remodeling and cell apoptosis through downregulating protein tyrosine phosphatase 1B in rats with myocardial infarction. J Cardiovasc Pharmacol. 2019;74(5):474–81.
Zhang L, Ebenezer PJ, Dasuri K, Fernandez-Kim SO, Francis J, Mariappan N, Gao Z, Ye J, Bruce-Keller AJ, Keller JN. Aging is associated with hypoxia and oxidative stress in adipose tissue: implications for adipose function. Am J Physiol Endocrinol Metab. 2011;301(4):e599–607.
Zhang N, Fu Z, Linke S, Chicher J, Gorman JJ, Visk D, Haddad GG, Poellinger L, Peet DJ, Powell F, Johnson RS. The asparaginyl hydroxylase factor inhibiting HIF-1α is an essential regulator of metabolism. Cell Metab. 2010;11(5):364–78.
Zhang X, Lam KS, Ye H, Chung SK, Zhou M, Wang Y, Xu A. Adipose tissue-specific inhibition of hypoxia-inducible factor 1α induces obesity and glucose intolerance by impeding energy expenditure in mice. J Biol Chem. 2010;285(43):32869–77.
Zhong J, Krawczyk SA, Chaerkady R, Huang H, Goel R, Bader JS, Wong GW, Corkey BE, Pandey A. Temporal profiling of the secretome during adipogenesis in humans. J Proteome Res. 2010;9(10):5228–38.
Zhu Q, Hu F. Antagonism of miR-429 ameliorates anoxia/reoxygenation injury in cardiomyocytes by enhancing MO25/LKB1/AMPK mediated autophagy. Life Sci. 2019;235:116842.
Acknowledgements
The authors appreciate all the scientists whose works contribute to this review article. We also apologize to scientists whose works were not included in this review due to space constriction.
Funding
This work was supported by grants from Ministry of Science and Technology (MOST 106-2320-B-006-072 -MY3 and MOST 107-2321-B-006-014) and National Health Research Institutes, Taiwan (NHRI-EX106-10516BI).
Author information
Authors and Affiliations
Contributions
SJT conceived the project. All authors wrote the manuscript. Particular contribution of each section was made by: PSC (cancer), PLH (cardiovascular), ICP (metabolic diseases), WTC (chronic kidney disease), CYW (preeclampsia), and SCL (endometriosis). The author(s) read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declare there is no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, PS., Chiu, WT., Hsu, PL. et al. Pathophysiological implications of hypoxia in human diseases. J Biomed Sci 27, 63 (2020). https://doi.org/10.1186/s12929-020-00658-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-020-00658-7