
Abstract
Background
Diffuse large B-cell lymphoma (DLBCL), an aggressive and heterogenic malignant entity, is still a challenging clinical problem, since around one-third of patients are not cured with primary treatment. Next-generation sequencing (NGS) technologies have revealed common genetic mutations in DLBCL. We devised an NGS multi-gene panel to discover genetic features of Chinese nodal DLBCL patients and provide reference information for panel-based NGS detection in clinical laboratories.
Methods
A panel of 116 DLBCL genes was designed based on the literature and related databases. We analyzed 96 Chinese nodal DLBCL biopsy specimens through targeted sequencing.
Results
The most frequently mutated genes were KMT2D (30%), PIM1 (26%), SOCS1 (24%), MYD88 (21%), BTG1 (20%), HIST1H1E (18%), CD79B (18%), SPEN (17%), and KMT2C (16%). SPEN (17%) and DDX3X (6%) mutations were highly prevalent in our study than in Western studies. Thirty-three patients (34%) were assigned as genetic classification by the LymphGen algorithm, including 12 cases MCD, five BN2, seven EZB, seven ST2, and two EZB/ST2 complex. MYD88 L265P mutation, TP53 and BCL2 pathogenic mutations were unfavorable prognostic biomarkers in DLBCL.
Conclusions
This study presents the mutation landscape in Chinese nodal DLBCL, highlights the genetic heterogeneity of DLBCL and shows the role of panel-based NGS to prediction of prognosis and potential molecular targeted therapy in DLBCL. More precise genetic classification needs further investigations.
Similar content being viewed by others


Background
Diffuse large B-cell lymphoma (DLBCL) is the commonest type of adult lymphoma worldwide, comprising 30%–40% of non-Hodgkin lymphoma (NHL) [1]. In China, DLBCL accounts for 37.9% of cases of NHL [2]. Although approximately 70% of DLBCL cases are curable using frontline immunochemotherapy with R-CHOP (rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone) [3, 4], one-third of patients are refractory to primary treatment or relapse after therapy [5]. One explanation for this incomplete therapeutic success is the heterogeneity of this disease. Gene expression profiling (GEP) has uncovered two major ‘‘cell-of-origin’’ (COO) subtypes arising from B cells in different phases of differentiation, namely germinal center B-cell-like (GCB) DLBCL and activated B-cell-like (ABC) DLBCL, which have distinct biology and survival rates [6]. As GEP is poorly available in routine clinical practice, the COO on basis of immunohistochemistry was founded by the Hans algorithm [7]. Immunohistochemical biomarkers (CD10, BCL6, and MUM1) are used to define the GCB and non-GCB subgroups of DLBCL and forecast survival similar to the GEP [7].
In recent years, next-generation sequencing (NGS) has rendered new insights on the genomic features of DLBCL by discovering novel mutation targets via whole genome sequencing (WGS) and whole exome sequencing (WES) [8,9,10,11,12,13,14,15,16,17]. Based on the results of in-depth genomic analyses, new classifications for DLBCL have been proposed. These classifications divide DLBCL into distinct subtypes with discrete genetic signatures [14,15,16,17]. Staudt and colleagues [14, 15] developed the LymphGen algorithm, and identified six genetic subtypes of DLBCL, including MCD (MYD88 L265P and CD79B co-mutated), BN2 (BCL6 fusions and NOTCH2 mutations), N1 (NOTCH1 mutations), EZB (EZH2 mutations and BCL2 translocations), ST2 (SGK1 and TET2 mutations), and A53 (TP53 mutations and deletions).
As WGS or WES is still comparatively expensive and has lower sequencing depth, targeted sequencing is commonly used in currently ongoing studies. Furthermore, clinical laboratories have been launching disease-targeted sequencing tests based on multi-gene panels [18]. In the current study, we designed a panel of 116 genes for DLBCL, and sequenced 96 Chinese nodal DLBCL patients via targeted sequencing. Our purpose was to observe the mutation landscape of Chinese nodal DLBCL and their relationship with clinicopathological characteristics, and to provide certain reference information for panel-based NGS detection in clinical laboratories.
Materials and methods
Patients
A total of 277 frozen lymph node samples with DLBCL were collected from Fudan University Shanghai Cancer Center between November 2005 and August 2014. Clinical information was collected through a review of medical charts. The inclusion criteria included newly diagnosed with DLBCL, previously untreated, treated with at least four cycles of R-CHOP or CHOP regimen in the first-line therapy, and with available DNA of adequate quality extracted from frozen tumor samples. Patients with human immunodeficiency virus infection, primary mediastinal large B-cell lymphoma and incomplete clinical information were precluded from this study. The diagnosis of DLBCL was established according to the 2008 World Health Organization (WHO) classification [1]. Finally, 96 patients were enrolled for analysis. This study was approved by the Fudan University Shanghai Cancer Center Institutional Review Board, and was performed in accordance with the Declaration of Helsinki.
Multi-gene panel design
A panel was devised to capture 116 DLBCL genes (Table S1), based on the literature [8,9,10,11,12, 19,20,21,22,23,24,25,26,27,28,29], the Catalogue of Somatic Mutations in Cancer (COSMIC) database, and the FoundationOne Heme (Foundation Medicine, Cambridge, MA, USA). Probes for all coding exons of the 116 genes were designed via the SureDesign tool (Agilent Technologies, Santa Clara, CA, USA).
Illumina-based targeted sequencing
Genomic DNA from frozen tumor tissue was extracted according to the standard protocols of the QIAamp DNA Mini Kit (Qiagen, Hilden, Germany). DNA quality was examined by spectrophotometer (Nanodrop, Thermo Fisher Scientific Inc.) and agarose gel electrophoresis. The qualified genomic DNA was randomly fragmented, ligated with adapters, purified, and amplified by ligation-mediated PCR. Hybrid capture was conducted through the SureSelect Target Enrichment System (Agilent Technologies, Santa Clara, CA, USA). The resulting DNA libraries were then loaded on Illumina HiSeq 2500 platform (Illumina, San Diego, CA, USA) for sequencing. The mean reads mapping rate was 99.8%. The mean coverage sequencing depth on the official target reached 750.8X.
Bioinformatic analysis of DNA variants
Sequence reads from the Illumina HiSeq instrument were mapped to the reference human genome (hg19) via Burrows-Wheeler Aligner software with the default parameters. Picard was used to mark duplicates and followed by Genome Analysis Toolkit (GATK) to raise alignment accuracy. Single nucleotide variants (SNVs) and short insertion/deletions (Indels) were detected with GATK. The called variants were further recalibrated and filtered to output reliable variant calls, and then annotated with ANNOVAR. Variants were manually reviewed via the Integrative Genomics Viewer (IGV) and verified by Sanger sequencing.
Sanger sequencing
Sanger sequencing was conducted to validate some of the identified variants gained from the targeted sequencing. PCR primers were either sourced from published studies [30,31,32,33,34,35] or designed with Primer-BLAST. PCR products were purified and sequenced on an ABI 3130 sequencer (Applied Biosystems, Foster City, CA, USA).
Statistical analysis
Statistical analysis was performed with the SPSS 22.0 software package (SPSS, Chicago, IL, USA). Categorical variables were compared through the chi-square test or Fisher's exact test when applicable. Progression-free survival (PFS) was calculated from the date of diagnosis to the date of first disease progression, relapse, death resulting from any cause, or last follow-up. Overall survival (OS) was measured from the time of initial diagnosis to the time of death from any cause or last follow-up. The PFS and OS rates were estimated by the Kaplan–Meier method. Univariate analysis was assessed by the log-rank test. Multivariate analysis was carried out by means of Cox proportional hazards model. P values were adjusted for multiple testing using the Benjamini–Hochberg method. P < 0.05 or false discovery rate (FDR) < 0.05 was considered statistically significant.
Results
Patient characteristics
In total, 96 DLBCL patients were enrolled. Seventy-six patients accepted R-CHOP regimen and 20 patients received CHOP therapy. The median age was 55 years. The Ann Arbor stage classification was as follows: stage I-II in 57 (59.4%) patients, and stage III-IV in 39 (40.6%) patients. Eighty-eight patients (91.7%) were International Prognostic Index (IPI) 0–2, and 8 cases were IPI 3–5. The median follow-up time was 35.8 months (range: 3.5–122.7 months). In terms of the Hans algorithm, 36 cases (37.5%) were classified as GCB, while 60 cases (62.5%) were assigned as non-GCB. After frontline therapy, the objective response rate (ORR) was 84.4%, with complete remission (CR) rate of 69.8% (67/96) and partial remission (PR) rate of 14.6% (14/96). The 5-year PFS was 62.1%, and the 5-year OS was 70%.
Gene mutational status
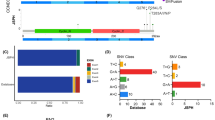
Sequence data were filtered using the database of dbSNP142 and 1000Genome to remove germline variants with more than 0.1% population frequency reported previously. Then, variants in exons and splice sites except synonymous SNVs were kept. Mutation patterns included insertion, deletion, splice site, nonsense and missense mutation. All the 96 patients had at least one mutation of the 116 genes. A total of 899 non-silent somatic mutations were discovered, including 677 missense, 26 insertion, 67 deletion, 73 nonsense, and 56 splice site mutations. The data of genes with mutation frequency ≥ 5% were analyzed by cBioPortal for an overview of mutations in these genes affecting DLBCL samples (Fig. 1) [36]. These mutations were concurrently annotated by OncoKB in cBioPortal (Fig. 1).

OncoPrint of non-silent mutations for the top genes in 96 Chinese DLBCL patients. Genes affected by non-silent mutations in at least 5 DLBCL samples (5% of cases) are listed. These mutations were annotated by OncoKB in cBioPortal on February 3, 2022. Each row represents a gene, and each column represents a DLBCL sample. Orange squares: inframe mutation (unknown significance); dark green squares: missense mutation (putative driver); light green squares: missense mutation (unknown significance); black squares: truncating mutation (putative driver); blue gray squares: truncating mutation (unknown significance); gray bars: no alterations. Nonsense, frameshift and splice site mutations are referred to as truncating mutations in the figure
Selected samples with identified mutations in BTK, CARD11, CD79A, CD79B, EZH2, ETV6, FBXW7, MEF2B, MYD88, NOTCH1, NOTCH2, SPEN and TNFAIP3 were sent for Sanger sequencing for verification. All results of Sanger sequencing were consistent with the targeted sequencing.
Among the 116 genes, no somatic mutations were found in 11 genes. They are AHR, AKT1, CCND2, CDKN2B, ID3, IDH1, KRAS, MAP3K7, RHOA, STAT5B, and TRAF3. The top five genes with more genetic variants were SOCS1 (86 SNVs), PIM1 (68 SNVs), KMT2D (36 SNVs), BTG1 (33 SNVs), and SGK1 (33 SNVs).
The gene mutation frequencies were shown in Table S2. As for mutation frequency, the top five genes were KMT2D (30%), PIM1 (26%), SOCS1 (24%), MYD88 (21%), and BTG1 (20%). There were 49 genes whose mutation frequency was ≥ 5%, and the mutation frequency of 21 genes was ≥ 10% (Fig. 1).
By comparing the mutation frequency between our study and published papers, we found that SPEN mutation frequency in this study (17%) was higher than that in Western research (1–6%), and the mutation frequency of DDX3X, which was not previously described as mutated in Western DLBCL cases, was 6% in our study (Table S2).
SPEN and DDX3X variants
Eighteen mutations in SPEN were identified in 16 DLBCL patients, and only four of them were present in the COSMIC database (v92). Among the 18 SPEN SNVs, 8 (44%) were nonsense mutations, 2 (11%) were deletions, and 8 (44%) were missense mutations (Table S4, Fig. 2a). Twelve of the 18 (67%) mutations were located in exon 11 of SPEN, two (11%) in exon 1, two (11%) in exon 7, one (6%) in exon 2, and one (6%) in exon 3.

Schematic diagrams summarizing the mutations identified in SPEN (a) and DDX3X (b). The schematic diagrams were performed with the aid of MutationMapper in cBioPortal. Green circles: missense mutation; black circles: truncating mutation. Nonsense, frameshift and splice site mutations are referred to as truncating mutations in the figure
Six mutations in DDX3X were found in 6 DLBCL cases, including 5 missense mutations and 1 splice site mutation (Table S3, Fig. 2b). Four of these mutations were present in the COSMIC database (v92). Four of the 6 SNVs clustered in the ATP-binding helicase domain (residues 211–403), one mutation was located close to the ATP-binding helicase domain, and one mutation was located in the C-terminal helicase domain (residues 414–575).
Genetic subtypes of DLBCL
The LymphGen classification tool [15] accommodates mutation-only data from exome or targeted sequencing, but if lacking copy number variant (CNV) data, the A53 subtype cannot be identified. We used the LymphGen tool (https://llmpp.nih.gov/lymphgen/index.php) to analyze our cohort. Twelve cases were assigned as MCD, five as BN2, seven as EZB, seven as ST2, and two as EZB/ST2 complex. Sixty-three cases (66%) remained unclassified (Fig. 3a). Each of the COO (Hans) subtypes involved several genetic subgroups, with GCB cases enriched for EZB and ST2, and non-GCB cases enriched for MCD and BN2 (Fig. 3b).

Genetic subtypes of DLBCL. a Prevalence of genetic subtypes of DLBCL classified by the LymphGen tool. b Prevalence of genetic subtypes in COO (Hans) subgroups
There was no overlay between cases with NOTCH1 and NOTCH2 mutations. Only one patient had both NOTCH1 and SPEN mutations. Mutual exclusivity analysis showed that NOTCH1 and NOTCH2 (FDR = 0.706), and NOTCH1 and SPEN (FDR = 0.706) tended to occur in a mutually exclusive way. NOTCH2 and SPEN (FDR = 0.706) tended to occur in a co-occurrence way. Twelve mutations in TP53 were identified in 9 cases, containing 2 insertion, 1 deletion, 5 missense, and 4 splice site mutations.
Pathway analysis
Seventy-four genes in the panel were grouped into specific pathways (Table 1): (i) B-cell receptor (BCR) signaling; (ii) Toll-like receptor (TLR) signaling; (iii)nuclear factor-κB (NF-κB) signaling; (iv) NOTCH signaling; (v) phosphatidylinositol 3-kinase (PI3K)/AKT signaling; (vi) Janus kinase/signal transducers and activators of transcription (JAK/STAT) signaling; (vii) mitogen-activated protein kinase (MAPK) signaling; (viii) sphingosine 1-phosphate receptor 2 (S1P2) signaling; (ix) epigenetic regulation; and (x) immunity.
Within the 96 specimens, 91 (95%) had at least one mutation in 74 pathway genes, including 34/36 in GCB (94%), and 57/60 in non-GCB (95%) (Table S4). On the whole, 570 somatic mutations were detected in 74 genes, including 412 missense, 17 insertion, 55 deletion, 58 nonsense, and 28 splice site mutations. There was no significant difference in the mutation frequencies of the 74 genes between the GCB and non-GCB subgroups (Table 1).
In the eight signaling pathways, the most frequently concurred pathways were PI3K/AKT and JAK/STAT signaling (18 concurrence out of 50 cases, 36%), and BCR and JAK/STAT signaling (17 concurrence out of 56 cases, 30%). TLR and MAPK signaling, and TLR and S1P2 signaling had none concurrence.
Potential biomarkers for targeted therapy
Our panel included 14 genes (CD79A, CD79B, MYD88, EZH2, CARD11, CREBBP, EP300, TNFAIP3, JAK3, SOCS1, STAT6, NOTCH1, NOTCH2, and SPEN) as therapeutic targets, and mutations of these genes could assist to stratify patients according to therapeutic options (Table S5) [28, 29, 37, 38]. Seventy-two cases (75%) had mutations in these genes, indicating that these patients might be candidates for corresponding clinical drug trials.
In our cohort, for CD79A, two missense mutations (Y188S and Y188D) in the ITAM domain were found in two cases. For CD79B, 17 patients had 23 variations, mainly situated in the ITAM domain. Eight missense mutations affected the tyrosine at position 196 (Y196). Twenty missense mutations in MYD88, mostly located in the TIR domain, were found in 20 patients. The most common variation was L265P (12/20; 60%), followed by S243N (4/20; 20%). For CARD11, 12 mutations were detected in 10 cases, mainly situated in the coiled-coil domain. Seven patients had mutations in TNFAIP3. For MEF2B, eight patients had eight mutations. In our study, five patients (5%) had both CD79B and MYD88 mutations, and 51 patients (53%) had mutations in these six genes.
Clinicopathological relevance
In this study, insertion, deletion, nonsense, and splice site mutations were considered to be pathogenic. To evaluate the pathogenicity of the missense mutations, the predictive software Polymorphism Phenotyping v2 (Polyphen-2) was used. These variations predicted as probably damaging or possibly damaging through Polyphen-2 were considered pathogenic. The pathogenic mutation frequencies of 41 genes were ≥ 5% (Table 2). No significant correlations between the 41 gene mutations and age, stage, or IPI were found. There was no correlation between genetic mutations and treatment response to frontline therapy in our study.
Univariate prognostic analysis was assessed for 41 genes in all 96 patients and 76 patients receiving the R-CHOP regimen separately (Table S6). Among all 96 patients, BCL2 pathogenic mutations were significantly associated with worse OS (FDR = 0.008568, Fig. 4a). In the R-CHOP group, MYD88 L265P–mutated patients had significantly lower PFS (FDR = 0.006771, Fig. 4b).

Kaplan–Meier curves for progression-free survival and overall survival of patients with DLBCL. a Survival analysis was performed on total patients according to BCL2 mutation status. b Survival analysis was performed on patients treated with R-CHOP according to the presence or absence of MYD88 L265P mutation
Important factors (KMT2D, MYD88, MYD88 L265P, TP53, and BCL2) from the univariate analyses for PFS and OS (Table S6), and age, stage, IPI, LDH, and COO (Hans) subtypes were included in the multivariate Cox regression analysis. In addition, treatment regimen was added as a factor in the all patients cohort. Among all patients, multivariate analysis demonstrated that the MYD88 L265P mutation (HR, 2.689; P = 0.017), TP53 pathogenic mutations (HR, 3.129; P = 0.015), stage III-IV (HR, 3.882; P < 0.001), and non-GCB (HR, 2.283; P = 0.042) were independently associated with shorter PFS. BCL2 pathogenic mutations (HR, 6.364; P = 0.007) were independently correlated with shorter OS. In the R-CHOP group, multivariate analysis revealed that the MYD88 L265P mutation (HR, 4.321; P = 0.003) and stage III-IV (HR, 2.665; P = 0.021) were independently correlated with worse PFS, and BCL2 pathogenic mutations (HR, 6.705; P = 0.016) were independently correlated with worse OS. Immunohistochemical data for BCL2 in 86 patients were analyzed. With the 50% cutoff, the expression of BCL2 was assessed for positivity or negativity. BCL2 was positive in 57% of the cases (n = 49), and negative in 43% of the cases (n = 37). IHC analysis revealed that BCL2 expression was not associated with PFS (P = 0.966) or OS (P = 0.736). There was no relationship between BCL2 expression and BCL2 mutation (Spearman rho = 0.037, P = 0.734).
Among the thirty-three patients assigned to the LymphGen subgroup, the treatment response of two MCD patients to frontline therapy was progressive disease (PD), and the response of the other patients was CR or PR. Fisher's exact test showed no difference in treatment response within genetic subgroups. We assessed the survival of patients classified into LymphGen subgroups. For all patients or the R-CHOP-treated patients, although not statistically significant, the PFS of MCD patients was inferior to that of non-MCD patients (P = 0.059 and P = 0.060, separately). Within all patients, ST2 patients had favorable PFS and OS compared with MCD patients (P = 0.059 and P = 0.061, separately), and had favorable OS compared with BN2 patients (P = 0.062). Among R-CHOP–treated patients, ST2 patients had significantly better PFS than MCD patients (P = 0.048). ST2 patients had favorable OS compared with MCD (P = 0.063) and BN2 (P = 0.062) patients.
Discussion
DLBCL remains a challenging clinical puzzle, as about one-third of patients not being cured by the R-CHOP regimen. The limitations to effective therapy are partly relative to the heterogeneity of this disease. In this study, we utilized a panel-based NGS strategy to identify the mutation landscape in 96 Chinese DLBCL patients.
The most recurrently mutated gene in our cohort was KMT2D (30%), in accordance with other NGS studies [8,9,10]. KMT2D, also known as MLL2, encodes a conserved histone methyltransferase that regulates gene transcription via methylating the lysine-4 position of histone H3 (H3K4) [39]. In our study, of 36 KMT2D SNVs, 25% were nonsense mutations, 25% were deletions, 8% were splice site mutations, and 42% were missense mutations. We did not find apparent hotspots. The truncated proteins generated from nonsense, deletion and splice site mutations lack the C-terminal SET domain needed for enzymatic activation, indicating that KMT2D is a tumor suppressor [8,9,10]. Recently, Zhang et al. [40] reported that KMT2D missense mutations affecting the C-terminal SET domain impaired KMT2D methyltransferase activity, resulting in reduction of H3K4 methylation. Moreover, Ortega-Molina et al. [41] showed that KMT2D mutations may promote lymphoma development by disturbing the expression of tumor repressor genes that regulate B cell–activating pathways. Clinically, we found that KMT2D pathogenic mutations were not related to survival in DLBCL patients, which was consistent with the findings of studies by Ortega-Molina et al. [41] and Dubois et al. [42]. Additional studies with more patients are needed to further analyze the clinical significance of KMT2D mutations.
SPEN was affected by 18 mutations in 17% of the sequenced patients, whose mutation frequency was higher than that in Western studies (1–6%). To our knowledge, this is the first report describing SPEN as a frequently mutated target in DLBCL. SPEN (aliases: MINT and SHARP), encodes a hormone inducible transcriptional repressor. This protein is characterized by four RNA recognition motifs at the N-terminus and a highly conservative SPOC domain at the C-terminus. It also contains several nuclear localization sequences, a region interacting with MSX2, and a region interacting with RBPJ [43]. SPEN has been shown to compete with the NOTCH endocellular domain for attaching to RBPJ and to repress the transactivation activity of NOTCH signaling [44, 45]. In addition, SPEN is engaged in transcriptional suppression in several systems other than the NOTCH signaling pathway, such as the MSX2 and Ras/MAPK signaling pathways [46, 47]. Recent studies have shown that SPEN is mutated in 5% of splenic marginal zone lymphoma cases [48], and SPEN functions as a tumor repressor and candidate biomarker of tamoxifen responsiveness in ERα-positive breast cancers [49].
DDX3X (aliases: DBX, DDX3 and CAP-Rf), an ATP-dependent RNA helicase gene, lies on chromosome X with functions in RNA transcription, RNA splicing, mRNA transport, translation, and cell cycle modulation [50]. Owing to its diverse role in RNA metabolism, DDX3X has received growing interest of its role in cancer. Predominantly missense mutations of DDX3X were identified in our DLBCL samples, which was similar to the mutational spectrum in medulloblastoma [51,52,53]. Whereas, the mutant spectrum of DDX3X in DLBCL was dissimilar to that in natural killer/T-cell lymphoma and chronic lymphocytic leukemia, in which mostly truncating mutations (nonsense, frameshift or splice site) have been identified [54, 55]. DDX3X has been reported to be both a tumorigenesis gene and a tumor repressor [56]. The function of DDX3X in DLBCL remains to be determined.
Staudt and colleagues [15] applied the LymphGen classification tool to DLBCL samples from an NCI patient cohort (n = 574), and the tool classified 63% of the cases. When we classified our cohort using the LymphGen tool [15], 33 cases (34%) were assigned as genetic classification, including 12 cases MCD, five BN2, seven EZB, seven ST2, and two EZB/ST2 complex. Sixty-three cases (66%) remained unclassified. This may be on account of the lack of CNV data, and information regarding BCL2 translocations and BCL6 translocations. These data need to be supplemented in the future. The distribution of genetic subtypes in COO (Hans) subgroups in our cohort was consistent with the distribution of subtypes in COO (GEP) subgroups in the study by Staudt and colleagues [15]. Despite functional similarities between NOTCH1 and NOTCH2, Staudt and colleagues speculated that the pathogenesis of N1 subtype was distinct from that of BN2 subtype based on the differences in genetics, phenotype, and clinic [14]. Although none were assigned as N1 subtype in our cohort, there was no overlap in specimens with NOTCH1 or NOTCH2 mutations.
Previous studies have shown the therapeutic potential of sotrastaurin, a selective protein kinase C inhibitor, in patients with CD79A/B mutant DLBCL, whereas CARD11 mutations rendered insensitive [57]. Furthermore, CARD11 mutation has been shown to confer DLBCL cell resistance to lenalidomide, an orally administered immunomodulatory drug [58]. A phase I/II clinical trial has revealed that DLBCL patients having both CD79B and MYD88 mutations were more responsive to the BTK inhibitor ibrutinib, whereas CARD11 mutations and TNFAIP3 inactivation (TNFAIP3 nonsense or frameshift mutation, TNFAIP3 double deletion, or low TNFAIP3 mRNA expression) were associated with inferior responses to ibrutinib [59]. In a phase II study, MEF2B-mutant patients with relapsed/refractory DLBCL had a higher response rate to panobinostat, an HDAC inhibitor, than those without MEF2B mutations [60].
Zhang et al. [11] compared the gene mutations from four Western DLBCL NGS studies. They discovered fairly modest overlaps, and even genes overlapped between different studies often varied, suggesting that DLBCL has considerable genetic heterogeneity [11]. Moreover, de Miranda et al. [13] identified 11 novel genes (TMSB4X, DCDC5, IGLL5, SLITRK3, CDKN2A, GPR37, LYN, OR10A2, PRDM15, TDRD6, and DDX3X) with recurrent mutations in Chinese DLBCL, with the addition of the findings of our study, underscoring the influence of ethnic diversity on somatic alterations.
Recently, a simplified 20-gene algorithm for genetic subtyping was established using targeted sequencing and FISH analysis [61]. DLBCL patients were divided into 6 genetic subtypes (MCD-like, BN2-like, N1-like, EZB-like, TP53mut, and not otherwise specified). This classification was based on mutation data of 18 genes (BTG1, CD70, CD79B, CREBBP, DTX1, EP300, EZH2, MPEG1, MTOR, MYD88, NOTCH1, NOTCH2, PIM1, STAT6, TBL1XR1, TNFAIP3, TNFRSF14, and TP53) and re-arrangement data of 2 genes (BCL2 and BCL6). The researchers found that R-CHOP combined with targeted agents (R-CHOP-X) based on the 6 genetic subtypes improved the CR rate, PFS and OS in patients with DLBCL in the GUIDANCE-01 trial.
There are several limitations to this study. First, the number of samples was small, and 91.7% of patients were in the low or low-intermediate risk group according to IPI. These factors led to the sample selection bias; therefore, further studies with a larger number of patients are needed. Second, we were unable to classify the A53 subtype (TP53 mutations and deletions) due to the lack of CNV data.
Conclusions
In conclusion, our work identified the genetic features of Chinese DLBCL patients. The most frequently mutated genes were KMT2D (30%), PIM1 (26%), SOCS1 (24%), MYD88 (21%), BTG1 (20%), HIST1H1E (18%), CD79B (18%), SPEN (17%), and KMT2C (16%). SPEN (17%) and DDX3X (6%) mutations were highly prevalent in our study than in Western studies. MYD88 L265P mutation, TP53 and BCL2 pathogenic mutations were unfavorable prognostic biomarkers in DLBCL. Thirty-three cases (34%) were assigned as genetic classification by the LymphGen algorithm. Additional studies with more specimens will be demanded to further analyze the prognostic significance of each genetic subtype. Seventy-two patients (75%) had mutations in potentially targeted therapeutic genes, and 51 cases (53%) had mutations that potentially predicted drug response or resistance. To achieve the aim of precise treatment in DLBCL, a consensus gene panel incorporating somatic mutations with proven diagnostic, prognostic and/or therapeutical relevancy must be designed.
Availability of data and materials
The raw sequence data during the current study have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA005331) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human.
References
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, et al. WHO classification of tumours of haematopoietic and lymphoid tissues, 4th ed. Lyon: International Agency for Research on Cancer (IARC); 2008.
Li X, Li G, Gao Z, Zhou X, Zhu X. The relative frequencies of lymphoma subtypes in China: a nationwide study of 10002 cases by the Chinese lymphoma study group. Ann Oncol. 2011;224:141.
Coiffier B, Thieblemont C, Van Den Neste E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d’Etudes des Lymphomes de l’Adulte. Blood. 2010;116(12):2040–5.
Pfreundschuh M, Kuhnt E, Trumper L, Osterborg A, Trneny M, Shepherd L, et al. CHOP-like chemotherapy with or without rituximab in young patients with good-prognosis diffuse large-B-cell lymphoma: 6-year results of an open-label randomised study of the MabThera International Trial (MInT) Group. Lancet Oncol. 2011;12(11):1013–22.
Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. Hematology Am Soc Hematol Educ Program. 2011;2011:498–505.
Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403(6769):503–11.
Hans CP, Weisenburger DD, Greiner TC, Gascoyne RD, Delabie J, Ott G, et al. Confirmation of the molecular classification of diffuse large B-cell lymphoma by immunohistochemistry using a tissue microarray. Blood. 2004;103(1):275–82.
Morin RD, Mendez-Lago M, Mungall AJ, Goya R, Mungall KL, Corbett RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298–303.
Pasqualucci L, Trifonov V, Fabbri G, Ma J, Rossi D, Chiarenza A, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43(9):830–7.
Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109(10):3879–84.
Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, et al. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1398–403.
Morin RD, Mungall K, Pleasance E, Mungall AJ, Goya R, Huff RD, et al. Mutational and structural analysis of diffuse large B-cell lymphoma using whole-genome sequencing. Blood. 2013;122(7):1256–65.
de Miranda NF, Georgiou K, Chen L, Wu C, Gao Z, Zaravinos A, et al. Exome sequencing reveals novel mutation targets in diffuse large B-cell lymphomas derived from Chinese patients. Blood. 2014;124(16):2544–53.
Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, et al. Genetics and pathogenesis of diffuse large B-cell lymphoma. N Engl J Med. 2018;378(15):1396–407.
Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, et al. A probabilistic classification tool for genetic subtypes of diffuse large B cell lymphoma with therapeutic implications. Cancer Cell. 2020;37(4):551–68.
Chapuy B, Stewart C, Dunford AJ, Kim J, Kamburov A, Redd RA, et al. Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat Med. 2018;24(5):679–90.
Lacy SE, Barrans SL, Beer PA, Painter D, Smith AG, Roman E, et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: a Haematological Malignancy Research Network report. Blood. 2020;135(20):1759–71.
Rehm HL. Disease-targeted sequencing: a cornerstone in the clinic. Nat Rev Genet. 2013;14(4):295–300.
Lenz G, Davis RE, Ngo VN, Lam L, George TC, Wright GW, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319(5870):1676–9.
Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459(7247):717–21.
Davis RE, Ngo VN, Lenz G, Tolar P, Young RM, Romesser PB, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463(7277):88–92.
Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181–5.
Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470(7332):115–9.
Slack GW, Gascoyne RD. Next-generation sequencing discoveries in lymphoma. Adv Anat Pathol. 2013;20(2):110–6.
Rossi D, Ciardullo C, Gaidano G. Genetic aberrations of signaling pathways in lymphomagenesis: revelations from next generation sequencing studies. Semin Cancer Biol. 2013;23(6):422–30.
Blombery PA, Dickinson M, Westerman DA. Molecular lesions in B-cell lymphoproliferative disorders: recent contributions from studies utilizing high-throughput sequencing techniques. Leuk Lymphoma. 2014;55(1):19–30.
Roschewski M, Dunleavy K, Wilson WH. Diffuse large B cell lymphoma: molecular targeted therapy. Int J Hematol. 2012;96(5):552–61.
Intlekofer AM, Younes A. Precision therapy for lymphoma–current state and future directions. Nat Rev Clin Oncol. 2014;11(10):585–96.
Roschewski M, Staudt LM, Wilson WH. Diffuse large B-cell lymphoma-treatment approaches in the molecular era. Nat Rev Clin Oncol. 2014;11(1):12–23.
Bohers E, Mareschal S, Bouzelfen A, Marchand V, Ruminy P, Maingonnat C, et al. Targetable activating mutations are very frequent in GCB and ABC diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2014;53(2):144–53.
Kim Y, Ju H, Kim DH, Yoo HY, Kim SJ, Kim WS, et al. CD79B and MYD88 mutations in diffuse large B-cell lymphoma. Hum Pathol. 2014;45(3):556–64.
Bodor C, Grossmann V, Popov N, Okosun J, O’Riain C, Tan K, et al. EZH2 mutations are frequent and represent an early event in follicular lymphoma. Blood. 2013;122(18):3165–8.
Ying CY, Dominguez-Sola D, Fabi M, Lorenz IC, Hussein S, Bansal M, et al. MEF2B mutations lead to deregulated expression of the oncogene BCL6 in diffuse large B cell lymphoma. Nat Immunol. 2013;14(10):1084–92.
Chanudet E, Huang Y, Ichimura K, Dong G, Hamoudi RA, Radford J, et al. A20 is targeted by promoter methylation, deletion and inactivating mutation in MALT lymphoma. Leukemia. 2010;24(2):483–7.
Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331(6016):435–9.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269): l1.
Miao Y, Medeiros LJ, Li Y, Li J, Young KH. Genetic alterations and their clinical implications in DLBCL. Nat Rev Clin Oncol. 2019;16(10):634–52.
Wang L, Li LR, Young KH. New agents and regimens for diffuse large B cell lymphoma. J Hematol Oncol. 2020;13(1):175.
Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705.
Zhang J, Dominguez-Sola D, Hussein S, Lee JE, Holmes AB, Bansal M, et al. Disruption of KMT2D perturbs germinal center B cell development and promotes lymphomagenesis. Nat Med. 2015;21(10):1190–8.
Ortega-Molina A, Boss IW, Canela A, Pan H, Jiang Y, Zhao C, et al. The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat Med. 2015;21(10):1199–208.
Dubois S, Viailly PJ, Mareschal S, Bohers E, Bertrand P, Ruminy P, et al. Next-generation sequencing in diffuse large B-cell lymphoma highlights molecular divergence and therapeutic opportunities: a LYSA study. Clin Cancer Res. 2016;22(12):2919–28.
VanderWielen BD, Yuan Z, Friedmann DR, Kovall RA. Transcriptional repression in the Notch pathway: thermodynamic characterization of CSL-MINT (Msx2-interacting nuclear target protein) complexes. J Biol Chem. 2011;286(17):14892–902.
Kuroda K, Han H, Tani S, Tanigaki K, Tun T, Furukawa T, et al. Regulation of marginal zone B cell development by MINT, a suppressor of Notch/RBP-J signaling pathway. Immunity. 2003;18(2):301–12.
Oswald F, Kostezka U, Astrahantseff K, Bourteele S, Dillinger K, Zechner U, et al. SHARP is a novel component of the Notch/RBP-Jkappa signalling pathway. Embo J. 2002;21(20):5417–26.
Newberry EP, Latifi T, Towler DA. The RRM domain of MINT, a novel Msx2 binding protein, recognizes and regulates the rat osteocalcin promoter. Biochemistry. 1999;38(33):10678–90.
Rebay I, Chen F, Hsiao F, Kolodziej PA, Kuang BH, Laverty T, et al. A genetic screen for novel components of the Ras/Mitogen-activated protein kinase signaling pathway that interact with the yan gene of Drosophila identifies split ends, a new RNA recognition motif-containing protein. Genetics. 2000;154(2):695–712.
Rossi D, Trifonov V, Fangazio M, Bruscaggin A, Rasi S, Spina V, et al. The coding genome of splenic marginal zone lymphoma: activation of NOTCH2 and other pathways regulating marginal zone development. J Exp Med. 2012;209(9):1537–51.
Legare S, Cavallone L, Mamo A, Chabot C, Sirois I, Magliocco A, et al. The estrogen receptor cofactor SPEN functions as a tumor suppressor and candidate biomarker of drug responsiveness in hormone-dependent breast cancers. Cancer Res. 2015;75(20):4351–63.
Rosner A, Rinkevich B. The DDX3 subfamily of the DEAD box helicases: divergent roles as unveiled by studying different organisms and in vitro assays. Curr Med Chem. 2007;14(23):2517–25.
Jones DT, Jager N, Kool M, Zichner T, Hutter B, Sultan M, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488(7409):100–5.
Pugh TJ, Weeraratne SD, Archer TC, Pomeranz KD, Auclair D, Bochicchio J, et al. Medulloblastoma exome sequencing uncovers subtype-specific somatic mutations. Nature. 2012;488(7409):106–10.
Robinson G, Parker M, Kranenburg TA, Lu C, Chen X, Ding L, et al. Novel mutations target distinct subgroups of medulloblastoma. Nature. 2012;488(7409):43–8.
Jiang L, Gu ZH, Yan ZX, Zhao X, Xie YY, Zhang ZG, et al. Exome sequencing identifies somatic mutations of DDX3X in natural killer/T-cell lymphoma. Nat Genet. 2015;47(9):1061–6.
Ojha J, Secreto CR, Rabe KG, Van Dyke DL, Kortum KM, Slager SL, et al. Identification of recurrent truncated DDX3X mutations in chronic lymphocytic leukaemia. Br J Haematol. 2015;169(3):445–8.
Schroder M. Human DEAD-box protein 3 has multiple functions in gene regulation and cell cycle control and is a prime target for viral manipulation. Biochem Pharmacol. 2010;79(3):297–306.
Naylor TL, Tang H, Ratsch BA, Enns A, Loo A, Chen L, et al. Protein kinase C inhibitor sotrastaurin selectively inhibits the growth of CD79 mutant diffuse large B-cell lymphomas. Cancer Res. 2011;71(7):2643–53.
Zhang LH, Kosek J, Wang M, Heise C, Schafer PH, Chopra R. Lenalidomide efficacy in activated B-cell-like subtype diffuse large B-cell lymphoma is dependent upon IRF4 and cereblon expression. Br J Haematol. 2013;160(4):487–502.
Wilson WH, Young RM, Schmitz R, Yang Y, Pittaluga S, Wright G, et al. Targeting B cell receptor signaling with ibrutinib in diffuse large B cell lymphoma. Nat Med. 2015;21(8):922–6.
Assouline SE, Nielsen TH, Yu S, Alcaide M, Chong L, MacDonald D, et al. Phase 2 study of panobinostat with or without rituximab in relapsed diffuse large B-cell lymphoma. Blood. 2016;128(2):185–94.
Zhang MC, Tian S, Fu D, Wang L, Cheng S, Yi HM, et al. Genetic subtype-guided immunochemotherapy in diffuse large B cell lymphoma: The randomized GUIDANCE-01 trial. Cancer Cell. 2023;41(10):1705–16.
Acknowledgements
We thank WuXi AppTec (Shanghai, China) for providing the technical service for targeted next generation sequencing and bioinformatic analysis.
Funding
This study was supported by grants from the National Natural Science Foundation of China (Nos. 81470353 and 81870155), Innovation Group Project of Shanghai Municipal Health Commission (2019CXJQ03), Shanghai Science and Technology Development Fund (19MC1911000), Shanghai Municipal Key Clinical Specialty (shslczdzk01301), and Innovation Program of Shanghai Science and Technology Committee (20Z11900300).
Author information
Authors and Affiliations
Contributions
X.Z. designed and supervised the research study, and revised the manuscript. B.C. designed the NGS panel, analyzed and interpreted the data, and wrote the first draft of the manuscript. C.S. and R.B. performed the Sanger sequencing. Z.L., Y.J., W.C., and M.S. collected clinical data and patient samples. B.Y. and X.L. revised the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was approved by the Fudan University Shanghai Cancer Center Institutional Review Board, and complied with the Declaration of Helsinki. Due to the retrospective nature of the study, informed consent was waived by the Fudan University Shanghai Cancer Center Institutional Review Board.
Consent for publication
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cao, B., Sun, C., Bi, R. et al. Mutation landscape in Chinese nodal diffuse large B-cell lymphoma by targeted next generation sequencing and their relationship with clinicopathological characteristics. BMC Med Genomics 17, 84 (2024). https://doi.org/10.1186/s12920-024-01866-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12920-024-01866-y