
Abstract
Background
Balanced reciprocal chromosomal translocations (RCTs) are the ones of the most common structural aberrations in the population, with an incidence of 1:625. RCT carriers usually do not demonstrate changes in phenotype, except when the translocation results in gene interruption. However, these people are at risk of production of unbalanced gametes during meiosis, as a result of various forms of chromosome segregation. This may cause infertility, non-implantation of the embryo, shorter embryo or foetus survival, as well as congenital defects and developmental disorders in children after birth.
The increasing popularity of cytogenetic molecular techniques, such as microarray-based CGH (aCGH), contributed to the improved detection of chromosomal abnormalities in patients with intellectual disability, however, these modern techniques do not allow the identification of the balanced in potential carriers. Therefore, classical chromosome analysis with GTG technique still plays an important role in the identification of balanced rearrangements in every case of procreation failure.
Case presentation
In this article, a family with multiple occurrences of 17p13.3 duplication syndrome in the offspring and multiple miscarriages resulting from carrying of the balanced reciprocal translocation t(7;17)(p22;p13.2) by proband father is presented.
The aCGH diagnostics allowed the identification of an unbalanced fragment responsible for the occurrence of clinical signs in the female patient, while karyotyping and FISH using specific probes allowed the localization of the additional material in the patient chromosomes, and identified the type of this translocation in the carriers.
Conclusions
Identification of a balanced structural aberration in one of the partners allows direct diagnostics for the exclusion or confirmation of genetic imbalance in the foetus via traditional invasive prenatal diagnostics. It is also possible to use an alternative method, Preimplantation Genetic Diagnosis (PGD) after in vitro fertilization, which prevents initiating pregnancy if genetic imbalance is detected in the embryo.
Similar content being viewed by others

Background
The incidence of balanced, i.e. reciprocal chromosomal translocations, Robertsonian translocations or inversions, in the general population is 0.7% and increases to 4.8% in couples after two miscarriages and to 5.2% in couples after three miscarriages [1]. In couples in which a pregnancy that ended with stillbirth or birth of a child with malformations occurred, the risk of carrying by one of the partners is higher and can reach 16% [2]. Carrying of balanced aberrations usually does not affect the carrier life span or health status. However, unbalanced gametes can be formed during meiotic divisions, which leads to miscarriages, stillbirths or births of disabled children [1, 3]. Development of new techniques of molecular cytogenetics, i.e. array Comparative Genomic Hybridization (aCGH) and Multiple Ligation-dependent Probe Amplification (MLPA), allowed for analysis of small regions of imbalance and for discovery of new genetic syndromes associated with microdeletions or microduplications that were not previously identified using classical cytogenetic methods [3, 4].
Identification of a balanced structural aberration in one of the partners allows direct diagnostics for the exclusion or confirmation of genetic imbalance in the fetus via traditional invasive prenatal diagnostics. It is also possible to use an alternative method, Preimplantation Genetic Diagnosis (PGD) after in vitro fertilization, which prevents initiating pregnancy if genetic imbalance is detected in an embryo [1, 5].
In this article, a family with multiple occurrences of 17p13.3 duplication syndrome in the offspring and multiple miscarriages resulting from carrying of the balanced reciprocal translocation t(7;17)(p22;p13.2) by proband father is presented.
Case presentation
An unrelated couple, woman aged 29 and man aged 33 years, were consulted for preconception guidance at the Genetic Outpatient Clinic before their first pregnancy due to the familial history of miscarriages and psychomotor/intellectual delay. Physical examination of both partners did not reveal any phenotypic abnormalities or chronic diseases.
In the man’s family, three out of four of his adult siblings, his father’ brother, and two out of three children of healthy father’s sister, showed intellectual disability. Moreover, his mother had five spontaneous miscarriages in the first trimester of pregnancies.
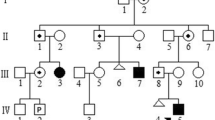
The woman from the consulted couple also had a familial history of intellectual disability; two her siblings were intellectually disabled as well as children of one them. However, the family did not consent to undergo genetic tests (Fig. 1).

Pedigree of a man’s family, a consulted couple. Legend: - white symbol (square or circle) - a healthy person, a symbol (square or circle) with the black dot - carrier of the balanced translocation t(7;17), black symbol (square or circle) - a person with mental retardation. Triangle – miscarriage, black diamond - intrauterine death, − n.t. - not tested genetically, − arrows - consulted couple
Three disabled siblings of the man were consulted in genetic outpatients clinic.
First patient (IV/21) - a man, aged 40 years at the cytogenetic examination, height 176 cm, weight 90 kg, born in the 38th week of pregnancy, weighted 3200 g, obtained Apgar score of 8.
The patient attended a school for children with moderate and severe intellectual disability. At the time of diagnosis he did not read or write, but understood spoken language. Willing to help with household activities.
Second patient (IV/22) - a woman, aged 39 years at the time of cytogenetic examination, height 168 cm, weight 90 kg. Born in the 36th week of pregnancy, weighted 2800 g, obtained Apgar score of 7.
Third patient (IV/25) - a woman, aged 31 years, height 170 cm, weight 70 kg. Born in the 36th week of pregnancy, weighted 2700 g, obtained Apgar score of 7.
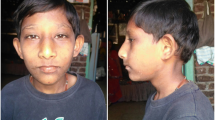
In the these three siblings, excessive hypotonia and delayed psychomotor development were found. They started to sit at 14 and walk at 20 months of age. Sleep disorders and very good memory were observed particularly in the female patients, while all three siblings exhibited development of speech comparable to that of their healthy siblings. Currently, their speech is unclear, vocabulary is poor and sentences are simple. In the female patients, premature puberty occurred, at the age of 9 years, followed by hirsutism remaining to date. Bed-wetting was present until 15 years of age. Both female patients graduated from a special school and can read and write.
In all three disabled siblings psychiatric problems started in early childhood, with serious depression during puberty. In all three, dysmorhpic features are present, i.e. triangular face with slight asymmetry, hypertelorism, down-slanting palpebral fissures, broad nasal root and rounded nasal tip, prognathism, high palate, low-set ears and hypodontia. In all, excessive weight is observed, as well as increasing scoliosis of the Th–L segment. All have shortened 3rd, 4th and 5th toes of both feet. All three also demonstrate higher pain threshold, which leads to frequent, deliberately caused self-injuries.
The material for the study was peripheral blood. Blood cells were cultured for 72 h with PHA, with the culture being carried out and harvested routinely. The cytogenetic slides were stained using the GTG technique, which was supplemented with fluorescent in situ hybridization (FISH). The following molecular probes were used: specific for the critical region of Miller–Dieker syndrome and for the critical region of Smith–Magenis syndrome [MD Miller-Dieker LIS (17p13)/Smith-Magenis RAI (17p11) [Kreatech], specific for TP53 gene (TP53 Deletion Probe, Cytocell), specific for subtelomere of the short arm of chromosome 7 (Subtelomere Specific Probe 7pter, Cytocell) and for subtelomere of the short arm of chromosome 17 (17pter Subtelomere, Leica). The FISH analysis was carried out according to the instructions of manufacturers of the molecular probes used.
GTG analysis was carried out in both partners (IV/27 and IV/34) and in one intellectually disabled sister of the male partner (patient IV/25). Normal karyotypes were found in both partners. In patient IV/25 an addition to the short arm of chromosome 7 was identified by GTG. aCGH examination using a whole-genome oligonucleotide microarray with the mean resolution of 60 kbp revealed a duplication of 4.2 Mbp in chromosome 17 short arm (dup17p13.3p13.2).
Array comparative genomic hybridization (array-CGH) was performed using commercially available array (CytoSure, Constitutional v3 (8x60k), Oxford Gene Technology (OGT), Oxfordshire, UK), according to the manufacturer’s protocol. The CytoSure Interpret Software (OGT) was used for genomic copy-number analysis.
The detected change was confirmed by FISH technique using the probe specific for the critical region of Miller–Diecker syndrome (MDS)(17p13.3—LIS1). The analysis revealed three signals of the MDS critical region and two normal signals of the Smith - Magenis syndrome (SMS) critical region (17p11.2—RAI1). The additional signal from the LIS1 (PAFAH1B1) gene was located on the short arm of chromosome 7 (7p22), which confirmed the presence of an unbalanced t(7;17) in the patient. Her karyotype was established as 46,XX,der(7)t(7;17)(p22;p13.2) (Fig. 2a, b, c). Additionally, a FISH test with the probe specific for the TP53 gene (17p13.1) was done, which revealed the presence of two normal copies of TP53 located on chromosomes 17 (Fig. 2d). The same change of MDS critical region, responsible for 17p13.3 duplication syndrome, was also found in the patient’s disabled brother. The older disabled sister (patient IV/22), presenting similar clinical symptoms, did not consent to genetic testing.

a Karyogram of a patient IV/21 with intellectual disability and a karyotype 46,XY,der(7)t(7;17)(p22;p13.2). The arrow indicates the abnormal chromosome 7. b Metaphase spread of the patient IV/21 stained using FISH technique with a probe specific for the critical region of Miller–Dieker syndrome (17p13.3—LIS1, red) and the critical region of Smith–Magenis syndrome (17p11.2—RAI1, green). Both signals are visible on short arms of both chromosomes 17. An additional, third LIS1 signal is visible on chromosome 7 short arm. The arrow indicates abnormal chromosome 7. c Metaphase spread of the patient IV/21 stained using FISH technique with a probe specific for the subtelomere of chromosome 7 short arm (red) and a probe specific for the subtelomere of chromosome 17 short arm (green). The arrow indicates abnormal chromosome 7 with two red/green signals from chromosome 7 and chromosome 17 subtelomeres. d Metaphase spread of the aforementioned patient (IV/21) stained using FISH technique with a probe specific for TP53 gene (17p13.1, red) and chromosome 17 centromere (green). These two signals are present on both chromosomes 17
After obtaing the results of aCGH and FISH examinations in disabled patients, cytogenetic preparations of proband, his parents and his healthy brother underwent FISH examination with probes specific for the subtelomeres of chromosomes 7 and 17 short arms and for the critical regions of Miller–Dieker syndrome (LIS1) and Smith–Magenis syndrome (RAI1), as well as probes specific for TP53 (17p13.1).
Balanced translocation between chromosomes 7 and 17 was excluded in the proband and confirmed in his father and healthy brother (IV/26)(Fig. 3a, b, c, d).

a Karyogram of the proband father III/13, carrier of the balanced translocation; t(7;17)(p22;p13.2). The arrows indicate abnormal chromosomes 7 and 17. b Metaphase spread of patient III/13 stained by FISH technique with a probe specific for the critical region of Miller–Dieker syndrome (17p13.3—LIS1, red) and the critical region of Smith–Magenis syndrome (17p11.2—RAI1, green). The arrows indicate the abnormal chromosomes 7 and 17. c Metaphase spread of the patient III/13 stained by FISH technique with a probe specific for the subtelomere of chromosome 7 short arm (red) and a probe specific for the subtelomere of chromosome 17 short arm (green). The longer arrow indicates the abnormal chromosome 7 with two signals from chromosome 7 and chromosome 17 subtelomeres. The shouter arrow indicates chromosome 17 without subtelomere signal. d Metaphase spread of the patient III/13 stained by FISH technique with a probe specific for the TP53 gene (17p13.1, red) and chromosome 17 centromere (green). The number and localization of signals are normal
Discussion & conclusions
RCTs are the ones of the most common structural aberrations in the population, with an incidence of 1:625 [1, 2, 5]. They arise from breaks of usually two chromosomes and the subsequent reciprocal lossless transfer of genetic material between these chromosomes. RCT carriers usually do not demonstrate changes in phenotype, except when the translocation results in gene disruption [2, 5].
However, these people are at risk of production of unbalanced gametes during meiosis, as a result of various forms of chromosome segregation. This may cause infertility, non-implantation of the embryo, shorter embryo or fetus survival, as well as congenital defects and developmental disorders in children after birth [2, 5].
The probabilities of different unfavorable pregnancy outcomes for carriers of particular RCTs depend strongly on the type, size, and genetic content of unbalanced chromosomal segments involved in RCT [1, 2, 5].
FISH and aCGH tests carried out in the analyzed family allowed the diagnosis of duplication of chromosome 17 short arm covering the region 17p13.3p13.2 of 4.2 Mbp, containing the region of 17p13.3 duplication syndrome (OMIM 613215) and enabled the identification of asymptomatic carriers in the family as well as the exclusion of the carrier status in the proband.
The presence of high density low copy repeats (LCRs) on chromosome 17 short arm promotes the occurrence of submicroscopic rearrangements [6, 7].
Microduplications in 17p13.3 occur in the same gene region that when deleted causes MDS, therefore this region is sometimes referred to as the MDS critical region. Duplications that cause 17p13.3 microduplication syndrome have various underlying mechanisms, various sizes, and include a variety of genes [3, 4, 8].
The following genes are found in17p13.3 chromosome region, according NCBI database; BHLHA9, YWHAE, MYO1C, INPP5K, PITPNA, SLC43A2, SCARF1, RILP, PRPF8, TLCD2, WDR81, SERPINF2, SERPINF1, SMYD4, RPA1, RTN4RL1, DPH1/OVCA1, OVCA2, HIC1, SMG6, SRR, TSR1, SGSM2, MNT, METTL16, PAFAH1B1 [8] (https://www.ncbi.nlm.nih.gov/gene/). The phenotypes generally associated with microduplications of 17p13.3. include developmental and psychomotor delay, behavioral problems and autism spectrum disorder (ASD), structural brain abnormalitis, and distinct physical features [8].
The deletions and duplications overlapping the PAFAH1B1 and YWHAE genes located in the 17p13.3 region are associated with different clinical phenotypes.
Deletion of only PAFAH1B1 gene causes isolated lissencephaly, while deletion of both of the aforementioned genes causes Miller–Dieker syndrome [6, 9, 10]. Isolated duplication of the PAFAH1B1 gene is associated with mild mental disability and hypotonia, while isolated duplication of the YWHAE gene causes intellectual disability and autism [7, 10].
Although the duplication identified in the proband (patient IV/25) covered a region of 4.2 Mbp, clinical signs such as impairment of psychomotor development, autistic disturbances, dysmorphic face, syndactyly of the 3rd and 4th toes, hypotonia and obesity indicated phenotypic type I of the 17p13.3 duplication syndrome, as proposed by Bruno et al. [10, 11]. According to this classification, type I is characterized by the presence of duplication of the YWHAE gene that has an impact on the development and maturation of neural network. In type II, duplication always comprises the PAFAH1B1 gene and partially also the CRK and YWHAE genes. These changes (type II) are associated with hypotonia, disturbances of psychomotor development, dysmorphy, microcephaly and growth cessation [11, 12]. According to Bruno et al., duplication of the PAFAH1B1 gene with the concurrent duplication or deletion of the YWHAE and CRK genes leads to a milder neurologic phenotype than duplication of the PAFAH1B1 gene alone, which may indicate a possible interaction between the CRK, YWHAE and PAFAH1B1 genes as the key molecular pathways that control the development and migration of cerebral cortex neurons [11,12,13]. Similarly, Bi et al. suggest that dysmorphic face is present in patients with duplication of the YWHAE gene, but not present in patients without its duplication [13]. According to these authors, duplication of the YWHAE gene is also associated with macrosomia, but only if the additional material includes the CRK gene that participates in the regulation of cell growth and differentiation [11, 13].
Previous studies reported patients with intellectual disability as well as with psychomotor development disorder in whom the dup17p13.3 syndrome was diagnosed [8].
Previous studies showed the connection of the microdplication syndrome 17q13.3 with limb defects and cleft palate [14, 15]. In the studies of Klopocki et al., in 30% of patients with split food malformation with long bone deficiency 17p13.3 microduplications were found, encompassing BHLHA9 gene [14]. However, the BHLHA9 is located within chromosome 17p13.3, but immediately outside the MDS critical region [8, 14, 15]. Thus, it can be concluded that the proband (IV/25) carried both genes in double copies.
The proband’s father and healthy brother were diagnosed as carriers of the balanced reciprocal chromosomal translocation t(7;17)(p22;p13.2) associated with the risk of production of gametes with a duplication or deletion of 17p13.3 region. In the case of the proband father, carrying of the translocation resulted in the intellectual disability of three liveborn children and in five miscarriages in his wife. However, due to cytogenetic diagnosis proband’s brother and his partner, who do not have children as yet, and did not suffer from miscarriages, may undergo preimplanatation cytogenetic diagnostics or prenatal cytogenetic diagnostics.
Previous literature reports showed that using new diagnostic techniques such as MLPA, aCGH or NGS allowed for establishing of phenotype-genotype correlations. The use of FISH with commercially available probes to recognize smaller deletions may result in a wrong diagnosis [8, 14, 15].
Our research showes possibilities and difficulties in diagnosing of healthy couple with history of intellectual disability in the family. Whole family involvement and determination of a consulted couple allowed to establish the diagnosis for symptomatic adult sibling.
In standard clinical practice, conventional cytogenetic methods that due to their low resolution do not allow the identification of submicroscopic unbalances, are still frequently used. A more exact characterization of these rearrangements is necessary for the proper genetic diagnosis of a patient and determination of the carrier status and genetic risk in the family members. The increasing popularity of cytogenetic molecular techniques, such as microarray-based CGH (aCGH), contributes to the improved detection in patients with intellectual disability, however, these modern techniques do not allow the identification of the balanced in potential carriers [1, 4, 7]. Therefore, classical chromosome analysis with GTG technique still plays an important role in the identification of balanced rearrangements in every case of procreation failure.
In the case of the consulted couple, family diagnostics was possible after the identification of a small chromosomal abnormality in the proband’s disabled sister (IV/25). The aCGH diagnostics allowed the identification of an unbalanced fragment responsible for the occurrence of clinical signs in the female patient, while karyotyping and FISH using specific probes allowed the localization of the additional material in the patient chromosomes, and identified the type of this translocation in the carriers [1, 2, 5, 8].
Abbreviations
- aCGH:
-
array Comparative Genomic Hybridization
- FISH:
-
Fluorescent in situ Hybridization
- GTG:
-
GTG technique
- LCRs:
-
low copy repeats
- LIS1:
-
Lissencephaly 1
- MDS:
-
Miller - Dieker syndrome
- MLPA:
-
Multiple Ligation - dependent Probe Amplification
- PAFAH1B1:
-
Platelet- Activating Platelet-Activating Factor Acetylhydrolase, Isoform 1B
- PGD:
-
Preimplantation Genetic Diagnosis
- PHA:
-
Phytohemagglutinin
- RAI1:
-
Retinoic Acid -Inducible 1
- RCTs:
-
Reciprocal Chromosomal Translocations
- SMS:
-
Smith - Magenis syndrome
- Th – L:
-
Thoracic and lumbar section of the spine
- TP53:
-
Tumor protein gene
References
De Krom G, Arens YH, Coonen E, Van Ravenswaaij-Arts CM, Meijer-Hoogeveen M, Evers JL, Van Golde RJ, De Die-Smulders CE. Recurrent miscarriage in translocation carriers: no differences in clinical characteristics between couples who accept and couples who decline PGD. Hum Reprod. 2015;30:484–9.
Kwinecka-Dmitriew B, Zakrzewska M, Latos-Bieleńska A, Skrzypczak J. Frequency of chromosomal aberrations in material from abortions. Ginekol Pol. 2010;81:896–901.
Vissers LE, Stankiewicz P, Yatsenko SA, Crawford E, Creswick H, Proud VK, de Vries BB, Pfundt R, Marcelis CL, Zackowski J, Bi W, van Kessel AG, Lupski JR, Veltman JA. Complex chromosome 17p rearrangements associated with low-copy repeats in two patients with congenital anomalies. Hum Genet. 2007;121:697–709.
Roos L, Jonch AE, Kjaergaard S, Taudorf K, Simonsen H, Hamborg-Peterson B, Brondum-Nielsen K, Kurchhoff M. A new microduplication syndrome encompassing the region of the Miller-Dieker (17p13 deletion) syndrome. J Med Genet. 2009;46:703–10.
Wu T, Yin B, Zhu Y, Li G, Ye L, Chen C, Zeng Y, Liang D. Molecular cytogenetic analysis of early spontaneous abortions conceived from varying assisted reproductive technology procedures. Mol Cytogenet. 2016;9:79–84.
Yuan B, Harel T, Gu S, Liu P, Burglen L, Chantot-Bastaraud S, Gelowani V, Beck CR, Carvalho CM, Cheung SW, Coe A, Malan V, Munnich A, Magoulas PL, Potocki L, Lupski JR. Nonrecurrent 17p11.2p12 rearrangement events that result in two concomitant genomic disorders: the PMP22-RAI1 contiguous gene duplication syndrome. Am J Hum Genet. 2015;97:691–707.
Capra V, Mirabelli-Badenier M, Stagnaro M, Rossi A, Tassano E, Gimelli S, Gimelli G. Identification of a rare 17p13.3 duplication including the BHLHA9 and YWHAE genes in a family with developmental delay and behavioural problems. BMC Med Genet. 2012;13:93–9.
Blazejewski SM, Bennison SA, Smith TH, Toyo-Oka K. Neurodevelopmental genetic diseases associated with microdeletions and microduplications of chromosome 17p13.3. Front Genet. 2018;9:1–18.
Outwin E, Carpenter G, Bi W, Withers A, Lupski JR, O'Driscoll M. Increased RPA1 gene dosage affects genomic stability potentially contributing to 17p13.3 duplication syndrome. PLoS Genet. 2011;7:1–13.
Bruno DL, Anderlid BM, Lindstrand A, van Ravenswaaij-Arts C, Ganesamoorthy D, Lundin J, Martin CL, Douglas J, Nowak C, Adam MP, Kooy RF, Van der Aa N, Reyniers E, Vandeweyer G, Stolte-Dijkstra I, Dijkhuizen T, Yeung A, Delatycki M, Borgström B, Thelin L, Cardoso C, van Bon B, Pfundt R, de Vries BB, Wallin A, Amor DJ, James PA, Slater HR, Schoumans J. Further molecular and clinical delineation of co-locating 17p13.3 microdeletions and microduplications that show distinctive phenotypes. J Med Genet. 2010;47:299–311.
Armour CM, Bulman DE, Jarinova O, Rogers RC, Clarkson KB, DuPont BR, Dwivedi A, Bartel FO, McDonell L, Schwartz CE, Boycott KM, Everman DB, Graham GE. 17p13.3 microduplications are associated with split-hand/foot malformation and long-bone deficiency (SHFLD). Eur J Hum Genet. 2011;19:1144–51.
Pilz DT, Dalton A, Long A, Jaspan T, Maltby EL, Quarrell OWJ. Detecting deletions in the critical region for lissencephaly on 17p13.3 using fluorescent in situ hybridisation and a PCR assay identifying a dinucleotide repeat polymorphism. J Med Genet. 1995;32:275–8.
Bi W, Sapir T, Shchelochkov OA, Zhang F, Withers MA, Hunter JV, Levy T, Shinder V, Peiffer DA, Gunderson KL, Nezarati MM, Shotts VA, Amato SS, Savage SK, Harris DJ, Day-Salvatore DL, Horner M, Lu XY, Sahoo T, Yanagawa Y, Beaudet AL, Cheung SW, Martinez S, Lupski JR, Reiner O. Increased LIS1 expression affects human and mouse brain development. Nat Genet. 2009;41:168–77.
Klopocki E, Lohan S, Doelken SC, Stricker S, Ockeloen CW, Soares Thiele de Aguiar R, Lezirovitz K, Mingroni Netto RC, Jamsheer A, Shah H, Kurth I, Habenicht R, Warman M, Devriendt K, Kordass U, Hempel M, Rajab A, Mäkitie O, Naveed M, Radhakrishna U, Antonarakis SE, Horn D, Mundlos S. Duplications of BHLHA9 are associated with ectrodactyly and tibia hemimelia inherited in non-Mendelian fashion. J Med Genet. 2012;49:119–25.
Carter TC, Sicko RJ, Kay DM, Browne ML, Romitti PA, Edmunds ZL, Liu A, Fan R, Druschel CM, Caggana M. Copy-number variants and candidate gene mutations in isolated split hand/foot malformation. J Hum Genet. 2017;62:877–84.
Availability of data and materials
All data generated or analysed during this study are included in this published article [and its supplementary information files].
Author information
Authors and Affiliations
Contributions
MP made substantial contributions to conception and design, acquisition of data and analysis and interpretation of data. MP, OH and KJ been involved in drafting the manuscript or revising it critically for important intellectual content. EŁ, KJ, BN and MBG contributed to sample preparation and worked out almost all of the technical details. All authors discussed the results and contributed to the final manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Research have been performed in accordance with the Declaration of Helsinki and accepted standards of ethics. The consent of the relevant bioethics committee was obtained: Bioethics Committee of Nicolaus Copernicus University in Toruń at the Collegium Medicum im. Ludwik Rydygier in Bydgoszcz. All authors signed a written informed consent for the publication of the article. All patients signed written informed consent for take part in the study.
Consent for publication
All patients signed written informed consent for the publication of their clinical data. For patients with intellectual disabilities, written consent for publication was obtained by the parents/legal guardians.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pasińska, M., Łazarczyk, E., Jułga, K. et al. Multiple occurrence of psychomotor retardation and recurrent miscarriages in a family with a submicroscopic reciprocal translocation t(7;17)(p22;p13.2). BMC Med Genomics 11, 69 (2018). https://doi.org/10.1186/s12920-018-0384-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-018-0384-4