
Abstract
Background
The autosomal recessive non-syndromic deafness DFNB28 is characterized by prelingual sensorineural hearing loss. The disease is related with mutations in TRIOBP (Trio- and F-actin-Binding Protein) gene, which has three transcripts referred to as TRIOBP-5, TRIOBP − 4 and TRIOBP-1. Among them, TRIOBP-5/− 4 are expressed in the inner ears and crucial for maintaining the structure and function of the stereocilia.
Methods
The proband is a 26-year-old Chinese female. She and her younger brother have being suffered from severe deafness since birth, whereas her parents, who are cousins, have normal communication ability. Hearing impairment of the two siblings was determined by pure tone audiometry. Whole Exome Sequencing (WES) was performed on the genomic DNA of the proband and Sanger sequencing was conducted on the DNA samples of the four family members.
Results
Tests of pure tone hearing thresholds showed a severe to profound symmetric hearing loss for the proband and her younger brother. Moreover, a novel TRIOBP c.1342C > T (p.Arg448*) variant was identified by WES in the DNA sample of the proband and confirmed by Sanger sequencing in DNA of the family members.
Conclusions
The TRIOBP c.1342C > T (p.Arg448*) variant is predicted to disrupt TRIOBP-5 and TRIOBP-4, which may lead to the congenital deafness. The results will broaden the spectrum of pathogenic variants in TRIOBP gene. The characteristics of deafness in the family imply that marriage between close relatives should be avoided.
Similar content being viewed by others

Background
As an autosomal recessive non-syndromic deafness, DFNB28 is characterized by pre-lingual sensorineural hearing loss. The disease is related with mutations in a gene named TRIOBP (Trio- and F-actin-Binding Protein) [1,2,3,4,5]. The TRIOBP gene in human and mouse encode three mRNA transcripts designated as TRIOBP-5, TRIOBP-4 and TRIOBP-1, respectively. TRIOBP-5 is the longest one covering both TRIOBP-4 and TRIOBP-1. TRIOBP-4 lies upstream of TRIOBP-1 without overlapping with TRIOBP-1. In human, TRIOBP-5, TRIOBP-4 and TRIOBP-1 code 2365, 1144 and 652 amino acids, respectively [5]. The amino acid sequences of TRIOBP-5/− 4 contain two repeat motifs, which are referred to as R1 (amino acid residues 357–500) and R2 (amino acid residues 684–896). The R1 motif is the major actin-binding domain of TRIOBP-4, while the R2 motif’s binding with actin filaments is nonspecific [4, 6]. TRIOBP-5 or TRIOBP-1 has a pleckstrin homology domain (PH) and several coiled-coil domains (CC) [5]. In human and mouse, TRIOBP-5/− 4 are mainly expressed in the inner ears and retinas, and TRIOBP-1 plays an important role in embryonic development [2,3,4].
Many TRIOBP mutations associated with human DFNB28 have been identified [1, 2, 5, 7,8,9,10]. Though the variants cover the region from exon 4 to exon 23 (corresponding to amino acid residues from 52 to 2334), most of them are localized within exon 7 (exon 6 previously, corresponding to amino acid residues from 210 to1316), impairing the function of TRIOBP-5 and TRIOBP-4. Therefore, the exon 7 of TRIOBP-5 is considered as the hotspot for variations [5]. The predicted protein changes due to variants in exon 7 of TRIOBP-5 are summarized mainly as the followings: (p.Gln268fs), (p.Gln297*), (p.Arg347*), (p.Arg474*), (p.Arg523*), (p.Gln581*), (p.Gln740*), (p.Arg785fs), (p.Arg788*), (p.Arg861*), (p.Arg885fs), (p.Arg920*), (p.Ala998Thr), (p.Gly1019Arg), (p.P1030Lfs), (p.Ile1065Val), (p.Arg1068*), (p.Asp1069fs), (p.Arg1078fs), (p.Arg1078Cys), (p.Arg1117*), (p.Met1151Val), (p.Leu1154fs), (p.Glu1156*), (p.Arg1221Gln) and (p.Glu1314Asp). Among them, the variants of (p.Arg861*), (p.Arg920*), (p.Ile1065Val) and (p.Met1151Val) were found in the Chinese population [8, 9]. In this study, we further identified a novel TRIOBP c.1342C > T (p.Arg448*) variant in a Chinese family. The mutation is supposed to disrupt the isoforms of TRIOBP-5 and TRIOBP-4, leading to the congenital deafness of the family members.
Clinical report
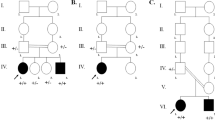
A four-generation family is included in this study. All the family members have normal hearing with exception of two cases complaining of deafness in the fourth generation (Fig. 1). The proband (IV1), a 26-year-old female, has being suffered from severe deafness since birth. She was married and went to see a doctor in the Reproductive Medicine Center of the Yantai Yuhuangding Hospital for advice if she could have a healthy child. Her younger brother (the family member IV2) also suffered from serious hearing loss. Their parents are close relatives (cousins). No other symptoms were complained by the family members. This study was approved by the medical ethics committee of the Yantai Yuhuangding Hospital, Qingdao University. Informed consent was obtained from each of the subjects.

Identification of the individuals with hearing impairment. a. Pedigree analyses to identify the family members with hearing loss. The proband is indicated by an arrow. Genotypes are indicated as heterozygous 1342C > T (Heter1342C > T) for III34 and III45, and homozygous 1342C > T (Homo1342C > T) for (IV1 and IV2). b. Pure-tone audiograms to identify the hearing affected individuals. The individuals (IV1 and IV2) show severe to profound symmetric hearing impairment
Methods
Test of brainstem evoked response audiometry
Clinical examination of the ear, nose and throat was performed on the two patients (IV1, IV2) and their parents (III4, III5) in the Department of Otolaryngology-HNS of Yuhuangding Hospital to exclude acquired causes of hearing impairment. Brainstem evoked response audiometry (BERA) was performed on the two patients (IV1, IV2) by using a computer-aided evoked potential system (Intelligent Hearing Systems, Miami, FL, USA). Auditory thresholds were obtained for stimuli of pure tone-bursts at 0.25, 0.5, 1, 2, 4, and 8 kHz in decibel sound pressure level (dB SPL).
Identification of the gene variants
The peripheral blood samples were taken from the four subjects (IV1, IV2, III4, III5). Genomic DNA was isolated using the QIAGEN Universal DNA Purification Kit and diluted to a concentration of 50 ng/μl. The genome variants of the proband were identified by WES and confirmed by Sanger sequencing in Yin Feng Biotechnology Company (Jinan, China). The main procedures are described as followings: (1) DNA quality testing: After the DNA was extracted from the sample, the concentration was tested by Qubit® 3.0 Flurometer (Life Technologies, CA, USA), the purity was examined by a NanoPhotometer (IMPLEN, CA, USA) and the integrity was detected by 1% agarose gel electrophoresis; (2) Library construction: Library of small fragment preparation was conducted following the methods and procedures described in SureSelectXT Target Enrichment System (G7530–90000). The exons were then captured by a gene chip, eluted, recovered and enriched. (3) Testing the quality of the library. Agilent 2100 Bioanalyzer system was used to determine the insert sizes. Bio-Rad CFX 96 quantitative fluorescence PCR instrument and Bio-Rad KIT iQ SYBR GRN were used for Q-PCR to accurately determine the effective concentration of the library (> 10 nM). (4) Sequencing: double-end sequencing program (PE150) was performed on NovaSeq 6000 platform and sequence reads of 150 bp were received. (5) Raw data filtering and data comparison: reads containing primer/adaptor (> 5 bp) were removed and low-quality reads (N content in any sequencing read exceeds 5% of the read base number) were filtered; paired reads were removed when the number of low-quality bases(Q ≤ 19) in any read exceeded 50% of the read base number; the repeat sequences were also removed. (6) Variant analysis software (GATK) [11] was used to extract the potential sites of SNP and InDel in the whole genome, compared with the reference database Human_GRCh38_dbSNP141. (7) The variants were firstly confirmed by Sanger sequencing in the genomic DNA of the proband (IV1), and then identified in the DNA of the other subjects (IV2, III4, III5). (8) Primer 5 software was applied to design the primers for PCR amplification and sequencing the gene fragments spanning a TRIOBP (NM_001039141.2) variant (TRIOBP-F, 5′-CTCACGAAGCACCCAACTGGATAA-3′; TRIOBP-R, 5′-GAGGTTCTGGAGGCTCTGGGATTG-3′), a HOMER2 (NM_199330.2) variant (HOMER2-F, 5′-CTGCCTTGTGGTGGTGTGT A-3′; HOMER2-R, 5′-CAAACGTT GCTGAGTCTGCC-3′) and a TMC2 (NM_080751.2) variant (TMC2-F, 5′-TGGTTCTTCAGTGGCATCGT-3′; TMC2-R, 5′-ACTATCTGGGTAATTGATGT GAGT-3′), respectively.
Bioinformatics analysis
Single-nucleotide variants (SNVs) and insertions/deletions (indels) were compared with the databases of the 1000 Genomes Project, the Exome Aggregation Consortium database (ExAC), Genome Aggregation Database (gnomAD) and allele frequency in the Gene Mutation Database of Chinese (CNGMD). Pathogenicity of variants was classified according to the guidelines of American College of Medical Genetics and Genomics (ACMG). The novelty of gene variations was evaluated by using the ClinVar software.
Results
Analysis of the clinical manifestations
A four-generation family with autosomal recessive hereditary deafness was included in this study. All the family members had normal communication ability, except for the proband and her younger brother who showed serious hearing problems. Pure tone hearing threshold test revealed a severe (61–80 dB SPL) to profound (> 80 dB SPL) symmetric hearing impairment for the two siblings (Fig. 1).
Identification of the pathogenic variant
WES was performed on the genomic DNA from the proband. Variant rate less than or equal to 0.01 in CNGMD was used as the frequency cut-offs. A TRIOBP c.1342C > T (p.Arg448*) variant with an allele frequency of 0.00002 in ExAC database was further identified. The gnomAD frequency of the variant for SAS is 0.0000 (https://www.ncbi.nlm.nih.gov/snp/rs773152243). The TRIOBP c.1342C > T (p.Arg448*) variant is likely pathogenic according to the ACMG guidelines for classification. To verify the results, Sanger sequencing was performed on the DNA of the four family members. It was found that the two siblings carried a homozygous TRIOBP c.1342C > T (p.Arg448*) variant, and each of their parents carried a heterozygous TRIOBP c.1342C > T (p.Arg448*) variant (Fig. 2). The TRIOBP c.1342C > T variant was found to meet the requirement of autosomal recessive disorders, that is, homozygous variant or heterozygous variant in the offspring of parents with a heterozygous variant.

Identification of the pathogenic TRIOBP variant in the proband’s family. a. Sequence of the wild type TRIOBP gene fragment spanning the variant site (1342C). b. Identification of the TRIOBP variant. The proband (IV1) and her younger brother (IV2) carry a homozygous 1342C > T variant, and her mother (III45) and father (III54) carry a heterozygous 1342C > T variant, as indicated by stars. c. Localization of the pathogenic TRIOBP variant. Schematic representations of the genomic structure and alternative transcript classes of TRIOBP: TRIOBP-5, TRIOBP-4 and TRIOBP-1 [1, 5, 7]. The novel c.1342C > T variant is located in exon 7 of TRIOBP-5 gene and alters the coding codon (Arg448) to a stop codon. Arrows, alternative translation start sites; *alternative stop codon; PH, pleckstrin homology domain; CC, coiled-coil domains
In addition to the TRIOBP c.1342C > T variant, a heterozygous HOMER2 c.826A > G (p.Ile276Val) variant with an AF of 0.002610 and a heterozygous TMC2 c.834 + 2 T > G variant in the noncoding splicing region were also identified. According to the ACMG standards and guidelines, the TMC2 and HOMER2 variants were classified as VUS (variants of uncertain significance). Sanger sequencing results revealed that the subjects of IV1, IV2 and III5 were carriers of a heterozygous HOMER2 c.826A > G variant and the subjects of IV1 and III4 were carriers of a heterozygous TMC2 c.834 + 2 T > G variant. The results indicate that the compound heterozygous variants of HOMER2 c.826A > G and TMC2 c.834 + 2 T > G in the proband are inherited from her parents.
Discussion
The cochlear hair cells in the inner ears detect sound by deflection of stereocilia. At the base of the stereocilia, the actin filaments are packed to form rootlets, which are boundled by TRIOBP and extend into the cell body [12, 13]. The rootlets act as pivots for the deflecton of the stereocilia. Mice deficient in TRIOBP-5 result in thin rootlets [14], and mice loss of of TRIOBP-5/− 4 fail to develop rootlets [1, 13]. In the present study, the TRIOBP gene of the family members harbors a c.1342C > T (p.Arg448*) variant, which may give rise to a truncated protein and result in disruption of the rootlets of stereocilia. This may be the main reason of hearing loss in the two siblings.
It is known that the R1 motif of TRIOBP-5/− 4 is the major actin-binding domain [6]. There are also iregular actin-binding sites in the CC domains or PH domains in TRIOBP-5/− 1 [5, 15, 16]. In this study, the TRIOBP proteins are predicted to be truncated within the R1 motif due to the mutation. Therefore, the R1 and R2 motifs of TRIOBP-5/− 4, and the PH and CC domains of TRIOBP-5 are predicted to be devoid of function [5], which may impair the actin binding activity of the TRIOBP proteins.
TRIOBP is not only a structure protein, but also a signal molecule. Study has shown that TRIOBP interacts with Trio protein to form complexes that coordinate actin remodeling. The Trio protein is derived from Dbl-homology guanine nucleotide exchange factors (DH-GEFs), which can also serve as transcription factors [3, 17, 18]. Thus, the TRIOBP c.1342C > T (p.Arg448*) variant may also impair the DH-GEFs related pathwaws, contributing to the development of deafness in the individuals.
There were reports that variants in HOMER2 and TMC1 genes may cause autosomal dominant non-syndromic deafness [19,20,21]. In addition to the homozygous TRIOBP c.1342C > T (p.Arg448*) mutation, a heterozygous HOMER2 c.826A > G (p.Ile276Val) variant and a heterozygous TMC2 c.834 + 2 T > G variant were also identified in the genome of the proband in this study. The TRIOBP variant is likely pathogenic, and the HOMER2 and TMC2 variants are VUS. The Homozygous TRIOBP c.1342C > T (p.Arg448*) variant is consistent with the main clinical phenotypes of the patients, whereas the compound heterozygous HOMER2 c.826A > G (p.Ile276Val) and TMC2 c.834 + 2 T > G variants may have little clinical significance, as the parents of the proband show normal communication ability.
Conclusions
In the present study, a novel TRIOBP c.1342C > T (p.Arg448*) variant was identified in a Chinese family. The mutation is predicted to produce a truncated TRIOBP-5 and TRIOBP-4, leading to severe to profound congenital deafness. The findings of this study will broaden the spectrum of the pathogenic variants in TRIOBP gene. The characteristics of deafness in the family imply that marriage between close relatives should be avoided to prevent genetic disorders.
Availability of data and materials
The datasets generated and analysed during the current study are available in the [NCBI SRA] repository, [Accession number: PRJNA633183; Web link: https://www.ncbi.nlm.nih.gov/Traces/study/?acc=PRJNA633183].
Abbreviations
- TRIOBP:
-
Trio- and F-actin-binding protein
- DFNB28:
-
Deafness of autosomal recessive type 28
- WES:
-
Whole exome sequencing
- PH:
-
Pleckstrin homology domain
- CC:
-
Coiled-coil domains
- BERA:
-
Brainstem evoked response audiometry
- dB SPL:
-
Decibel sound pressure level
- SNVs:
-
Single-nucleotide variants
- indels:
-
Insertions/deletions
- ExAC:
-
Exome aggregation consortium database
- gnomAD:
-
Genome aggregation database
- CNGMD:
-
The gene mutation database of Chinese
- ACMG:
-
American college of medical genetics and genomics
- AF:
-
Alternative allele frequency
- VUS:
-
Variant of uncertain significance
- DH-GEFs:
-
Dbl-homology guanine nucleotide exchange factors
- HOMER2:
-
Homer scaffolding protein 2
- TMC1:
-
Transmembrane channel-like 1.
References
Kitajiri S, Sakamoto T, Belyantseva IA, Goodyear RJ, Stepanyan R, Fujiwara I, Bird JE, Riazuddin S, Ahmed ZM, Hinshaw JE, et al. Actin-bundling protein TRIOBP forms resilient rootlets of hair cell stereocilia essential for hearing. Cell. 2010;141(5):786–98.
Riazuddin S, Khan SN, Ahmed ZM, Ghosh M, Caution K, Nazli S, Kabra M, Zafar AU, Chen K, Naz S, et al. Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness. Am J Hum Genet. 2006;78(1):137–43.
Shahin H, Walsh T, Sobe T, Abu Sa'ed J, Abu Rayan A, Lynch ED, Lee MK, Avraham KB, King MC, Kanaan M. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet. 2006;78(1):144–52.
Park S, Lee H, Kim M, Park J, Kim SH. Emerging roles of TRIO and F-actin-binding protein in human diseases. Cell Commun Signal. 2018;16(1):29.
Pollak A, Lechowicz U, Murcia Pienkowski VA, Stawinski P, Kosinska J, Skarzynski H, Oldak M, Ploski R. Whole exome sequencing identifies TRIOBP pathogenic variants as a cause of post-lingual bilateral moderate-to-severe sensorineural hearing loss. BMC Med Genet. 2017;18(1):142.
Bao J, Bielski E, Bachhawat A, Taha D, Gunther LK, Thirumurugan K, Kitajiri S, Sakamoto T. R1 motif is the major actin-binding domain of TRIOBP-4. Biochemistry. 2013;52(31):5256–64.
Wesdorp M, van de Kamp JM, Hensen EF, Schraders M, Oostrik J, Yntema HG, Feenstra I, Admiraal RJC, Kunst HPM, Tekin M, et al. Broadening the phenotype of DFNB28: mutations in TRIOBP are associated with moderate, stable hereditary hearing impairment. Hear Res. 2017;347:56–62.
Gu X, Guo L, Ji H, Sun S, Chai R, Wang L, Li H. Genetic testing for sporadic hearing loss using targeted massively parallel sequencing identifies 10 novel mutations. Clin Genet. 2015;87(6):588–93.
Zou S, Mei X, Yang W, Zhu R, Yang T, Hu H. Whole-exome sequencing identifies rare pathogenic and candidate variants in sporadic Chinese Han deaf patients. Clin Genet. 2020;97(2):352–6.
Shang H, Yan D, Tayebi N, Saeidi K, Sahebalzamani A, Feng Y, Blanton S, Liu X. Targeted next-generation sequencing of a deafness gene panel (MiamiOtoGenes) analysis in families unsuitable for linkage analysis. Biomed Res Int. 2018;2018:3103986.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303.
Flock A, Cheung HC. Actin filaments in sensory hairs of inner ear receptor cells. J Cell Biol. 1977;75(2 Pt 1):339–43.
Boutet de Monvel J, Petit C. Wrapping up stereocilia rootlets. Cell. 2010;141(5):748–50.
Katsuno T, Belyantseva IA, Cartagena-Rivera AX, Ohta K, Crump SM, Petralia RS, Ono K, Tona R, Imtiaz A, Rehman A, et al. TRIOBP-5 sculpts stereocilia rootlets and stiffens supporting cells enabling hearing. JCI Insight. 2019;4(12):e128561.
Yao L, Janmey P, Frigeri LG, Han W, Fujita J, Kawakami Y, Apgar JR, Kawakami T. Pleckstrin homology domains interact with filamentous actin. J Biol Chem. 1999;274(28):19752–61.
Sobue K, Sellers JR. Caldesmon, a novel regulatory protein in smooth muscle and nonmuscle actomyosin systems. J Biol Chem. 1991;266(19):12115–8.
Yano T, Yamazaki Y, Adachi M, Okawa K, Fort P, Uji M, Tsukita S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J Cell Biol. 2011;193(2):319–32.
Seipel K, O'Brien SP, Iannotti E, Medley QG, Streuli M. Tara, a novel F-actin binding protein, associates with the Trio guanine nucleotide exchange factor and regulates actin cytoskeletal organization. J Cell Sci. 2001;114(Pt 2):389–99.
Lu X, Wang Q, Gu H, Zhang X, Qi Y, Liu Y. Whole exome sequencing identified a second pathogenic variant in HOMER2 for autosomal dominant non-syndromic deafness. Clin Genet. 2018;94(5):419–28.
Azaiez H, Decker AR, Booth KT, Simpson AC, Shearer AE, Huygen PL, Bu F, Hildebrand MS, Ranum PT, Shibata SB, et al. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet. 2015;11(3):e1005137.
Askew C, Rochat C, Pan B, Asai Y, Ahmed H, Child E, Schneider BL, Aebischer P, Holt JR. Tmc gene therapy restores auditory function in deaf mice. Sci Transl Med. 2015;7(295):295ra108.
Acknowledgements
We thank Dr. Zhaoli Du in Yin Feng Biotechnology Company (Jinan, China) for her assistance in bioinformatics analysis.
Funding
This project was supported by the National Natural Science Foundation of China (No. 81271092, 81570927 and 81771020), Grant for Scientific and Technological Development in Shandong (2014GSF118083) and Research Initiation Grant of Binzhou Medical University (BY2012KYQD01). The funding bodies played no role in the design of the study and collection, analysis and interpretation of data, and in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: FH and XW; performed the experiments: BZ, LY, YW, YX and WS; clinical data collection: LY and XW; analyzed the data: FH, XW, BZ and WS; wrote the paper: FH and BZ. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was approved by the medical ethics committee of the Yantai.
Yuhuangding Hospital, Qingdao University. Written informed consent was.
obtained from each of the participants.
Consent for publication
Written informed consent for publication of the participants’ clinical details was obtained from each of the participants.
Competing interests
The authors declare that they have no competing interests with regard to this study.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, B., Yu, L., Wang, Y. et al. A novel mutation in TRIOBP gene leading to congenital deafness in a Chinese family. BMC Med Genet 21, 121 (2020). https://doi.org/10.1186/s12881-020-01055-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01055-5