
Abstract
Background
Autosomal dominant hypertension with brachydactyly type E syndrome caused by pathogenic variants in the PDE3A gene was first reported in 2015. To date, there are only a few reports of this kind of syndrome. Other patients still lack a genetic diagnosis.
Case presentation
Whole-exome sequencing was performed in an 18-year-old female proband with a clinical diagnosis of hypertension with brachydactyly syndrome. Quantitative real-time PCR was used to identify pathogenic copy number variations (CNVs). After bioinformatics analysis and healthy control database filtering, we revealed a heterozygous missense PDE3A variant (c.1346G > A, p.Gly449Asp). The variant was absent in the ExAC database and located in a highly evolutionarily conserved cluster of reported PDE3A pathogenic variants. Importantly, this variant was predicted to affect protein function by both SIFT (score = 0) and PolyPhen-2 (score = 1). After Sanger sequencing, the variant was determined to be absent in the healthy parents of the proband as well as 800 ethnically and geographically matched healthy controls.
Conclusion
We present a report linking a de novo PDE3A variant to autosomal dominant hypertension with brachydactyly type E syndrome.
Similar content being viewed by others
Background
In 1973, Biliginturan et al. [1] reported a large no consanguineous Turkish family showing brachydactyly associated with hypertension, and the authors clinically described the phenotype of the disorder in detail for the first time. Schuster et al. [2] discovered severe autosomal dominant hypertension and brachydactyly in a unique Turkish lineage mapped to human chromosome 12p. In 2015, Maass et al. [3] identified mutations in the PDE3A gene at 12p12.2 in six families with hypertension with brachydactyly type E syndrome (HTNB) (OMIM: 112410). HTNB is an autosomal dominant disease characterized by brachydactyly type E (BDE) and severe hypertension [2, 4, 5]. Patients with HTNB feature salt-independent and sharply increasing blood pressure with age from childhood [4]. They usually suffer from stroke (haemorrhagic or thrombotic) if untreated, usually before the age of 50 years [6]. HTNB patients usually have a stockier build, shorter stature and rounder face than healthy persons [6, 7].
Cyclic nucleotide phosphodiesterases (PDEs) regulate intracellular signalling by hydrolysing cyclic AMP (cAMP) and cyclic GMP (cGMP) [8, 9]. They thereby play key roles in regulating the amplitude and duration of many cellular processes and, consequently, myriad biological responses in healthy persons and persons with disease [9, 10]. PDE3A, a cGMP-inhibited cAMP PDE, plays important roles in cardiovascular function by regulating vascular smooth muscle contraction and relaxation [9,10,11].
In 2015, Maasset al. revealed for the first time that the PDE3A gene at 12p12.2 is the causative gene for HTNB [3]. The authors demonstrated that gain-of-function PDE3A variants (p.Thr445Ala; p.Thr445Ser; p.Thr445Asn; p.Ala447Thr; p.Ala447Val; and p.Gly449Val) upregulate protein kinase A (PKA)-mediated PDE3A phosphorylation, therefore increasing cAMP hydrolytic activity and producing lower intercellular cAMP concentrations in vascular smooth muscle cells (VSMCs) [3]. On the one hand, the variants promoted peripheral vascular constriction and increased VSMC proliferation, thus causing hypertension; on the other hand, they regulated chondrogenesis and resulted in brachydactyly [3]. In 2016, Boda et al. identified another pathogenic variant (p.Ser446Pro) in the same cluster region in exon 4 of the PED3A gene causing HTNB in a Japanese family [6]. In 2018, Hauer et al. identified a pathogenic variant (p.Gly449Asp) by whole-exome sequencing for short stature [12].
Here, we present another report linking the PDE3A variant to HTNB. We identified a de novo PDE3A variant in the pathogenic cluster in a Han Chinese family with HTNB.
Case presentation
The ethics committee of Tongji Hospital approved our study. Written informed consent was obtained from the patient. Our experiments followed the principles expressed in the Declaration of Helsinki.
To identify the genetic cause in the proband, total DNA was isolated from the blood of each participant using a QIAamp DNA Mini Kit (Qiagen, Germany). Then, we checked the purified DNA using electrophoresis to avoid fragmental DNA degradation and RNA pollution.
We performed WES using an Ion AmpliSeq™ Exome Panel (Hi-Q) (57.7 Mb target region) on an Ion Proton Sequencer (Life Technologies, Thermo Fisher, USA). Library construction and next-generation sequencing were performed as we previously described [13].
We initially processed the data with the Ion Torrent platform-specific software Torrent Suite v5.0.4 to generate reads and trim adapter sequences. Then, reads were aligned to the hg19/GRCh37 human reference genome (https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&position=lastDbPos) to analyse coverage status, and the consensus on-target reads were used to call variants using recommended germline parameters. Finally, the annotation was performed with Ion Reporter 5.0 (Life Technologies, Thermo Fisher, USA). The datasets generated and/or analysed during the current study are available in the SRA - NCBI repository, the Sequence Read Archive (SRA) accession number is: SRR11974541 (https://www.ncbi.nlm.nih.gov/sra/?term=SRR11974541).
To identify reported pathogenic variants, we searched the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/) and the HGMD database (http://www.hgmd.cf.ac.uk/ac/search.php). To filter non-pathogenic variants, we removed low-quality data (coverage less than 15-fold) and variants with a minor allele frequency (MAF) greater than 0.1% in the Exome Sequencing Project database (http://evs.gs.washington.edu/EVS/), the 1000 Genomes Project database (http://browser.1000genomes.org/) and the ExAC database (http://exac.broadinstitute.org/). We removed intronic, up/downstream, and synonymous variants. Furthermore, we filtered out the variants in our exome database of 100 healthy Chinese. Then, we predicted the impact scores of the missense variants using both PolyPhen-2 scores (http://genetics.bwh.harvard.edu/pph2/) and SIFT scores (http://sift.jcvi.org). We also evaluated their evolutionary conservation across multiple species by using phyloP scores. Finally, we filtered out variants in genes unrelated to hypertension. For WES data filtering procedures, a filtering tree illustrated the step-by-step narrowing down of candidate gene/variants for the illustrated NGS data analysis (Supplementary Fig. S1).
We performed Sanger sequencing of all identified pathogenic variants and low-coverage regions of PDE3A by using an Applied Biosystems 3500xl sequencer (Applied Biosystems). PCR amplification and BigDye reactions were optimized using Taq™ Hot Start enzyme (TaKaRa) with a BigDye Terminator Cycle Sequencing Kit. After validation in the patient, the potential pathogenic variant was also directly Sanger sequenced in 800 unrelated ethnically and geographically matched healthy controls. We performed Sanger sequencing-based (21 loci) paternity/maternity tests for the three family members.
The multiplex parallel AmpliSeq technique of the Ion Torrent platform allowed us to analyse CNVs directly based on the sequencing data by using the CNV workflow in Ion Reporter™ software 5.0. We performed whole-exome CNV analysis using exome sequencing data of the proband and healthy controls that we previously sequenced. All identified CNVs were validated by gDNA real-time PCR analysis for each exon. We also performed real-time PCR-based CNV analysis of each exon of the PDE3A gene. The primers are listed in Table 1.
To investigate the influence of the identified pathogenic variants, protein structural modelling was performed using homologous protein structure in SPDBV 4.10 online software (http://spdbv.vital-it.ch/).
Statistical analysis was performed using SPSS software version 20.0. Differences between groups were test for significance using the Pearson chi-square test, and a two-sided P value less than 0.05 was considered statistically significant.
An 18-year-old female of short stature (152 cm) was referred to our hospital with a two-year history of hypertension. She underwent 24-h blood pressure measurement. Her systolic pressure ranged from 150 to 220 mmHg and diastolic pressure ranged from 80 to 120 mmHg. She exhibited short fingers, short toes and normal intelligence. The radiographic images of her fingers and toes revealed BDE (Fig. 1). Renal ultrasonography and computed tomography angiography (CTA) of her renal arteries did not present obvious anomalies. Duplex tests of plasma renin activity and aldosterone, catecholamine measurements, liver and kidney functions, and electrolytes were all in the normal range. These results imply primary hypertension instead of a known cause of secondary hypertension. She underwent funduscopic examination and an echocardiogram. Both examinations presented normal results. There was no family history of hypertension or stroke. Both of her parents presented normal blood pressure (~ 130/70 mmHg) and normal stature (father: 175 cm, mother: 168 cm) and did not show obvious anomalies during physical or laboratory examinations.

Photos and radiographic images of the patient’s hands and right foot showing short fingers and toes associated with brachydactyly type E (BDE)
By WES, we obtained an output of 34,062,121 mapped reads, 93.49% of which were on target. In conclusion, 99.54% of all 293,903 target amplicons were covered at least once, and 94.55% of amplicons were covered at least 20 times. The average base-read depth was 108-fold.
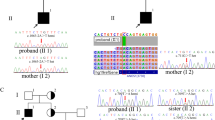
After filtering and Sanger sequencing validation, we found a heterozygous missense variant, c.1346G > A (p.Gly449Asp), in exon 4 of the PDE3A gene (Fig. 2a), according to the guidelines [14]. No other variants or suspicious CNVs were detected in the coding region of known hypertension-related genes. The identified variant was successfully validated in the proband by Sanger sequencing; however, it was absent in her parents, which implied a de novo inheritance mode (Fig. 2b). Furthermore, it was absent in the 800 unrelated matched healthy controls, which implied that it was not a rare SNP in Chinese. Moreover, the identified pathogenic variant was in neither the ESP database nor the ExAC database. The variant was in a highly evolutionarily conserved region across multiple species (phyloP score = 2.7). PDE3A-p.Gly449Asp was predicted to be a protein function-affecting variant (SIFT score = 0, PolyPhen-2 score = 1).

The pathogenic variant and pedigree description. a Sanger sequencing of the family. Coloured blocks show the evolutionary conservation of the cluster across multiple species. b The family tree of the HTNB pedigree. Males and females are indicated by squares and circles, respectively. The filled symbol represents the HTNB-affected individual. The arrow shows the proband. +/− represents the heterozygous PDE3A p.Gly449Asp variant. −/− represents the wild type
Discussion and conclusions
We report a PDE3A variant related to HTNB. Our finding implies that the c.1346G > A (p.Gly449Asp) variant, in exon 4 of the PDE3A gene, may be the genetic cause in the patient of a Han Chinese family with HTNB.
To date, nine pathogenic variants (c.1333A > G, p.Thr445Ala; c.1334C > G, p.Thr445Ser; c.1334C > A, p.Thr445Asn; c.1336 T > C, p.Ser446Pro; c.1339G > A, p.Ala447Thr; c.1340C > T, p.Ala447Val; c.1333_1335de, p.Thr445del; c.1346G > T, p.Gly449Val; and c.1346G > A, p.Gly449Asp) have been reported in a highly conserved cluster region in exon 4 of the PDE3A gene (Fig. 3a) [3, 6]. These variants are adjacent to the Ser428 and Ser438 residues, which can be phosphorylated by protein kinase C (PKC) or protein kinase A (PKA), thereby uncovering the two cryptic sites for phosphorylation and, consequently, hyperactivating the enzyme and resulting in lower cAMP levels [3, 11].

Functional analysis of the pathogenic variant PDE3A-p.Gly449Asp. a The peptide sequence of PDE3A exon 4, which harbours the pathogenic variant cluster (red-coloured region), and the phosphorylation sites Ser428 and Ser438 (shown in a green colour) near the altered residues are shown. Reported pathogenic variants causing autosomal dominant hypertension with brachydactyly type E syndrome are shown in red. Our novel pathogenic variant is shown in blue. b In silico structural modelling of the two variants (p.Gly449Val-reported, p.Gly449Asp-novel) compared with the wild type in the PDE3A protein
Our variant was located in the same cluster as these reported pathogenic variants and at the same position as the c.1346G > T (p.Gly449Val) variant (Fig. 3a). In protein structural modelling, these two variants presented similar structures (Fig. 3b), which implied that the two substitutions have similar functions. Based on this finding, we postulate a similar mechanism, in which our variant uncovers the Ser428 and Ser438 cryptic sites for phosphorylation and hyperactivated PDE3A. Thus, we believe that our variant causes HTNB via a mechanism similar to that reported in previous studies [3, 11]. Therefore, milrinone is a recommended choice for such patients. Further functional studies and additional genetic validation of this variant should be performed in the future.
In conclusion, we identified a de novo PDE3A variant in a Han Chinese family with HTNB. Our finding enriches the pathogenic spectrum of PDE3A-related hypertension and will facilitate future genetic diagnosis.
Availability of data and materials
The hg19/GRCh37 human reference genome (https://genome.ucsc.edu/cgi-bin/hgTracks?db=hg19&position=lastDbPos) was used as the reference dataset in this study. The datasets generated and/or analysed during the current study are available in the SRA - NCBI repository, the Sequence Read Archive (SRA) accession number is: SRR11974541 (https://www.ncbi.nlm.nih.gov/sra/?term=SRR11974541).
Abbreviations
- HTNB:
-
Hypertension with brachydactyly type E syndrome
- WES:
-
Whole-exome sequencing
- CNV:
-
Copy number variation
- MAF:
-
Minor allele frequency
References
Bilginturan N, Zileli S, Karacadag S, Pirnar T. Hereditary brachydactyly associated with hypertension. J Med Genet. 1973;10(3):253–9.
Schuster H, Wienker TE, Bahring S, Bilginturan N, Toka HR, Neitzel H, Jeschke E, Toka O, Gilbert D, Lowe A, et al. Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat Genet. 1996;13(1):98–100.
Maass PG, Aydin A, Luft FC, Schachterle C, Weise A, Stricker S, Lindschau C, Vaegler M, Qadri F, Toka HR, et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet. 2015;47(6):647–53.
Luft FC, Toka O, Toka HR, Jordan J, Bahring S. Mendelian hypertension with brachydactyly as a molecular genetic lesson in regulatory physiology. Am J Physiol Regul Integr Comp Physiol. 2003;285(4):R709–14.
Bahring S, Rauch A, Toka O, Schroeder C, Hesse C, Siedler H, Fesus G, Haefeli WE, Busjahn A, Aydin A, et al. Autosomal-dominant hypertension with type E brachydactyly is caused by rearrangement on the short arm of chromosome 12. Hypertension. 2004;43(2):471–6.
Boda H, Uchida H, Takaiso N, Ouchi Y, Fujita N, Kuno A, Hata T, Nagatani A, Funamoto Y, Miyata M, et al. A PDE3A mutation in familial hypertension and brachydactyly syndrome. J Hum Genet. 2016;61(8):701–3.
Pereda A, Garin I, Garcia-Barcina M, Gener B, Beristain E, Ibanez AM, Perez de Nanclares G. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J Rare Dis. 2013;8:141.
Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35(2):91–100.
Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13(4):290–314.
Movsesian M. Novel approaches to targeting PDE3 in cardiovascular disease. Pharmacol Ther. 2016;163:74–81.
Houslay M. Hypertension linked to PDE3A activation. Nat Genet. 2015;47(6):562–3.
Hauer NN, Popp B, Schoeller E, Schuhmann S, Heath KE, Hisado-Oliva A, Klinger P, Kraus C, Trautmann U, Zenker M, et al. Clinical relevance of systematic phenotyping and exome sequencing in patients with short stature. Genet Med. 2018;20(6):630–8.
Li Z, Zhou C, Tan L, Chen P, Cao Y, Li C, Li X, Yan J, Zeng H, Wang DW, et al. Variants of genes encoding collagens and matrix metalloproteinase system increased the risk of aortic dissection. Sci China Life Sci. 2017;60(1):57–65.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24.
Acknowledgements
We thank the family who cooperated with the evaluation of the subjects and consented to participate in this study.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Project Nos. 81700413, 8157020983) and the National Key R&D Program of China (2017YFC0909400). The funding bodies played no direct role in the design of the study, the collection, analysis, or interpretation of data, or the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
XL performed the genomic DNA and RNA isolation, real-time PCR and Sanger sequencing and participated in the clinical sample and data collection. ZL and PC performed whole-exome sequencing, analysed, and interpreted the data. DWW1 designed the experiments and critically revised the manuscript. YW and DWW2 wrote the manuscript and made final revisions to the manuscript. All authors participated in the proofreading. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The publication of this manuscript has been reviewed and approved by the ethics committees of Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology and The First Affiliated Hospital of Nanjing Medical University. We obtained written informed consent from all participants for participation in this study.
Consent for publication
We obtained written informed consent from all subjects for publication of this study.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Illustration for the filtering process of WES. (PPTX 39 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Li, X., Li, Z., Chen, P. et al. Whole-exome sequencing identifies a de novo PDE3A variant causing autosomal dominant hypertension with brachydactyly type E syndrome: a case report. BMC Med Genet 21, 144 (2020). https://doi.org/10.1186/s12881-020-01077-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01077-z