
Abstract
This review examines the biology of the Fat mass- and obesity-associated gene (FTO), and the implications of genetic association of FTO SNPs with obesity and genetic aging. Notably, we focus on the role of FTO in the regulation of methylation status as possible regulators of weight gain and genetic aging. We present a theoretical review of the FTO gene with a particular emphasis on associations with UCP2, AMPK, RBL2, IRX3, CUX1, mTORC1 and hormones involved in hunger regulation. These associations are important for dietary behavior regulation and cellular nutrient sensing via amino acids. We suggest that these pathways may also influence telomere regulation. Telomere length (TL) attrition may be influenced by obesity-related inflammation and oxidative stress, and FTO gene-involved pathways. There is additional emerging evidence to suggest that telomere length and obesity are bi-directionally associated. However, the role of obesity risk-related genotypes and associations with TL are not well understood. The FTO gene may influence pathways implicated in regulation of TL, which could help to explain some of the non-consistent relationship between weight phenotype and telomere length that is observed in population studies investigating obesity.
Similar content being viewed by others
Background
Genetic background is important in understanding metabolic pathways that regulate adiposity [1, 2]. Telomere length attrition may be influenced by obesity-related inflammation and oxidative stress. There is emerging evidence to suggest that telomere length (TL) and obesity are bi-directionally associated [3,4,5]. However, the role of regulatory obesity genotypes and associations with TL are not well understood. Across genetic association studies of obesity, there is considerable obesity phenotype variance [6, 7]. Factors such as gene drift, environment, migration or ethnicity may in part be responsible for such obesity phenotype variance [8,9,10]. Alternatively, TL-obesity relationship could theoretically be influenced by gene methylation levels, which in turn could affect genes, due to the characteristics of, for example, permitting changes which affect gene function but at the same time do not modify DNA sequences. The fat mass and obesity associated (FTO) gene is a likely example of a regulatory “master switch” gene that influences epigenetic control over a number of key regulatory pathways in obesity regulation. We have recently demonstrated that FTO genotype is not associated with FTO methylation levels [11]. In order to understand how FTO may be associated with TL it is important to consider the role of the FTO gene in the context of its function, regulation and obesity.
FTO has been correlated with metabolic syndrome and diabetes risk [12, 13]. The genetic link to metabolic balance and adiposity homeostasis is important for chronic diseases and also regulation of telomere length [9, 13, 14], since there is an association between shorter telomeres and increased body mass index, increased adiposity, and increasing waist to hip ratio and visceral excess fat accumulation [15]. Additionally, the biochemical abnormalities of obesity, for example, abnormal glycemic and lipidemic profiles, lead to organ dysfunction resembling the accelerated aging process [15]. An emerging hypothesis links obesity, shorter telomeres and accelerated aging [16], however, the manner in which the FTO gene fits into this hypothesis remains to be fully elucidated.
GWAS and FTO associations with obesity
The FTO gene expresses a 505 amino acid protein that shares sequence motifs with Fe(II)- and 2-oxoglutarate (2-OG) dependent dioxygenases [17, 18]. This enzyme family can repair alkylated DNA and RNA by oxidative demethylation, an important mechanistic process in epigenetic regulation of genes [18,19,20,21,22], suggesting that FTO may be involved in DNA/RNA demethylation [21].
FTO has been clearly identified as an obesity associated gene via Genome Wide Association Study (GWAS) [23,24,25]. The application of GWAS led to the understanding that a number of variants in the first intron of the FTO gene may be associated with adiposity, for example, each additional copy of the rs9939609 risk allele is associated with increased BMI of ~0.4 kg/m2 [23,24,25]. Subsequently, at least 75 obesity susceptibility loci [9, 26] have been described across the genome. However, FTO appears to have a greater effect on obesity compared to all other obesity loci and this has been confirmed through replication studies throughout the life span and across ethnicities [27,28,29,30,31,32].
The FTO rs9939609 SNP is the most commonly reported population obesity gene in association studies. The risk allele (A allele) of rs9939609 is associated with greater total energy (food intake), and increased protein and fat intake in children and adults [33,34,35,36]. FTO SNPs, mainly located in intron 1, have been reported to be associated with individual variation in appetite rating scales, loss of control over eating, as well as eating in the absence of hunger [33, 35, 37,38,39,40,41,42,43,44,45,46,47]. It has been observed that children and adolescents that carry the FTO rs9939609 A allele (AA or AT genotype) self-select more energy dense components of a test meal [45, 46]. In adults, the A allele carriers are more likely to demonstrate a loss of control of eating [46], altered postprandial satiety levels [39], increased feeling of hunger [47] and reduced fullness self-ratings [48]. The FTO rs9939609 AA genotype is associated with activity of neural substrates associated with food-cue reactivity [49,50,51].
FTO associations with amino acids
Amino acids play a role in central regulation of food intake - the understanding how amino acids may influence FTO signaling is not well understood. FTO catalyzes the oxidation reaction of methyl DNA/RNA substrates, together with co-factors 2-OG, Fe (II) and oxygen [52]. Early studies presumed that FTO likely functions as an intracellular sensor by assessing the concentration of 2-OG, which is a key intermediate of the citric-acid cycle. Studies using mouse and human cell lines demonstrated that by restricting total amino acids in the medium there was significant downregulation of mRNA and protein expression of FTO in vitro [53], consistent with FTO being a sensor of amino acids, instead of possibly being a 2-OG, within cells [54].
The importance of amino acids are starting to be recognized in metabolic systems, for example, a reduction in leucine in mouse models increases hepatic insulin sensitivity via general control non-depressible 2 (GCN2) /mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathways [55]. These pathways involve nutrient-responsive protein kinases and are important for proper regulation of glucose metabolism in mammals at both the hormonal and cellular level (see below) [56]. Phenylalanine is also a significant circulating amino acid that declines with weight loss [57], although there is a lack of evidence to suggest a direct interaction with FTO pathways. For example, fasting over 72 h results in an increase in phenylalanine and an associated decrease in skeletal mTOR activity and cell growth signaling [58].
FTO signaling pathways
FTO regulates energy sensors in the central nervous system
Global energy sensors in the central nervous system include mammalian target of rapamycin (mTOR), AMP-activated protein kinase (AMPK), and uncoupling protein 2 (UCP2) [59]. Interestingly, these three sensors are all likely to interact with the FTO gene both directly or indirectly. FTO-overexpressed cells are insensitive to amino acid deficiency under the regulation of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway, a major regulator of cell growth and basic catabolic mechanisms [60, 61]. As eluded to above, while there is an association between mTOR and phenylalanine in fasting states, FTO obesity-risk SNPs are reported to affect AKT expression of interacting protein (AKTIP) in an allele-dosage manner by altering the binding site of the transcription factor Cut Like Homeobox 1 (CUX1) [62]. This influences the phosphorylation of AKT’ regulation sites, likely modulating AKT activity via an AKT-AKTIP direct interaction [63].
FTO-downstream mTORC1 influences obesity
The mTORC1 complex itself plays a complex role in obesity. For example, DEP-domain containing mTOR-interacting protein (DEPTOR) functions to suppress mTORC1’s function [64]. Consequently, this inhibits the mTORC1 negative feedback loop, resulting in increased insulin signaling and Akt/PKB activation [64]. Mechanisms underlying FTO expression regulation by amino acids and interactions with mTORC1 are not entirely clear. A primary hypothesis is that FTO is an amino acid (AA) sensor coupling AA levels to mTORC1 [65]. mTORC1 is involved in cell cycle regulation, for example, nutritional signaling through target of rapamycin complex 1 (TORC1) in yeasts permits a cellular check point regulatory choice for a cell to either continue replication in the presence of DNA damage or cell cycle arrest to maintain genetic stability [66]. Furthermore, because of the role of mTORC1 in genetic stability, it is possible that mTORC1 will also have an influence on telomere regulation [67]. FTO SNPs have been clearly associated with greater food intake and increased hunger, but not with decreased resting energy expenditure or low physical activity in human population studies [68]. This paradox of FTO function might be explained by the interaction of FTO with diverse functions of mTORC1.
The metabolism of glucose and glutamine, and primary carbon sources utilized by mitochondria, is directly regulated by mTORC1 [69]. The activity of the mTOR kinase itself, an essential component of mTORC1, is increased by cellular adenosine triphosphate (ATP) levels [70]. Additionally, FTO-variant linked obesity may be associated with altered metabolic functions through activation of downstream metabolic mediators including AMPK [71]. The gastrointestinal tract “hunger hormone”, ghrelin, alters hypothalamic mitochondrial respiration in neurons in an UCP2-dependent manner. This process is driven by a hypothalamic fatty acid oxidation pathway, that involves AMPK, CPT1 and free radical scavenging by UCP2 [72]. Furthermore, mTORC1 activation is associated with increased oxygen consumption [73]. In summary, we hypothesize that global energy sensors, AMPK, UCP2 and mTORC1, are likely to be influenced by FTO gene regulation, supporting the hypothesis that FTO functions in the process of obesity.
IRX 3 and IRX 5 bridge FTO and obesity
An alternate explanation for the discrepancy in FTO SNP polymorphisms and regulation of energy expenditure is via the effect of the obesity-associated FTO region on expression regulation of homeobox gene IRX3 (Iroquois homeobox protein 3) and IRX5. A SNP within intron 1 of the FTO gene alters the expression of IRX3 and IRX5 to regulate adipocyte thermogenesis via influencing adipocyte differentiation [74]. Indeed, Irx3-deficient mice were found to lose 25 to 30% of their weight, due to an increase in basal metabolic rate. The hypothalamic expression of a dominant-negative form of Irx3 reproduces the metabolic phenotypes of Irx3-deficient mice [75], suggesting hypothalamic expression of FTO possibly plays an important role in adiposity regulation via Irx3 pathways.
FTO and hunger-related hormone signaling
Interestingly, mTORC1 is a major intracellular target for hormones and nutrients that regulate food intake and body weight in the hypothalamus [73]. Additionally, hypothalamic CUX1 expression influences leptin receptor trafficking, resulting in altered leptin signaling in mice, which modulates eating behavior [76]. These findings provide more evidence to explain the diverse influence of FTO SNPs on energy balance regulation.
Both mouse Fto and human FTO mRNA are expressed ubiquitously, but FTO expression is higher in the brain and specifically the hypothalamus [18, 77]. This regional distribution for expression is intriguing because the hypothalamus plays a key role in the regulation of both energy balance and control of food intake.
There is a lack of support for the idea that human FTO expression is regulated at the transcriptional level in a leptin-dependent manner. Leptin inhibits hunger, and if it has an effect it appears to be indirect. The relationship between leptin and FTO expression has been explored extensively in rodents, but no clear consensus emerges concerning this relationship. For example, leptin reduces FTO expression in the hypothalamus by activating the STAT3 signaling pathway, in which the Leptin Receptor Long Isoform (LepRb) is also required [78]. Paradoxically, Fto knock-out mice develop features of metabolic syndrome that are normally observed in leptin deficient mice [37]. Mice studies have identified that a fed state is associated with increased hypothalamic Fto mRNA expression, while extended caloric restriction reduces hypothalamic Fto protein expression in Leptin-knockout mice (Lepob) [18, 79]. This latter response is absent in Leptin receptor-mutated mice (Leprdb) [79]. In contrast, Fto expression is increased within the hypothalamus of food-restricted and food-deprived rats [80]. Interestingly, an alternate observation reveals an over-expression of Fto mRNA and protein in the rat hypothalamus due to 48 h fasting [81], whilst 40% reduced Fto expression in rat arcuate nucleus of the hypothalamus increases food intake by 16% [82]. These cumulative findings may be explained by a difference in mRNA versus protein FTO expression, as a consequence of complex FTO gene regulatory mechanisms.
In the transgenic Fto-overexpressing mouse model, greater food intake induces a gene-dose-response increase in body fat mass [83]. Weight gain in humans may be because of an endocrine balance shift from the satiety hormone leptin toward the hunger-promoting hormone ghrelin. Intriguingly, Karra et al. observed rs9939609 AA and TT genotypes showed divergent neural activity in response to circulating ghrelin, which is a key mediator of ingestive behavior [49]. In summary, these data support the concept of fine tuning of hypothalamic neurons to facilitate metabolic regulation by altering their own activities or the activities of upstream/downstream targets in response to hormonal and nutrient signals.
FTO and TL regulation
Fe(II)- and 2-OG dependent dioxygenase family and TL regulation
Fe(II)- and 2-OG dependent dioxygenase family members are involved in diverse processes, including DNA and RNA repair, fatty acid metabolism, posttranslational modifications and demethylation of CpG islands [84, 85]. CpG methylation is associated with gene silencing or CpG demethylation with increased transcription. It is possible that a methylation or demethylation status switch that alters expression of TL regulation-related genes, via 2-OG dependent dioxygenase, is involved in telomere attrition process. The relationship between FTO, a 2-OG dependent dioxygenase, and telomere length regulation is still not elucidated.
One known 2-OG dioxygenase family of proteins that has been associated with regulating TL is the Ten-Eleven-Translocation (TET) proteins, Tet1, Tet2 and Tet3 [86]. Tet proteins act as DNA demethylation enhancers that can influence telomere homeostasis. Functionally, mouse embryonic stem cells deficient for all three Tet proteins (Tet triple knockout) have been shown to exhibit increased telomere-sister chromatid exchange and elongated telomeres [86]. Similar to Tet, the 2-OG dependent dioxygenase catalytic activity of FTO may regulate gene transcription or TL regulation via nucleic acid demethylation. For example, DNA methylation can directly affect transcription factor binding, or indirectly change post-translational histone packaging and modulation of chromatin conformation and function [87, 88]. Identification of epigenetic modifications may aid the exploration of genotype-phenotype interactions in metabolic disease relevant to obesity and telomere regulation. Gerken et al. has made the valid observation that breakdown of genomic repair processes may be associated with susceptibility to obesity and metabolic syndrome [18].
FTO regulates TL indirectly
Interestingly, FTO may influence TL regulation via expression of upstream/downstream flanking genes. For example, FTO rs8050136 (without altering FTO expression) correlates with the expression of retinoblastoma-like 2 protein (Rbl2) gene [89]. The Rbl2 gene is approximately 270,000 base pairs distant from FTO [89]. Rbl2 inhibits Dnmt3a,3b expression, and interestingly this interaction influences telomere regulation, involving significant genomic hypomethylation, including in the subtelomeric regions. Telomeric phenotypes characterized by increased telomere recombination and length are observed [90]. The capacity of Rbl1 and Rbl2 to regulate telomere function has been further explored using mouse embryonic fibroblasts deficient in Rbl1 and Rbl2, where markedly elongated telomeres were observed in the absence of increased telomerase activity and the retention of their end-capping function. Taken together, these data confirm the role of the Rb1 family in the regulation of telomere length in mammalian cells [91], suggesting that FTO may influence mammalian TL regulation processes.
Conclusion
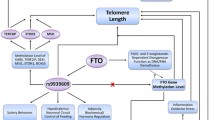
We have previously reviewed the bi-directional interactions between diabetes and short telomeres [16]. We suggest in this review the possibility that FTO genotypes may be associated with genetic aging, i.e. shorter telomeres (Fig. 1). Dlouha et al. reported that carriers of at least one FTO risky (rs17817449 G) allele, is associated with shorter telomeres in middle age women [14]. Additionally, Zhou and colleagues found the relationship between FTO obesity-related risk allele (rs9939609 A) and shorter telomere length only in the high, but not low, FTO methylation levels in non-diabetics [11].

FTO gene interacts with telomere length and obesity. FTO interacts with uncoupling protein 2 (UCP2), AMP-activated protein kinase (AMPK), retinoblastoma-like 2 protein (RBL2), Iroquois homeobox protein 3 (IRX3), cut like homeobox 1 (CUX1) and mammalian target of rapamycin complex 1 (mTORC1). These interactions are important for dietary behavior regulation and cellular nutrient sensing. Additionally, the hypothesis is presented that the FTO genotype may influence telomere regulation. Bold arrow means there is published evidence; dotted arrow means there is rational speculation but without published evidence
Importantly, we have discussed downstream genes and bi-directional feedback loops that influence obesity outcomes across FTO genotypes. FTO feedback loops may in part also be involved in telomere regulation. Certainly we have previously highlighted a role for UCP2 in telomere regulation [92] and currently in this review we relate UCP2 to obesity regulation. Bell et al. identified an FTO obesity susceptibility haplotype is associated with increases in methylation of the FTO gene [88]. Further well-designed and detailed studies in humans and animals are required to explore biochemical and functional roles of FTO genotypes and interactions with FTO epigenetic modification. We suspect these epigenetic FTO interactions with the FTO gene will have modifiable effects on obesity and telomere attrition.
Abbreviations
- 2-OG:
-
2-oxoglutarate
- AA:
-
Amino acid
- AKT/PKB:
-
protein kinase B
- AKTIP:
-
AKT interacting protein
- AMPK:
-
AMP-Activated protein kinase
- ATP:
-
Adenosine triphosphate
- CUX1:
-
Cut like homeobox 1
- DEPTOR:
-
DEP-domain containing mTOR-interacting protein
- FTO:
-
Fat mass and obesity-associated (FTO) gene
- GCN2:
-
General control non-depressible 2
- GWAS:
-
Genome wide association study
- Lepob:
-
Leptin-knockout mice
- LepRb:
-
Leptin Receptor Long Isoform
- Leprdb:
-
Leptin receptor-mutated mice
- mTOR:
-
mammalian target of rapamycin
- mTORC1:
-
mammalian target of rapamycin complex 1
- RPGRIP1L:
-
Retinitis pigmentosa GTPase regulator interacting protein 1 like
- TET:
-
Ten-Eleven-Translocation
- TL:
-
Telomere length
- TORC1:
-
Target of rapamycin complex 1
- UCP2:
-
Uncoupling protein 2
References
Wardle J, Carnell S, Haworth CM, Plomin R. Evidence for a strong genetic influence on childhood adiposity despite the force of the obesogenic environment. Am J Clin Nutr. 2008;87(2):398–404.
Speakman JR, Levitsky DA, Allison DB, Bray MS, de Castro JM, Clegg DJ, Clapham JC, Dulloo AG, Gruer L, Haw S, et al. Set points, settling points and some alternative models: theoretical options to understand how genes and environments combine to regulate body adiposity. Dis Model Mech. 2011;4(6):733–45.
Guzzardi MA, Iozzo P, Salonen MK, Kajantie E, Eriksson JG. Maternal adiposity and infancy growth predict later telomere length: a longitudinal cohort study. Int J Obes. 2016;40(7):1063–9.
Zannolli R, Mohn A, Buoni S, Pietrobelli A, Messina M, Chiarelli F, Miracco C. Telomere length and obesity. Acta Paediatr. 2008;97(7):952–4.
Carulli L, Anzivino C, Baldelli E, Zenobii MF, Rocchi MB, Bertolotti M. Telomere length elongation after weight loss intervention in obese adults. Mol Genet Metab. 2016;118(2):138–42.
Li H, Wu Y, Loos RJ, Hu FB, Liu Y, Wang J, Yu Z, Lin X. Variants in the fat mass- and obesity-associated (FTO) gene are not associated with obesity in a Chinese Han population. Diabetes. 2008;57(1):264–8.
Yao Y, Wen Y, Du T, Sun N, Deng H, Ryan J, Rao S. Meta-analysis indicates that SNP rs9939609 within FTO is not associated with major depressive disorder (MDD) in Asian population. J Affect Disord. 2016;193:27–30.
Elks CE, den Hoed M, Zhao JH, Sharp SJ, Wareham NJ, Loos RJ, Ong KK. Variability in the heritability of body mass index: a systematic review and meta-regression. Front Endocrinol (Lausanne). 2012;3:29.
Loos RJ, Yeo GS. The bigger picture of FTO: the first GWAS-identified obesity gene. Nat Rev Endocrinol. 2014;10(1):51–61.
Valdes AM, Andrew T, Gardner JP, Kimura M, Oelsner E, Cherkas LF, Aviv A, Spector TD. Obesity, cigarette smoking, and telomere length in women. Lancet. 2005;366(9486):662–4.
Zhou Y, Simmons D, Lai D, Hambly BD. McLachlan CS: rs9939609 FTO genotype associations with FTO methylation level influences body mass and telomere length in an Australian rural population. Int J Obes. 2017; doi:10.1038/ijo.2017.127.
O'Neill S, O'Driscoll L. Metabolic syndrome: a closer look at the growing epidemic and its associated pathologies. Obes Rev. 2015;16(1):1–12.
Merkestein M, Sellayah D. Role of FTO in adipocyte development and function: recent insights. Int J Endocrinol. 2015;2015:521381.
Dlouha D, Pitha J, Lanska V, Hubacek JA. Association between FTO 1st intron tagging variant and telomere length in middle aged females. 3PMFs study. Clin Chim Acta. 2012;413(15–16):1222–5.
Tzanetakou IP, Katsilambros NL, Benetos A, Mikhailidis DP. Perrea DN: "is obesity linked to aging?": adipose tissue and the role of telomeres. Ageing Res Rev. 2012;11(2):220–9.
Zhou Y, Ning Z, Lee Y, Hambly BD, McLachlan CS. Shortened leukocyte telomere length in type 2 diabetes mellitus: genetic polymorphisms in mitochondrial uncoupling proteins and telomeric pathways. Clin Transl Med. 2016;5(1):8.
Sebert S, Salonurmi T, Keinanen-Kiukaanniemi S, Savolainen M, Herzig KH, Symonds ME, Jarvelin MR. Programming effects of FTO in the development of obesity. Acta Physiol (Oxf). 2014;210(1):58–69.
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, et al. The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Science. 2007;318(5855):1469–72.
Sanchez-Pulido L, Andrade-Navarro MA. The FTO (fat mass and obesity associated) gene codes for a novel member of the non-heme dioxygenase superfamily. BMC Biochem. 2007;8:23.
Trewick SC, McLaughlin PJ, Allshire RC. Methylation: lost in hydroxylation? EMBO Rep. 2005;6(4):315–20.
Jia G, Yang CG, Yang S, Jian X, Yi C, Zhou Z, He C. Oxidative demethylation of 3-methylthymine and 3-methyluracil in single-stranded DNA and RNA by mouse and human FTO. FEBS Lett. 2008;582(23–24):3313–9.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Dina C, Meyre D, Gallina S, Durand E, Korner A, Jacobson P, Carlsson LM, Kiess W, Vatin V, Lecoeur C, et al. Variation in FTO contributes to childhood obesity and severe adult obesity. Nat Genet. 2007;39(6):724–6.
Frayling TM, Timpson NJ, Weedon MN, Zeggini E, Freathy RM, Lindgren CM, Perry JR, Elliott KS, Lango H, Rayner NW, et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science. 2007;316(5826):889–94.
Scuteri A, Sanna S, Chen WM, Uda M, Albai G, Strait J, Najjar S, Nagaraja R, Orru M, Usala G, et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007;3(7):e115.
Day FR, Loos RJ. Developments in obesity genetics in the era of genome-wide association studies. J Nutrigenet Nutrigenomics. 2011;4(4):222–38.
Lu Y, Loos RJ. Obesity genomics: assessing the transferability of susceptibility loci across diverse populations. Genome Med. 2013;5(6):55.
Jacobsson JA, Schioth HB, Fredriksson R. The impact of intronic single nucleotide polymorphisms and ethnic diversity for studies on the obesity gene FTO. Obes Rev. 2012;13(12):1096–109.
Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Lango Allen H, Lindgren CM, Luan J, Magi R, et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42(11):937–48.
Yang J, Loos RJ, Powell JE, Medland SE, Speliotes EK, Chasman DI, Rose LM, Thorleifsson G, Steinthorsdottir V, Magi R, et al. FTO genotype is associated with phenotypic variability of body mass index. Nature. 2012;490(7419):267–72.
Rzehak P, Scherag A, Grallert H, Sausenthaler S, Koletzko S, Bauer CP, Schaaf B, von Berg A, Berdel D, Borte M, et al. Associations between BMI and the FTO gene are age dependent: results from the GINI and LISA birth cohort studies up to age 6 years. Obes Facts. 2010;3(3):173–80.
Graff M, Ngwa JS, Workalemahu T, Homuth G, Schipf S, Teumer A, Volzke H, Wallaschofski H, Abecasis GR, Edward L, et al. Genome-wide analysis of BMI in adolescents and young adults reveals additional insight into the effects of genetic loci over the life course. Hum Mol Genet. 2013;22(17):3597–607.
Speakman JR, Rance KA, Johnstone AM. Polymorphisms of the FTO gene are associated with variation in energy intake, but not energy expenditure. Obesity (Silver Spring). 2008;16(8):1961–5.
Timpson NJ, Emmett PM, Frayling TM, Rogers I, Hattersley AT, McCarthy MI, Davey Smith G. The fat mass- and obesity-associated locus and dietary intake in children. Am J Clin Nutr. 2008;88(4):971–8.
Steemburgo T, Azevedo MJ, Gross JL, Milagro FI, Campion J, Martinez JA. The rs9939609 polymorphism in the FTO gene is associated with fat and fiber intakes in patients with type 2 diabetes. J Nutrigenet Nutrigenomics. 2013;6(2):97–106.
Lee HJ, Kim IK, Kang JH, Ahn Y, Han BG, Lee JY, Song J. Effects of common FTO gene variants associated with BMI on dietary intake and physical activity in Koreans. Clin Chim Acta. 2010;411(21–22):1716–22.
Do R, Bailey SD, Desbiens K, Belisle A, Montpetit A, Bouchard C, Perusse L, Vohl MC, Engert JC. Genetic variants of FTO influence adiposity, insulin sensitivity, leptin levels, and resting metabolic rate in the Quebec family study. Diabetes. 2008;57(4):1147–50.
Haupt A, Thamer C, Staiger H, Tschritter O, Kirchhoff K, Machicao F, Haring HU, Stefan N, Fritsche A. Variation in the FTO gene influences food intake but not energy expenditure. Exp Clin Endocrinol Diabetes. 2009;117(4):194–7.
den Hoed M, Westerterp-Plantenga MS, Bouwman FG, Mariman EC, Westerterp KR. Postprandial responses in hunger and satiety are associated with the rs9939609 single nucleotide polymorphism in FTO. Am J Clin Nutr. 2009;90(5):1426–32.
Liu G, Zhu H, Lagou V, Gutin B, Stallmann-Jorgensen IS, Treiber FA, Dong Y, Snieder H. FTO variant rs9939609 is associated with body mass index and waist circumference, but not with energy intake or physical activity in European- and African-American youth. BMC Med Genet. 2010;11:57.
Jonassaint CR, Szatkiewicz JP, Bulik CM, Thornton LM, Bloss C, Berrettini WH, Kaye WH, Bergen AW, Magistretti P, Strober M, et al. Absence of association between specific common variants of the obesity-related FTO gene and psychological and behavioral eating disorder phenotypes. Am J Med Genet B Neuropsychiatr Genet. 2011;156B(4):454–61.
Ibba A, Pilia S, Zavattari P, Loche A, Guzzetti C, Casini MR, Minerba L, Loche S. The role of FTO genotype on eating behavior in obese Sardinian children and adolescents. J Pediatr Endocrinol Metab. 2013;26(5–6):539–44.
Roswall N, Angquist L, Ahluwalia TS, Romaguera D, Larsen SC, Ostergaard JN, Halkjaer J, Vimaleswaran KS, Wareham NJ, Bendinelli B, et al. Association between Mediterranean and Nordic diet scores and changes in weight and waist circumference: influence of FTO and TCF7L2 loci. Am J Clin Nutr. 2014;100(4):1188–97.
Cecil JE, Tavendale R, Watt P, Hetherington MM, Palmer CN. An obesity-associated FTO gene variant and increased energy intake in children. N Engl J Med. 2008;359(24):2558–66.
Wardle J, Llewellyn C, Sanderson S, Plomin R. The FTO gene and measured food intake in children. Int J Obes. 2009;33(1):42–5.
Tanofsky-Kraff M, Han JC, Anandalingam K, Shomaker LB, Columbo KM, Wolkoff LE, Kozlosky M, Elliott C, Ranzenhofer LM, Roza CA, et al. The FTO gene rs9939609 obesity-risk allele and loss of control over eating. Am J Clin Nutr. 2009;90(6):1483–8.
Rutters F, Lemmens SG, Born JM, Bouwman F, Nieuwenhuizen AG, Mariman E, Westerterp-Plantenga MS. Genetic associations with acute stress-related changes in eating in the absence of hunger. Patient Educ Couns. 2010;79(3):367–71.
Dougkas A, Yaqoob P, Givens DI, Reynolds CK, Minihane AM. The impact of obesity-related SNP on appetite and energy intake. Br J Nutr. 2013;110(6):1151–6.
Karra E, O'Daly OG, Choudhury AI, Yousseif A, Millership S, Neary MT, Scott WR, Chandarana K, Manning S, Hess ME, et al. A link between FTO, ghrelin, and impaired brain food-cue responsivity. J Clin Invest. 2013;123(8):3539–51.
Kuhn AB, Feis DL, Schilbach L, Kracht L, Hess ME, Mauer J, Bruning JC, Tittgemeyer M. FTO gene variant modulates the neural correlates of visual food perception. NeuroImage. 2016;128:21–31.
Wiemerslage L, Nilsson EK, Solstrand Dahlberg L, Ence-Eriksson F, Castillo S, Larsen AL, Bylund SB, Hogenkamp PS, Olivo G, Bandstein M, et al. An obesity-associated risk allele within the FTO gene affects human brain activity for areas important for emotion, impulse control and reward in response to food images. Eur J Neurosci. 2016;43(9):1173–80.
Gulati P, Yeo GS. The biology of FTO: from nucleic acid demethylase to amino acid sensor. Diabetologia. 2013;56(10):2113–21.
Cheung MK, Gulati P, O'Rahilly S, Yeo GS. FTO expression is regulated by availability of essential amino acids. Int J Obes. 2013;37(5):744–7.
Ma M, Harding HP, O'Rahilly S, Ron D, Yeo GS. Kinetic analysis of FTO (fat mass and obesity-associated) reveals that it is unlikely to function as a sensor for 2-oxoglutarate. Biochem J. 2012;444(2):183–7.
Xiao F, Yu J, Guo Y, Deng J, Li K, Du Y, Chen S, Zhu J, Sheng H, Guo F. Effects of individual branched-chain amino acids deprivation on insulin sensitivity and glucose metabolism in mice. Metabolism. 2014;63(6):841–50.
Engin A. Human protein kinases and obesity. Adv Exp Med Biol. 2017;960:111–34.
Geidenstam N, Magnusson M, Danielsson APH, Gerszten RE, Wang TJ, Reinius LE, Mulder H, Melander O, Ridderstrale M. Amino acid signatures to evaluate the beneficial effects of weight loss. Int J Endocrinol. 2017;2017:6490473.
Vendelbo MH, Moller AB, Christensen B, Nellemann B, Clasen BF, Nair KS, Jorgensen JO, Jessen N, Moller N. Fasting increases human skeletal muscle net phenylalanine release and this is associated with decreased mTOR signaling. PLoS One. 2014;9(7):e102031.
Lopez M, Alvarez CV, Nogueiras R, Dieguez C. Energy balance regulation by thyroid hormones at central level. Trends Mol Med. 2013;19(7):418–27.
Tan VP, Miyamoto S. Nutrient-sensing mTORC1: integration of metabolic and autophagic signals. J Mol Cell Cardiol. 2016;95:31–41.
Gulati P, Cheung MK, Antrobus R, Church CD, Harding HP, Tung YC, Rimmington D, Ma M, Ron D, Lehner PJ, et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc Natl Acad Sci U S A. 2013;110(7):2557–62.
Stratigopoulos G, Burnett LC, Rausch R, Gill R, Penn DB, Skowronski AA, LeDuc CA, Lanzano AJ, Zhang P, Storm DR, et al. Hypomorphism of Fto and Rpgrip1l causes obesity in mice. J Clin Invest. 2016;126(5):1897–910.
Remy I, Michnick SW. Regulation of apoptosis by the Ft1 protein, a new modulator of protein kinase B/Akt. Mol Cell Biol. 2004;24(4):1493–504.
Caron A, Labbe SM, Lanfray D, Blanchard PG, Villot R, Roy C, Sabatini DM, Richard D, Laplante M. Mediobasal hypothalamic overexpression of DEPTOR protects against high-fat diet-induced obesity. Mol Metab. 2016;5(2):102–12.
Gulati P, Avezov E, Ma M, Antrobus R, Lehner P, O'Rahilly S, Yeo GS. Fat mass and obesity-related (FTO) shuttles between the nucleus and cytoplasm. Biosci Rep. 2014:34(5).
Klermund J, Bender K, Luke B. High nutrient levels and TORC1 activity reduce cell viability following prolonged telomere dysfunction and cell cycle arrest. Cell Rep. 2014;9(1):324–35.
Sahin E, DePinho RA. Axis of ageing: telomeres, p53 and mitochondria. Nat Rev Mol Cell Biol. 2012;13(6):397–404.
Speakman JR. The 'Fat mass and obesity Related' (FTO) gene: mechanisms of impact on obesity and energy balance. Curr Obes Rep. 2015;4(1):73–91.
Kim SG, Hoffman GR, Poulogiannis G, Buel GR, Jang YJ, Lee KW, Kim BY, Erikson RL, Cantley LC, Choo AY, et al. Metabolic stress controls mTORC1 lysosomal localization and dimerization by regulating the TTT-RUVBL1/2 complex. Mol Cell. 2013;49(1):172–85.
Cota D, Proulx K, Smith KA, Kozma SC, Thomas G, Woods SC, Seeley RJ. Hypothalamic mTOR signaling regulates food intake. Science. 2006;312(5775):927–30.
Pitman RT, Fong JT, Billman P, Puri N. Knockdown of the fat mass and obesity gene disrupts cellular energy balance in a cell-type specific manner. PLoS One. 2012;7(6):e38444.
Andrews ZB, Liu ZW, Walllingford N, Erion DM, Borok E, Friedman JM, Tschop MH, Shanabrough M, Cline G, Shulman GI, et al. UCP2 mediates ghrelin's action on NPY/AgRP neurons by lowering free radicals. Nature. 2008;454(7206):846–51.
Catania C, Binder E, Cota D. mTORC1 signaling in energy balance and metabolic disease. Int J Obes. 2011;35(6):751–61.
Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, et al. FTO obesity variant circuitry and adipocyte Browning in humans. N Engl J Med. 2015;373(10):895–907.
Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, et al. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014;507(7492):371–5.
Hulea L, Nepveu A. CUX1 transcription factors: from biochemical activities and cell-based assays to mouse models and human diseases. Gene. 2012;497(1):18–26.
Peters T, Ausmeier K, Ruther U. Cloning of fatso (Fto), a novel gene deleted by the fused toes (Ft) mouse mutation. Mamm Genome. 1999;10(10):983–6.
Wang P, Yang FJ, Du H, Guan YF, Xu TY, Xu XW, Su DF, Miao CY. Involvement of leptin receptor long isoform (LepRb)-STAT3 signaling pathway in brain fat mass- and obesity-associated (FTO) downregulation during energy restriction. Mol Med. 2011;17(5–6):523–32.
Stratigopoulos G, Padilla SL, LeDuc CA, Watson E, Hattersley AT, McCarthy MI, Zeltser LM, Chung WK, Leibel RL. Regulation of Fto/Ftm gene expression in mice and humans. Am J Physiol Regul Integr Comp Physiol. 2008;294(4):R1185–96.
Fredriksson R, Hagglund M, Olszewski PK, Stephansson O, Jacobsson JA, Olszewska AM, Levine AS, Lindblom J, Schioth HB. The obesity gene, FTO, is of ancient origin, up-regulated during food deprivation and expressed in neurons of feeding-related nuclei of the brain. Endocrinology. 2008;149(5):2062–71.
Vujovic P, Stamenkovic S, Jasnic N, Lakic I, Djurasevic SF, Cvijic G, Djordjevic J. Fasting induced cytoplasmic Fto expression in some neurons of rat hypothalamus. PLoS One. 2013;8(5):e63694.
Tung YC, Ayuso E, Shan X, Bosch F, O'Rahilly S, Coll AP, Yeo GS. Hypothalamic-specific manipulation of Fto, the ortholog of the human obesity gene FTO, affects food intake in rats. PLoS One. 2010;5(1):e8771.
Church C, Moir L, McMurray F, Girard C, Banks GT, Teboul L, Wells S, Bruning JC, Nolan PM, Ashcroft FM, et al. Overexpression of Fto leads to increased food intake and results in obesity. Nat Genet. 2010;42(12):1086–92.
Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4(3):152–6.
Loenarz C, Schofield CJ. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem Sci. 2011;36(1):7–18.
Lu F, Liu Y, Jiang L, Yamaguchi S, Zhang Y. Role of Tet proteins in enhancer activity and telomere elongation. Genes Dev. 2014;28(19):2103–19.
Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22.
Bell CG, Finer S, Lindgren CM, Wilson GA, Rakyan VK, Teschendorff AE, Akan P, Stupka E, Down TA, Prokopenko I, et al. Integrated genetic and epigenetic analysis identifies haplotype-specific methylation in the FTO type 2 diabetes and obesity susceptibility locus. PLoS One. 2010;5(11):e14040.
Jowett JB, Curran JE, Johnson MP, Carless MA, Goring HH, Dyer TD, Cole SA, Comuzzie AG, MacCluer JW, Moses EK, et al. Genetic variation at the FTO locus influences RBL2 gene expression. Diabetes. 2010;59(3):726–32.
Benetti R, Gonzalo S, Jaco I, Munoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, et al. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nat Struct Mol Biol. 2008;15(9):998.
Garcia-Cao M, Gonzalo S, Dean D, Blasco MA. A role for the Rb family of proteins in controlling telomere length. Nat Genet. 2002;32(3):415–9.
Zhou Y, Simmons D, Hambly BD, McLachlan CS. Interactions between UCP2 SNPs and telomere length exist in the absence of diabetes or pre-diabetes. Sci Rep. 2016;6:33147.
Acknowledgments
NA.
Funding
NA.
Availability of data and materials
Data and materials related to this work are available upon request.
Author information
Authors and Affiliations
Contributions
All authors contributed to this theoretical review, including drafting the manuscript and final refinement.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approve the manuscript for publication.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Y., Hambly, B.D. & McLachlan, C.S. FTO associations with obesity and telomere length. J Biomed Sci 24, 65 (2017). https://doi.org/10.1186/s12929-017-0372-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12929-017-0372-6