
Abstract
Background
Unbalanced translocations can cause developmental delay (DD), intellectual disability (ID), growth problems, dysmorphic features, and congenital anomalies. They may arise de novo or may be inherited from a parent carrying a balanced rearrangement. It is estimated that 1/500 people is a balanced translocation carrier. The outcomes of different chromosomal rearrangements have the potential to reveal the functional consequences of partial trisomy or partial monosomy and can help guide genetic counseling for balanced carriers, and other young patients diagnosed with similar imbalances.
Methods
We performed clinical phenotyping and cytogenetic analyses of two siblings with a history of developmental delay (DD), intellectual disability (ID) and dysmorphic features.
Results
The proband, a 38-year-old female, has a history of short stature, dysmorphic features and aortic coarctation. She underwent chromosomal microarray analysis, which identified partial monosomy of 4q and partial trisomy of 10p. Her brother, a 37-year-old male, has a history of more severe DD, behavioral problems, dysmorphic features, and congenital anomalies. Subsequently, karyotype confirmed two different unbalanced translocations in the siblings: 46,XX,der(4)t(4;10)(q33;p15.1) and 46,XY,der(10)t(4;10)(q33;p15.1), respectively. These chromosomal rearrangements represent two possible outcomes from a parent who is a carrier for a balanced translocation 46,XX,t(4;10)(q33;p15.1).
Conclusion
To our knowledge, this 4q and 10p translocation has not been described in literature. In this report we compare clinical features due to the composite effects of partial monosomy 4q with partial trisomy 10p and partial trisomy 4q with partial monosomy 10p. These findings speak to the relevance of old and new genomic testing, the viability of these segregation outcomes, and need for genetic counseling.
Similar content being viewed by others

Background
Unbalanced translocations can cause developmental delay (DD), intellectual disability (ID), growth problems, dysmorphic features, and congenital anomalies. They may arise de novo or may be inherited from a parent carrying a balanced rearrangement. It is estimated that 1/500 people is a balanced translocation carrier. The chance of a balanced translocation carrier having a live-born child with an abnormality varies by the specific chromosomal regions affected, but most risk figures range from 0 to 30% [1]. Balanced translocation carriers are typically asymptomatic but have a chance of having offspring with DDs and/or birth defects and an increased risk for miscarriage due to unbalanced segregation [1]. The outcomes of different chromosomal rearrangements have the potential to reveal the functional consequences of partial trisomy or partial monosomy.
The 4q deletion syndrome is a recurrent abnormality with an estimated incidence of 1:100,000 [2]. Features include DD, ID, growth failure, autism spectrum disorders, attention deficit disorders, as well as craniofacial, skeletal, digital, and cardiac anomalies [3,4,5,6,7,8]. On the other hand, duplications of 4q33-4q34 are associated with DD, mild-to-severe ID, growth delay, microcephaly, dysmorphic features, and digital anomalies [9]. Less common features include cardiac malformations, renal anomalies, cryptorchidism, umbilical hernia, and epilepsy. Deletions of 10p15.3 are associated with severe ID, DDs, craniofacial dysmorphism, hypotonia, brain anomalies, seizures, language impairment, and autistic behavior [10, 11]. Duplication of 10p15 has rarely been described in the literature.
The aim of this report is to present two siblings with a history of DD, ID and dysmorphic features, whose G-banded karyotype confirmed different reciprocal unbalanced translocations. These chromosomal rearrangements represent two possible outcomes from a parent who is a carrier of a balanced translocation. To our knowledge, this 4q and 10p translocation has not been described in literature. These findings speak to the viability of these segregation outcomes and can help guide genetic counseling for balanced carriers, and other young patients diagnosed with similar imbalances.
Methods
Participant characteristics
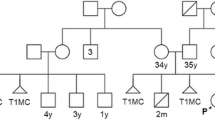
Two siblings with a history of DD, ID and dysmorphic features present for genetic counseling and evaluation. Parents are South Asian and non-consanguineous. Their first-born child is a phenotypically normal female with a reportedly normal karyotype and two healthy children. There was no family history of spontaneous abortions.
Chromosomal microarray analysis (CMA)
CMA was performed on peripheral blood for patient 1 at the UCLA Clinical Microarray Laboratory using clinically validated protocols. A whole genome Single Nucleotide Polymorphism (SNP) oligonucleotide array was used to assess for imbalances in the genomic DNA sample tested. The assay compared patient 1’s DNA to an internal reference and to an external reference from 380 normal controls using the Affymetrix Genome-Wide SNP Array CytoScan™ HD (ThermoFisher Scientific). This array platform contains 2.6 million markers for Copy Number Variant detection, of which 750,000 are genotype SNPs and 1.9 million are non–polymorphic probes, for whole genome coverage. The analysis was performed using the Affymetrix Chromosome Analysis Suite (ChAS) software, version 3.1.0.15 (r9069).
G-banded karyotype and fluorescence in situ hybridization (FISH)
Standard G-banded high-resolution chromosome analysis and metaphase FISH were performed for both patients and their mother at the UCLA Cytogenetics Laboratory following clinically validated protocols. Stimulated peripheral blood cultures were used. Giemsa-banded metaphase cells were analyzed and described according to ISCN 2016. Fluorescence in situ hybridization (FISH) was performed using the Vysis ToTelVysion Probe kit (Abbott Molecular) with probe cocktails specific to chromosomes 4p/q and 10p/q.
Results
Patient 1, a 38-year-old female, was born via vaginal delivery at 36 weeks gestation to a 35-year-old mother and a 42-year-old father. Her birth was complicated by umbilical rupture and bleeding. She had motor and speech delays, attributed to possible anoxic injury at birth. During pre-operative evaluation for dental surgery at the age of 37, electrocardiogram (ECG) showed a low atrial rhythm (negative P waves) and slight slurring of the QRS, consistent with Wolff-Parkinson-White pattern. Transthoracic echocardiogram showed mild coarctation of the aorta. At age 38, she has mild ID, takes the bus independently to volunteer, and attends a two-year community college part-time. Other diagnoses include iron deficiency, migraine without aura, and generalized anxiety disorder. Height is 155 cm (10% percentile). She has hypotelorism, palpebral fissure length 3 cm, mild proptosis, low set ears, midface hypoplasia, thin upper lip, micrognathia, dental crowding with orthodontics, high arched palate, fifth digit brachydactyly, soft tissue syndactyly of the 2–4 toes, levoscoliosis, right knee hyperreflexia, and multiple small nevi.
Patient 2, a 37-year-old male, is the brother of patient 1. He has a history of DD and behavioral difficulties. At 8 months of age he could not sit and started physical therapy. He sat and walked at 3 years. First words were at 4 years of age. He was in special education but mainstreamed by the fourth grade. He had trouble speaking full sentences and received speech and occupational therapy. He was diagnosed with unilateral cryptorchidism at 3 years of age, which was successfully treated with hormone therapy. He exhibits aggressive behavior, treated with quetiapine. At age 37, he attends a day program. On echocardiogram he has a mildly thickened but functionally trileaflet aortic valve that is not clinically significant. He has hirsutism, brachycephaly, triangular head, metopic ridge, synophrys, broad nasal bridge, mild proptosis, ptosis, low set posteriorly rotated ears, micrognathia, short neck with sloping shoulders, pectus excavatum, mild fifth finger clinodactyly, soft tissue syndactyly of the toes, and levoscoliosis.
Patient 1’s CMA showed a loss minimally spanning 19.4 Mb of 4q33→4q35.2 and a gain minimally spanning 5.2 Mb of 10p15.3→10p15.1 (arr[GRCh37] 4q33q35.2(171509635_190957473) × 1,10p15.3p15.1(100026_5313662) × 3) (Fig. 1). The loss on 4q involves 96 curated RefSeq genes, while the gain on 10p involves 38 curated RefSeq genes (gene content is provided in Additional file 1: Table S1). These findings indicate a terminal loss from the long arm of chromosome 4 and a terminal gain involving the short arm of chromosome 10. No additional copy number changes of known clinical significance or long contiguous stretches of homozygosity were identified.

Chromosomal microarray analysis (CMA) for Patient 1. A Whole genome view of CMA data, showing weighted log2 ratio (top panel) and allele difference (bottom panel). These data show a copy number loss on chromosome 4, with a corresponding decrease in allele difference, and a copy number gain on chromosome 10, with a corresponding increase in allele difference. B View of CMA data for the chromosome 4q region, highlighting log2 ration -0.5, decrease in allele difference (two tracks, normal pattern is 3 tracks) and smooth signal of 1 all representing a 19.4 Mb monoallelic terminal loss (red bar). C View of CMA data for the chromosome 10p region, highlighting log2 ration 0.5, increase in allele difference (four tracks tracks) and smooth signal of 3 all representing a 5.2 Mb terminal gain (blue bar). Screenshots were taken from the Chromosome Analysis Suite (ChAS) software from Affymetrix, Inc
Standard chromosome analysis identified an abnormal karyotype, with a derivative chromosome 4 resulting from an unbalanced (4;10) translocation, with breakpoints consistent with those observed by CMA: 46,XX,der(4)t(4;10)(q33;p15.1) (Fig. 2A). Metaphase FISH with subtelomere probes targeting chromosomes 4 and 10 confirmed the unbalanced translocation, with loss of a 4q-specific signal and gain of a 10p-specific signal: ish der(4)t(4;10)(q35.2−,p15.3+)(D4S2930-,10pTEL006+) (Fig. 2B).

Chromosome analysis and fluorescence in situ hybridization (FISH) for Patient 1 and 2. A G-band chromosome analysis for Patient 1 showed one normal copy of chromosome 4, a derivative chromosome 4, and two normal copies of chromosome 10. Standard ideograms are shown next to the normal chromosomes, as well as a hypothetical ideogram for the derivative chromosome 4. The red and blue bars highlight the regions that were found by CMA to be lost/gained. B Metaphase FISH for Patient 1, focusing on the derivative chromosome 4. This chromosome showed a 4p-specific signal but was missing a 4q-specific signal (left). In a separate hybridization, this chromosome was found to hybridize with a 10p-specific FISH probe (right). C Chromosome analysis for Patient 2 showed two normal copies of chromosome 4, one normal copy of chromosome 10, and a derivative chromosome 10. Standard ideograms are shown next to the normal chromosomes, as well as a hypothetical ideogram for the derivative chromosome 10. D Metaphase FISH for Patient 2, focusing on the derivative chromosome 10. This chromosome showed a 10q-specific signal but was missing a 10p-specific signal (left). In a separate hybridization, this chromosome was also found to hybridize with a 4q-specific FISH probe (right). E Chromosome 4 and 10 karyotype of the siblings’ mother
Patient 2 underwent G-banded karyotype and metaphase FISH. These studies found that he also carried an unbalanced (4;10) translocation but with a derivative chromosome 10: 46,XY,der(10)t(4;10)(q33;p15.1) (Fig. 2C). Metaphase FISH confirmed loss of a 10p-specific signal and gain of a 4q-specific signal: ish der(10)t(4;10)(q35.2+,p15.3−)(D4S2930+,10pTEL006−) (Fig. 2D).
Parental studies showed that mother carries a balanced translocation, 46,XX,t(4;10)(q33;p15.1) (Fig. 2E). The father’s karyotype was normal.
Discussion
This report highlights different features associated with two different outcomes of meiotic segregation in a balanced translocation carrier. The mother of our proband was found to carry a balanced (4;10) translocation. The daughter, patient 1, inherited the derivative chromosome 4, resulting in partial monosomy 4q (loss of 19.4 Mb) and partial trisomy 10p (gain of 5.2 Mb), with breakpoints defined by CMA. The son, patient 2, inherited the derivative chromosome 10, resulting in partial trisomy 4q and partial monosomy 10p. While patient 2 did not get a CMA, the breakpoints of his translocation can be inferred based on those identified in his sister, suggesting a 19.4 Mb gain of 4q and a 5.2 Mb loss of 10p. Together, these patients help expand the phenotypic spectrum associated with copy number variation involving 4q and 10p and identify possible candidate genes associated with these phenotypes. The clinical data for our patients compared to individuals reported in the literature and analyzed by CMA is summarized in Table 1.
Regions 4q32.3–q34.3 and 4q33–q34 have been implicated as critical regions accounting for 4q deletion and duplication phenotypes, respectively [8, 9]. Xu et al. reported an 8-month-old male with growth delay, skeletal anomalies, ventricular septal defect, secundum atrial septal defect thickened dysplastic pulmonary valve with stenosis and regurgitation, and a de novo 11.6 Mb deletion of 4q32.3q34.3 [8]. The authors concluded that the region spanning TLL1 (MIM# 606742), HPGD (MIM# 601688), and HAND2 (MIM# 602407) is critical for the development of congenital heart defects in the 4q deletion syndrome (Additional file 1: Table S2). The overlap between their patient’s deletion and that seen in our Patient 1 is approximately 7.0 Mb and involves 31 curated RefSeq genes, including HGPD and HAND2 but not TLL1. HGPD encodes 15-hydroxyprostaglandin dehydrogenase and may play a role in remodeling of the ductus arteriosus [12]. HAND2, heart and neural crest derivatives expressed 2, encodes a helix-loop-helix transcription factor that regulates several cell types during embryonic development [13]. Deletions of HAND2 have been associated with congenital heart defects [2].
Regarding the 4q duplication phenotypes, Thapa et al. proposed 4q33–q34 to be a critical region and suggested that dosage sensitivity of one or both of GLRA3 (MIM# 600421) and GPM6A (MIM# 601275) may be associated with DD and ID, while HAND2 may be critical for craniofacial development [9]. The minimal region of overlap between the two patients reported by Thapa et al. and the predicted gain in our Patient 2 is approximately 7.5 Mb and involves 33 curated RefSeq genes, including GLRA3, GPM6A, and HAND2. GLRA3 encodes the alpha-3 subunit of the neuronal glycine receptor essential for synaptogenesis while GPM6A [14]. GPM6A is a neuronal transmembrane protein that is thought to play a role in neurite growth, neuronal differentiation, and synapse formation. Gregor et al. reported a GPM6A duplication in an individual with learning disability, behavioral problems, and minor facial dysmorphism and studied the impact of dosage alterations of GPM6A on Drosophila melanogaster [15]. Their findings implicated the overexpression of GPM6A in the cognitive phenotype seen in their patient.
A 1.6 Mb region in 10p15.3 has been proposed as a critical region for ID and speech impairment, with two proposed candidate genes, UCN3 (MIM# 605901) and IL15RA (MIM# 601070) [10]. The region of overlap between the proposed 1.6 Mb critical region and the region deleted in our patient is approximately 622 kb but does not include either UCN3 or IL15RA. DeScipio et al., documented 19 individuals with submicroscopic deletions of 10p15.3 and found that ZMYND11 (MIM# 608668) was deleted in all individuals but one and DIP2C (MIM# 611380) was deleted in all but one other individual. They suggested that haploinsufficiency of these genes contributes to the features of the 10p15 deletion phenotype [11]. ZMYND11 has been associated with autosomal dominant ID and both ZMYND11 and DIP2C are expressed in the brain [16]. Both genes were deleted in our patient 2.
Stone et al. reported siblings with a paternally inherited derivative chromosome 9 resulting in a 10p14p15 duplication [17]. Features in this family included dysmorphic features, decreased IgG levels, mild learning problems and eye anomalies. Benzacken et al. described a patient with a de novo 10p14pter duplication presenting with significant hypotonia, DD, agenesis of the corpus callosum, glottic stenosis, and dysmorphic features [18]. In contrast, duplication involving the entire short arm of chromosome 10 has been well described and is associated with dysmorphic features, DD, ID, growth retardation, hypotonia, high-arched/ cleft palate, organ malformations, skeletal abnormalities, and clubfoot [19,20,21].
Translocations between 4q and 10p have previously been described in association with multiple congenital anomalies, but contain different breakpoints than our patients’. One report documented an unbalanced translocation between 4q35 and 10p11.23 resulting in a duplication of 10p11.23pter found in a child with severe failure to thrive, moderate delays, multiple congenital anomalies, facial dysmorphism, and craniosynostosis [22]. Another report identified a cryptic reciprocal translocation resulting in a 4q35 deletion and 10p15 duplication in proband and her maternal aunt who both had ID, dysmorphic features, and immunological symptoms [3]. Shah et al. reported a 4q34.3q35.2 11.5 Mb deletion and 10p15.3p12.1 29.3 Mb duplication in a patient with global DD, severe ID, Marfanoid phenotype, moderate aortic regurgitation due to progressive aortic root dilatation, and a “string of beads” appearance of the femoral artery typical of medial fibroplasia type of fibromuscular dysplasia [23]. The deletion of 4q reported in this patient was slightly larger but overlapping that seen in our patient 1 (breakpoints within 2 Mb of each other), while the 10p duplication was significantly larger (29.3 Mb versus 5.2 Mb).
Finally, these cases illustrate how balanced translocation carriers gives rise to unbalanced translocations in offspring, specifically, how quadrivalent chromosomes of a balanced translocation carrier during meiosis give rise to reciprocal unbalanced translocations (Fig. 3). Accurate and comprehensive family history information assists in diagnosis and tracking of genetic conditions throughout a family. Balanced carriers of chromosomal rearrangements can have reproductive histories that include infertility, recurrent pregnancy loss, fetal anomalies, and offspring with abnormalities. Therefore, collection of a targeted family history can identify individuals with increased risks to carry a chromosomal rearrangement based on their own reproductive history or the history of close relatives. Identification and evaluation of at-risk relatives allows for preconception consideration of various reproductive options, including the use of assistive reproductive technology, and the prevention of added negative psychological stress due to learning of an increased risk for abnormalities during an ongoing pregnancy.

Diagram depicting meiosis of a balanced translocation carrier resulting in reciprocal unbalanced translocations in the offspring. The balanced translocation chromosomes are paired, forming quadrivalent, then segregated during the meiosis I, into two different unbalanced translocations arrangements
Conclusions
To our knowledge, this is the first report of siblings with reciprocal unbalanced translocations between 4q33q35.2 and 10p15.3p15.1. Both have a history of DD, learning problems, and characteristic craniofacial features, with varying degrees of special needs. These findings speak to the viability of these segregation outcomes, their natural history and need of further characterization of CMA findings, and for genetic counseling of carriers of this translocation. Moreover, we compared clinical features due to the composite effects of unbalanced translocations. The outcomes of different chromosomal rearrangements have the potential to reveal the functional consequences of specific partial trisomy or partial monosomy.
Availability of data and materials
The study data has been deposited and made publicly available through dbVar at accession number nstd228 [24].
Abbreviations
- DD:
-
Developmental delay
- ID:
-
Intellectual disability
- CMA:
-
Chromosomal microarray analysis
- ECG:
-
Electrocardiogram
- SNP:
-
Single nucleotide polymorphism
- FISH:
-
Fluorescence in situ hybridization
References
Gardner RJM, Sutherland GR, Shaffer LG. Chromosome abnormalities and genetic counseling. 4th ed. Oxford: Oxford University Press; 2012.
Strehle EM, Yu L, Rosenfeld JA, Donkervoort S, Zhou Y, Chen TJ, et al. Genotype-phenotype analysis of 4q deletion syndrome: proposal of a critical region. Am J Med Genet A. 2012;158A(9):2139–51.
Cingoz S, Bisgaard AM, Bache I, Bryndorf T, Kirchoff M, Petersen W, et al. 4q35 deletion and 10p15 duplication associated with immunodeficiency. Am J Med Genet A. 2006;140(20):2231–5.
Caliebe A, Waltz S, Jenderny J. Mild phenotypic manifestations of terminal deletion of the long arm of chromosome 4: clinical description of a new patient. Clin Genet. 1997;52(2):116–9.
Eggermann K, Bergmann C, Heil I, Eggermann T, Zerres K, Schuler HM. Rare proximal interstitial deletion of chromosome 4q, del(4)(q13.2q21.22): new case and comparison with the literature. Am J Med Genet A. 2005;134A(2):226–8.
Strehle EM. Dysmorphological and pharmacological studies in 4q- syndrome. Genet Couns. 2011;22(2):173–85.
Strehle EM, Bantock HM. The phenotype of patients with 4q-syndrome. Genet Couns. 2003;14(2):195–205.
Xu W, Ahmad A, Dagenais S, Iyer RK, Innis JW. Chromosome 4q deletion syndrome: narrowing the cardiovascular critical region to 4q32.2-q34.3. Am J Med Genet A. 2012;158A(3):635–40.
Thapa M, Asamoah A, Gowans GC, Platky KC, Barch MJ, Mouchrani P, et al. Molecular characterization of distal 4q duplication in two patients using oligonucleotide array-based comparative genomic hybridization (oaCGH) analysis. Am J Med Genet A. 2014;164A(4):1069–74.
Lindstrand A, Malmgren H, Verri A, Benetti E, Eriksson M, Nordgren A, et al. Molecular and clinical characterization of patients with overlapping 10p deletions. Am J Med Genet A. 2010;152A(5):1233–43.
DeScipio C, Conlin L, Rosenfeld J, Tepperberg J, Pasion R, Patel A, et al. Subtelomeric deletion of chromosome 10p15.3: clinical findings and molecular cytogenetic characterization. Am J Med Genet A. 2012;158A(9):2152–61.
Coggins KG, Latour A, Nguyen MS, Audoly L, Coffman TM, Koller BH. Metabolism of PGE2 by prostaglandin dehydrogenase is essential for remodeling the ductus arteriosus. Nat Med. 2002;8(2):91–2.
Tamura M, Amano T, Shiroishi T. The Hand2 gene dosage effect in developmental defects and human congenital disorders. Curr Top Dev Biol. 2014;110:129–52.
Dutertre S, Becker CM, Betz H. Inhibitory glycine receptors: an update. J Biol Chem. 2012;287(48):40216–23.
Gregor A, Kramer JM, van der Voet M, Schanze I, Uebe S, Donders R, et al. Altered GPM6A/M6 dosage impairs cognition and causes phenotypes responsive to cholesterol in human and Drosophila. Hum Mutat. 2014;35(12):1495–505.
Coe BP, Witherspoon K, Rosenfeld JA, van Bon BW, Vulto-van Silfhout AT, Bosco P, et al. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet. 2014;46(10):1063–71.
Stone D, Ning Y, Guan XY, Kaiser-Kupfer M, Wynshaw-Boris A, Biesecker L. Characterization of familial partial 10p trisomy by chromosomal microdissection, FISH, and microsatellite dosage analysis. Hum Genet. 1996;98(4):396–402.
Benzacken B, Lapierre JM, Siffroi JP, Chalvon A, Tachdjian G. Identification and characterization of a de novo partial trisomy 10p by comparative genomic hybridization (CGH). Clin Genet. 1998;54(4):334–40.
Berend SA, Shaffer LG, Bejjani BA. Pure trisomy 10p involving an isochromosome 10p. Clin Genet. 1999;55(5):367–71.
Clement SJ, Leppig KA, Jarvik GP, Kapur RP, Norwood TH. Trisomy 10p: report of an unusual mechanism of formation and critical evaluation of the clinical phenotype. Am J Med Genet. 1996;65(3):197–204.
Kohannim O, Peredo J, Dipple KM, Quintero-Rivera F. Clinical findings associated with a de novo partial trisomy 10p11.22p15.3 and monosomy 7p22.3 detected by chromosomal microarray analysis. Case Rep Genet. 2011;2011:131768.
Wiktor A, Feldman GL, Kratkoczki P, Ditmars DM Jr, Van Dyke DL. 10p duplication characterized by fluorescence in situ hybridization. Am J Med Genet. 1994;52(3):315–8.
Shah P, Dawson A, Tam J, Semaniuk N, Hovanes K, Bailey K, et al. Distal trisomy 10p and 4q monosomy: associated with marfanoid features, syndromic aortic root dilatation and intellectual disability. J Clin Case Stud. 2016;1(1).
Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, et al. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res. 2013;41(Database issue):D936–41.
Acknowledgements
We thank the patients and their family for their willingness to contribute to this report.
Funding
Funding was provided by Center for Scientific Review (Grant Nos. K08HL133491, R03HL157012).
Author information
Authors and Affiliations
Contributions
JF and JW conceived and designed the study. JYL, MB and JAM-A acquired longitudinal history, performed detailed phenotype assessment, and drafted the clinical summary. NS and FQ-R analyzed and interpreted the molecular pathology studies and drafted the results and figures. JF summarized similar translocations in the literature and drafted the manuscript. JW substantially revised the manuscript. All authors approved the final manuscript and greed to be personally accountable for the author’s own contributions and to ensure that questions related to the accuracy or integrity of any part of the work, even ones in which the author was not personally involved, are appropriately investigated, resolved, and the resolution documented. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The UCLA Office of the Human Research Protection Program has determined that ethics approval for case reports are exempt from IRB review based on 45 CFR 46.104(d)(4).
Consent for publication
Written informed consent for publication of their clinical details was obtained from the patients. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1. Table S1.
Lists of genes contained within the genomic imbalances. Table S2. Genes that may contribute to the patients’ phenotypes.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Fan, J., Senaratne, T.N., Liu, J.Y. et al. Outcomes of two different unbalanced segregations from a maternal t(4;10)(q33;p15.1) translocation. BMC Med Genomics 16, 65 (2023). https://doi.org/10.1186/s12920-023-01491-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-023-01491-1