
Abstract
Background
Recently, copy number alteration (CNA) of 9p24.1 were demonstrated in 10% of diffuse large b-cell lymphoma (DLBCL), with gene expression and mutation profiles that were similar to those of primary mediastinal large B-cell lymphoma (PMBCL). However, their CNA-based profile and clinical impact still remain unclear.
Methods
Multiplex ligation-dependent probe amplification were employed to investigate the prevalence of JAK2/PD-L2 amplification in DLBCL and their CNA-based pattern of driver genes. The clinical outcome and characteristics were also analyzed.
Results
Using unsupervised hierarchical clustering, a small group of DLBCL (10.5%, 8/76) was clustered together with PMBCL as Cluster_2, demonstrating amplification of JAK2 (100%,8/8) and PD-L2 (75.0%,6/8). This subgroups of DLBCL demonstrated significant higher expression of PD-L1 than those with MYD88 L265P mutation(p = 0.024). And they exhibited dismal OS and PFS as compared with DLBCL_others(p = 0.003 and 0.001, respectively), which is similar to DLBCL with MYD88 L265P mutation.
Conclusions
DLBCL with amplification of JAK2/PD-L2 exhibits CNA pattern that is similar to PMBCL, and demonstrates unfavorable clinical outcome that resembles those with MYD88 L265P mutation. It is essential to identify this subgroup of DLBCL who may acquire more benefits from the JAK2 and PD-L1 signaling inhibition.
Similar content being viewed by others


Background
Diffuse large B-cell lymphoma (DLBCL) is a highly heterogeneous disease. Recently, several distinctive genetic subtypes were identified, including Schmitz R et al. study (MCD, BN2, N1 and EZB subtypes) and Chapuy B et al. study (C0 ~ C5 clusters) [1, 2]. Godfrey J et al. study also identified an unique biological subset of DLBCL with PD-L1 gene alterations, having high risk features [3]. Thus, the genetics of DLBCL relating to potential therapeutic targets for immune checkpoint inhibitors should be paid much more attention to.
Janus kinase 2 (JAK2), programmed cell death 1 ligand 1 (PD-L1/CD274/PDCD1LG1) and programmed cell death 1 ligand 2 (PD-L2/CD273/PDCD1LG2) are adjacent to each other on chromosome 9p24.1, playing key roles in host immune surveillance [4]. Amplification of 9p24.1 were frequently seen in cell lines of classical Hodgkin lymphoma (cHL) (100%, 6/6) and primary mediastinal large B-cell lymphoma (PMBCL) (100%,1/1), but much less in DLBCL cell lines (22%, 4/18, 4]. Correspondingly, PD1 ligands (PD-L1 and PD-L2) transcripts and proteins were more abundant in cHL and PMBCL cell lines than that in DLBCL cell lines [4]. Recently, Y Wang et al. study demonstrated that 10% of DLBCL had copy number alteration (CNA) of 9p24.1, with a gene expression and mutation profile similar to those of PMBCL [5]. However, their CNA-based profile and clinical impact still remain unclear.
Therefore, in this study, we employed multiplex ligation-dependent probe amplification (MLPA) to investigate the prevalence of JAK2/PD-L2 amplification in DLBCL, and their CNA-based pattern of driver genes, including BCL2, CDKN2A and TP53 [6]. And we analyzed their long-term survival outcome after treatment of R-CHOP-like regime.
Methods
Case selection
We collected consecutive cases of DLBCL and PMBCL in our clinical FFPE archives of excisional biopsy database between Jan 2009 and Oct 2010. And 77 cases of DLBCL and 4 cases of PMBCL were found. After confirmation, one case of DLBCL was diagnosed as PMBCL. Thus, 76 cases of DLBCL and 5 cases of PMBCL were acquired finally (see Additional file 1). All patients were diagnosed at National Cancer Center/National Clinical Research Center for Cancer/Cancer Hospital, Chinese Academy of Medical Sciences and Peking Union Medical College according to the revised 4th edition of the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues [7]. The data regarding treatment and prognosis were acquired by means of medical record consultation and telephone conversation.
Multiplex ligation-dependent probe amplification (MLPA)
Genomic DNA were extracted from formalin-fixed, paraffin-embedded (FFPE) blocks using QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA). Then DNA copy number quantification and MYD88 L265P mutation were detected using MLPA kit (MRC-Holland, Netherlands). The PCR products were detected on an ABI 3500 Genetic Analyzer (Applied Biosystems, USA), and the final result were analyzed using Coffalyser 9.4 software. The relative peak ratio (PRR) of probe larger than 1.3 was defined as amplification, and less than 0.7 was defined as deletion (see Additional file 2). Genes which had two or more probes covering two different exomes were put into final analysis, including JAK2, PD-L2, MDM2, REL, PUS10, BCL2, NFATC1, SPIB, FOXP1, NFKBIZ, BCL6, PRDM1, TNFAIP3, CDKN2A, PTEN, ING1 and TP53 [6]. The details of MLPA probes of driver genes in DLBCL are shown in the online supporting material (see Additional file 3). True amplification of one gene was regarded only when all probes of this gene exhibited amplification, and vice versa (see Additional file 2).
MYD88 L265P mutation was identified when the probe had a high peak. MYD88 wildtype didn’t show any peak (see Additional file 2).
Immunohistochemistry (IHC) staining of PD-L1(22C3)
IHC staining was performed on Dako Autostainer Link 48 (ASL48) platform. Each FFPE block were cut at a thickness of 4-μm, and then deparaffinized. Antigen retrieval were performed using the EnVision™ FLEX Target Retrieval Solution at low pH 6.0. Monoclonal PD-L1 (clone 22C3, Dako) were used as primary antibody, followed by incubation with EnVision™ FLEX+ Mouse LINKER, and then EnVision™ FLEX HRP reagent. Finally, the IHC was visualized by EnVision™ FLEX DAB. Each IHC slide contained a positive control (Lung carcinoma).
IHC score of PD-L1 were calculated by multiplying the percentage of positive cells with mean intensity (0, no staining, 1, weak staining, 2, moderate staining; 3, strong staining), which was reported in previous study [5]. The results were evaluated by an experienced hematopathologist (Xuemin).
Statistical analysis
The differences of clinicopathological characteristics among different groups were analyzed using Chi-square test, Fisher exact test or Kruskal-Wallis rank sum test. PD-L1 IHC score between different groups was analyzed using Wilcoxon test. Overall survival (OS) and progress-free survival (PFS) times were defined from the date of pathologic diagnosis to the date of the event or the last follow-up. The hazard ratio (HR) of each parameter was calculated by univariate Cox proportional regression analysis firstly, in which parameters with p < 0.05 were evaluated together using multivariate Cox proportional regression analysis. The survival curve were made according to Kaplan–Meier procedure. The day of last follow-up was March 1st 2019. All statistical analysis were two sided and p < 0.05 was defined as significance.
Unsupervised hierarchical clustering was carried out using Euclidean distance and complete method. Heatmap was plot using pheatmap package.
All above statistical analyses were run in R 3.4.1 statistic software.
Results
Unsupervised hierarchical clustering of CNAs of driver genes and its survival analysis in DLBCL and PMBCL patients
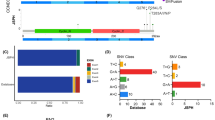
Based on array CGH, Lenz G et al. study previously identified specific CNAs in PMBCL which were different from ABC and GCB of DLBCL [6]. ABC DLBCLs often have CNAs in FOXP1, NFKBIZ, CDKN2A, CDKN2B, INF4a, BCL2, NFATC1 and SPIB, while GCB DLBCLs frequently harbor CNAs in REL, PTEN, MDM2, MIHG1 and ING1. PMBCL often demonstrate CNAs of JAK2 and PD-L2 [6]. Using unsupervised hierarchical clustering, we explored the CNA-based pattern of these genes in DLBCL and PMBCL. The result showed that a small group of DLBCL (10.5%, 8/76) was clustered together with PMBCL as Cluster_2, with amplification of JAK2 (100%,8/8) and PD-L2 (75.0%%,6/8)(Fig. 1a). This subgroup of DLBCL occurred at the site of cervical lymph node (3 cases), gastrointestinal tract (3 cases), nasal cavity (1 case) and spleen (1 cases) (Fig. 1a, Table 1)(Additional File 4). The frequency of JAK2 and PD-L2 amplification in the whole cohort of DLBCL were 10.5% (8/76) and 7.9% (6/76), while both of them were 100% in PMBCL (Fig. 1a) (see Additional file 1). Meanwhile, all cases in Cluster_3 harbored amplification of NFKBIZ which is essential for NF-κB activation in ABC DLBCL [10], but no amplification of NFKBIZ was found in Cluster_1.

Heatmap and survival analysis based on unsupervised hierarchical clustering, and status of JAK2/PD-L1 amplification and MYD88 mutation in TCGA dataset. a, Heatmap of CNA-based profiles of driver genes in DLBCL and PMBCL by using unsupervised hierarchical clustering. b, Survival curves and cox-regression analysis of OS and PFS among three CNA-based clusters after RCHOP-like treatment. c, Status of amplifications of JAK2, PD-L1(CD274) and PD-L2(PDCD1LG2), and mutation of MYD88 in DLBCL (TCGA, PanCancer Atlas) from cBioPortal [8, 9]
As to survival, DLBCL in Cluster_2 demonstrated significant worse OS (p = 0.016) and PFS (p = 0.008) as compared with DLBCL in Cluster_1(Fig. 1b). However, Cluster_1 and Cluster_3 didn’t reveal significant difference in survival (Fig. 1b). We also analyzed the OS and PFS between DLBCL with and without JAK2/PD-L2 amplification, and got statistical significance (see Additional file 5).
Of note, we found that DLBCL in Cluster_2 enriched for JAK2/PD-L2 amplification had less frequency of MYD88_L265P mutation (12.5%, 1/8) (Fig. 1a, Table 4), which was supported by the Cancer Genome Atlas data (TCGA, PanCancer Atlas) from cBioPortal [8, 9] (Fig. 1c).
JAK2/PD-L2 amplification identify a distinctive CNA-based pattern of DLBCL similar to that of PMBCL
Since DLBCL with JAK2/PD-L2 amplification had less frequency of MYD88 L265P mutation, our study separated DLBCL patients into three subgroups: DLBCL with JAK2/PD-L2 amplification (DLBCL_JAK2/PD-L2_amp), DLBCL with MYD88 L265P mutation (DLBCL_MYD88_L265P), and DLBCL without JAK2/PD-L2 amplification nor MYD88_L265P mutation (DLBCL_others) (Fig. 2a). Based on the unsupervised cluster result (Fig. 1a), one patient who had both JAK2/PD-L2 amplification and MYD88 L265P mutation was clustered into Cluster_2. Therefore, this patient was put into DLBCL_JAK2/PD-L2_amp subgroup accordingly. We also analyzed the data when this case was included in DLBCL_MYD88_L265P subgroup, and got the similar result (see Additional file 6).

Comparison of CNA-based pattern, PD-L1 expression and survival analysis among PMBCL and three subgroups of DLBCL. a, Comparison of CNA-based patterns of driver genes among PMBCL and three subgroups of DLBCL according to the status of JAK2/PD-L2 amplification and MYD88 L265P mutation. b, Comparison of PD-L1 expression (IHC score) among PMBCL and three subgroups of DLBCL. c, Survival curves and cox-regression analysis of OS and PFS among three subgroups of DLBCL after RCHOP-like treatment. d, Representative images of HE(× 200) and PD-L1(× 200) IHC in DLBCL_JAK2/PD-L2_amp and DLBCL_ MYD88_L265P
Unlike DLBCL_MYD88_L265P and DLBCL_others, DLBCL_JAK2/PD-L2_amp showed a distinctive pattern similar to that of PMBCL, with high frequency of REL and NFKBIZ amplifications, but no amplification of BCL2 and NFATC1 and no deletion of PRDM1 was found (Fig. 2a).
With respect to clinicopathologic characteristics, DLBCL_JAK2/PD-L2_amp tend to be younger than DLBCL_MYD88_L265P (p = 0.003) (Table 2). While, Hans model, international prognostic index (IPI) risk category and MYC break-apart didn’t show any significant differences (Table 2).
PD-L1 expression in DLBCL with JAK2/PD-L2 amplification was significantly higher than that in DLBCL with MYD88 L265P mutation
Totally, 32 cases were performed PD-L1 (22C3) IHC detection, including DLBCL_MYD88_L265P (14 cases), DLBCL_JAK2/PD-L2_amp (8 cases), DLBCL_others (5 cases) and PMBCL (5 cases). The result showed that PD-L1 expression in DLBCL_JAK2/PD-L2_amp was significantly higher than that in DLBCL_MYD88_L265P (p = 0.024) and DLBCL_others (p = 0.037) (Fig. 2b and d). While no significant difference was found between DLBCL_JAK2/PD-L2_amp and PMBCL (p = 0.768) (Fig. 2b).
JAK2/PD-L2 amplification identify a subgroup of DLBCL with unfavorable survival outcome similar to that of MYD88 L265P mutation
Trying to explore the survival indication of JAK2/PD-L2 amplification and MYD88 L265P mutation, 49 cases of DLBCLs who received RCHOP-like regiment with or without surgical resection were enrolled to performed cox proportional regression analysis of OS and PFS. The median follow-up time was 108 months (range, 3–192 months).
In the univariate analysis, as compared with DLBCL_others, DLBCLs with MYD88 L265P mutation had significantly worse OS and PFS (p = 0.025 and 0.007, respectively), and the same to DLBCLs with JAK2/PD-L2 amplification (p = 0.003 and 0.001, respectively). Meanwhile, IPI risk category were significantly associated with OS and PFS (Fig. 2c, Tables 3 and 4).
In the multivariate analysis, IPI risk category and three subgroups of DLBCL were put into analysis. As compared with DLBCL_others, DLBCL with MYD88 L265P mutation still showed poor OS and PFS (p = 0.005 and 0.001, respectively), and the same to DLBCL with JAK2/PD-L2 amplification for PFS and OS (p = 0.006 and 0.001, respectively). Meanwhile, IPI risk category was still an independent risk predictors for OS and PFS (Fig. 2c, Tables 3 and 4).
Either JAK2/PD-L2 amplification or MYD88 L265P mutation are frequently seen in relapse/refractory DLBCL with PFS less than 2 years
DLBCL with PFS less than 2 years was defined as primary relapse/refractory cases. Among these cases who treated by RCHOP-like regime, the frequency of JAK2 and PD-L2 amplification were 20%(3/15) and 13.3% (2/15). Meanwhile, the frequency of MYD88 L265P mutation were 33.3% (5/15). DLBCL with either JAK2/PD-L2 amplification or MYD88 L265P accounted for 46.7% (7/15).
Discussion
DLBCL presents with a wide spectrum of genetic aberration. Recently, Shi et al. study exhibited PD-L2 amplification in 75% PMBCL and 0% of DLBCL [11]. Chapuy et al. demonstrated 15% of 9p24.1 amplification in DLBCL [2]. Meanwhile, DLBCL with PD-L1 gene alterations was identified as a unique biological subgroup, having high risk features [3]. Y Wang et al. study demonstrated that 10% of DLBCL had CNA of 9p24.1, with gene expression and mutation profiles that were similar to those of PMBCL [5]. In our study, by using unsupervised hierarchical clustering, 10.5% (8/76) cases of DLBCL were clustered together with PMBCL as Cluster_2, indicating that they shared recurrent CNAs. They were enriched for JAK2 amplification and PD-L2 amplification (Fig. 1a).
Using Hans model, most of DLBCL in Cluster_2 were non-GCB (75%, 6/8), and tend to be younger than other groups of DLBCL (Table 2), which was consistent with prior study [5]. Therefore, coupled with Y Wang et al. study, we confirmed that DLBCL with JAK2/PD-L2 amplification is a unique subgroup resembling the PMBCL with respect to CNA pattern.
With regard to survival, increasingly data exhibited that the suppression of immune surveillance in DLBCL was associated with poor survival. Godfrey J et al. study has demonstrated that DLBCL with PD-L1 gene alterations showed high risk features [3]. Meta-analysis also showed that PD-L1 expression was associated with poor OS and adverse clinicopathologic features in DLBCL [12].
In Y Wang et al. study, 10% of DLBCL harbored CNA of 9p24.1, of which 65% were gains and 35% were amplifications [5]. And, as compared with those who have no gain of 9p24.1, DLBCL with 9p24 amplification had a trend of better EFS, while patients with only gain tend to have worse prognosis [5]. Unfortunately, they didn’t show any statistical significance [5]. In our study, 10.5% (8/76) of DLBCL were found that had CNA of JAK2. When JAK2 CNA was separated into gain (MLPA value between 1.7–2.0) and amplification (MLPA value > 2.0) as described [13], 50%(4/8) cases in DLBCL_JAK2/PD-L2_amp group were found that had JAK2 gain, which was slightly lower than that in Wang J et al. study [5] (as shown in Additional File 7). And both DLBCL with JAK2 gain and with amplification demonstrated significant poor prognosis as compared with rest of DLBCL (as shown in Additional File 7). More interesting, unlike Y Wang et al. study [5], 5 cases of PMBCL were included in our study as control, all of which demonstrating JAK2 gains, rather than amplifications (as shown in Additional File 7).
Of note, we found that DLBCL in Cluster_2 enriched for JAK2/PD-L2 amplification had less frequency of MYD88_L265P mutation (12.5%, 1/8) (Fig. 1a, Table 4), which was supported by the Cancer Genome Atlas data (TCGA, PanCancer Atlas) from cBioPortal [8, 9] (Fig. 1c).
MYD88 L265P is a poor indicator of survival for DLBCL [14] which may lead to primary refractory/relapsed disease. This is a gain-of-function driver mutation, occurring in 10% ~ 24% of DLBCL, but absent in PMBCL [14,15,16]. In our study, the frequency of MYD88 L265P in DLBCL and PMBCL were 21.1%(16/76) and 0%(0/5), which were in line with prior studies [14,15,16]. Of great interest, MYD88 L265P mutation occurred less frequently in Cluster_2 (12.5%, 1/8), which was supported by the data (TCGA, PanCancer Atlas) from cBioPortal [8, 9]. Thus, when we divided DLBCL patients into three subgroups (DLBCL_JAK2/PD-L2_amp, DLBCL_MYD88_L265P and DLBCL_others, both DLBCL_JAK2/PD-L2_amp and DLBCL_MYD88_L265P demonstrated dismal OS and PFS with a median follow-up of 9 "years, as compared with DLBCL_others. Therefore, DLBCL with JAK2/PD-L2 amplification was identified as a poor survival subgroup that is similar to DLBCL with MYD88 L265P mutation.
Meanwhile, we also compared the CNA patterns of driver genes among DLBCL_JAK2/PD-L2_amp, DLBCL_MYD88_L265P, DLBCL_others and PMBCL. DLBCL_JAK2/PD-L2_amp showed a distinctive pattern similar to PMBCL, with high frequency of REL and NFKBIZ amplifications, but no amplification of BCL2 and NFATC1 and no deletion of PRDM1 was found. The profile of DLBCL_MYD88_L265P was closed to DLBCL_others, showing relatively high frequency of CDKN2A deletion, NFATC1 amplification and BCL2 amplification.
In our study, 75.0%(6/8) of DLBCL_JAK2/PD-L2_amp harbored both JAK2 and PD-L2 amplifications simultaneously, indicating that they may also have the PD-L1 amplification, because PD-L1 located in the middle of JAK2 and PD-L2 at 9p24.1. Thus, we hypothesized that PD-L1 expression would be upregulated in this subgroup. As what we expected, using PD-L1 (22C3) IHC detection, PD-L1 expression in DLBCL_JAK2/PD-L2_amp was significantly higher than that in DLBCL_MYD88_L265P (p = 0.024) and DLBCL_others (p = 0.037) (Fig. 2b and d), but not in PMBCL (p = 0.768) (Fig. 2b). Meanwhile, PD-L1 expression could be enhanced not only by PD-L1 amplification but also by JAK2 activation [4, 17]. Therefore, DLBCL with JAK2/PD-L2 amplification was confirmed as an unique subtype that is different from DLBCL with MYD88 L265P and others.
Objective response rates (ORR) of PD-1 blockade therapy was 10–36% in unselected patients with relapsed/refractory DLBCL [18, 19]. The wide spectrum of ORR may be due to high heterogeneity of this subgroup. Ansell SM et al. study demonstrated 19% patients with 9p24.1 alteration in relapsed/refractory DLBCL [18]. In our cohort, the frequency of JAK2 and PD-L2 amplification in relapsed/refractory DLBCL were 20% and 13.3%, which were within the range of ORR in the prior studies [18, 19]. While, 33.3% (5/15) patients were found that had MYD88 L265P mutation who may not be suitable for anti-PD-1 therapy. Thus, the genetic analysis in refractory/relapsed DLBCL is required for future therapy selection to increase the ORR of immune checkpoint inhibitors.
JAK2 amplification could augment the expression of itself and PD-1 ligands (PD-L1 and PD-L2), enhancing the sensitivity to JAK2 kinase inhibitor [4]. Chemical JAK2 inhibition could reduce the RNA transcription and protein expression of PD-L1 [20]. Thus, selective inhibition of JAK2 would be a valuable complementary therapy for PD-L1 blockade.
Conclusions
DLBCL with amplification of JAK2/PD-L2 exhibits PMBCL-like CNAs pattern, and demonstrates unfavorable outcome resembling those with MYD88 L265P mutation. Thus, it is essential to identify this subgroup of DLBCL who may acquire more benefits from the JAK2 and PD-L1 signaling inhibition, and JAK2 amplification detection by MLPA would be feasible in routine practice. Meanwhile, the difference of survival outcome between our study and Wang J et al. study indicated that PMBCL-like DLBCL suggested by 9p24.1 CNA could be an intermixed subgroup, which required further exploration.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- DLBCL:
-
Diffuse large B-cell lymphoma
- PMBCL:
-
Primary mediastinal large B-cell lymphoma
- MLPA:
-
Multiplex ligation-dependent probe amplification
- TCGA:
-
The Cancer Genome Atlas
- IPI:
-
International Prognostic Index
- FFPE:
-
Formalin-fixed, paraffin-embedded
- OS:
-
Overall survival
- PFS:
-
Progress-free survival
- HR:
-
Hazard ratio
References
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396–407.
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, Lawrence MS, Roemer MGM, Li AJ, Ziepert M, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679–90.
Godfrey J, Tumuluru S, Bao R, Leukam M, Venkataraman G, Phillip J, Fitzpatrick C, McElherne J, MacNabb BW, Orlowski R, et al. PD-L1 gene alterations identify a subset of diffuse large B-cell lymphoma harboring a T cell-inflamed phenotype. Blood. 2019;133(21):2279–90.
Green MR, Monti S, Rodig SJ, Juszczynski P, Currie T, O'Donnell E, Chapuy B, Takeyama K, Neuberg D, Golub TR, et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116(17):3268–77.
Wang Y, Wenzl K, Manske MK, Asmann YW, Sarangi V, Greipp PT, Krull JE, Hartert K, He R, Feldman AL et al: Amplification of 9p24.1 in diffuse large B-cell lymphoma identifies a unique subset of cases that resemble primary mediastinal large B-cell lymphoma. Blood Cancer J 2019;9(9):73.
Lenz G, Wright GW, Emre NC, Kohlhammer H, Dave SS, Davis RE, Carty S, Lam LT, Shaffer AL, Xiao W, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci U S A. 2008;105(36):13520–5.
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J: WHO classification of Tumours of Haematopoietic and lymphoid tissues, revised 4th edn. Lyon: IARC; 2017.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1.
Nogai H, Wenzel SS, Hailfinger S, Grau M, Kaergel E, Seitz V, Wollert-Wulf B, Pfeifer M, Wolf A, Frick M, et al. IkappaB-zeta controls the constitutive NF-kappaB target gene network and survival of ABC DLBCL. Blood. 2013;122(13):2242–50.
Shi M, Roemer MG, Chapuy B, Liao X, Sun H, Pinkus GS, Shipp MA, Freeman GJ, Rodig SJ. Expression of programmed cell death 1 ligand 2 (PD-L2) is a distinguishing feature of primary mediastinal (thymic) large B-cell lymphoma and associated with PDCD1LG2 copy gain. Am J Surg Pathol. 2014;38(12):1715–23.
Qiu L, Zheng H, Zhao X. The prognostic and clinicopathological significance of PD-L1 expression in patients with diffuse large B-cell lymphoma: a meta-analysis. BMC Cancer. 2019;19(1):273.
Moelans CB, Monsuur HN, de Pinth JH, Radersma RD, de Weger RA, van Diest PJ. ESR1 amplification is rare in breast cancer and is associated with high grade and high proliferation: a multiplex ligation-dependent probe amplification study. Anal Cell Pathol (Amst). 2010;33(1):13–8.
Fernandez-Rodriguez C, Bellosillo B, Garcia-Garcia M, Sanchez-Gonzalez B, Gimeno E, Vela MC, Serrano S, Besses C, Salar A. MYD88 (L265P) mutation is an independent prognostic factor for outcome in patients with diffuse large B-cell lymphoma. Leukemia. 2014;28(10):2104–6.
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, Kohlhammer H, Xu W, Yang Y, Zhao H, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–9.
Dubois S, Viailly PJ, Bohers E, Bertrand P, Ruminy P, Marchand V, Maingonnat C, Mareschal S, Picquenot JM, Penther D, et al. Biological and clinical relevance of associated genomic alterations in MYD88 L265P and non-L265P-mutated diffuse large B-cell lymphoma: analysis of 361 cases. Clin Cancer Res. 2017;23(9):2232–44.
Gupta S, Cheville JC, Jungbluth AA, Zhang Y, Zhang L, Chen YB, Tickoo SK, Fine SW, Gopalan A, Al-Ahmadie HA, et al. JAK2/PD-L1/PD-L2 (9p24.1) amplifications in renal cell carcinomas with sarcomatoid transformation: implications for clinical management. Mod Pathol. 2019;32(9):1344–58.
Ansell SM, Minnema MC, Johnson P, Timmerman JM, Armand P, Shipp MA, Rodig SJ, Ligon AH, Roemer MGM, Reddy N, et al. Nivolumab for relapsed/refractory diffuse large B-cell lymphoma in patients ineligible for or having failed autologous transplantation: a single-arm, phase II study. J Clin Oncol. 2019;37(6):481–9.
Lesokhin AM, Ansell SM, Armand P, Scott EC, Halwani A, Gutierrez M, Millenson MM, Cohen AD, Schuster SJ, Lebovic D, et al. Nivolumab in patients with relapsed or refractory hematologic malignancy: preliminary results of a phase Ib study. J Clin Oncol. 2016;34(23):2698–704.
Hao Y, Chapuy B, Monti S, Sun HH, Rodig SJ, Shipp MA. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res. 2014;20(10):2674–83.
Acknowledgements
Not applicable.
Funding
This study was partly supported by the Beijing Municipal Science & Technology Commission (Grant Number: Z151100004015121), the Cancer Foundation of China (Grant Number: LC2014L13) and CAMS Innovation Fund for Medical Sciences (Grant Number: 2016-I2M-1-001) to perform FFPE tissues collection and MLPA detection, and was partly supported by the Cancer Foundation of China (Grant Number: LC2018B10) and PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (Grant Number: 3332016026) to conduct PD-L1 IHC staining and clinical follow-up, and collect FISH data of c-MYC.
Author information
Authors and Affiliations
Contributions
XX contributed to PD-L1 IHC staining, clinical follow-up, data analysis and manuscript writing. WH contributed to FFPE tissues collection, MLPA detection and clinical follow-up. TQ and LG provided experiment guidance and data interpretation. JY and NL contributed to study design, coordination, discussion and manuscript editing. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This is a retrospective study that was launched in November 2014. The cases enrolled in this project were diagnosed between Jan 2009 and Oct 2010, whose FFPE samples were used. The data regarding treatment and prognosis were acquired by means of medical record consultation and telephone conversation. Thus, the need for consent was waived by the Independent Ethics Committee of Cancer Hospital, Chinese Academy of Medical Sciences, National GCP Center for Anticancer Drugs (NCC2015ST-05).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
MLPA results and clinical follow-up data. The clinicopathological characteristics, clinical follow-up data and MLPA results are showed in this file.
Additional file 2: Figure S1.
Representative results of MLPA. Representative results of MLPA are showed in this figure.
Additional file 3: Table S1.
The details of MLPA probes of genes in DLBCL. The locations and lengths of MLPA probes of genes are showed in this table.
Additional file 4.
The detailed information of DLBCL with JAK2/PD-L2 amplification. The detailed data about clinicopathological characteristics, morphology, immunohistochemistry and treatments of DLBCL with JAK2/PD-L2 amplification are showed in this file.
Additional file 5: Figure S2.
the OS and PFS of DLBCL with or without JAK2/PD-L2_amp. the OS and PFS of DLBCL with or without JAK2/PD-L2_amp.
Additional file 6: Figure S3.
Comparison of CNA-based pattern and their survival outcome among PMBCL and three subgroups of DLBCL (one case of DLBCL with JAK2/PD-L2 amplification and MYD88 L265P mutation were included in DLBCL_MYD88_L265P group). a, Comparison of CNA-based patterns of driver genes among PMBCL and three subgroups of DLBCL according to the status of JAK2/PD-L2 amplification and MYD88 L265P mutation. b, Survival curves and cox-regression analysis of OS and PFS among three subgroups of DLBCL after RCHOP-like treatment.
Additional file 7: Figure S4.
The frequencies of JAK2 gain and amplification, and their survival analysis. a. The frequencies of JAK2 gain and amplification in DLBCL_JAK2/PD-L2_amp and PMBCL. b. the OS and PFS of DLBCL with JAK2 gain or with JAK2 amplification.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Xue, X., Huang, W., Qiu, T. et al. DLBCL with amplification of JAK2/PD-L2 exhibits PMBCL-like CNA pattern and worse clinical outcome resembling those with MYD88 L265P mutation. BMC Cancer 20, 816 (2020). https://doi.org/10.1186/s12885-020-07293-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-020-07293-3