
Abstract
Background
Genetic factors play an important role in hearing loss, contributing to approximately 60 % of cases of congenital hearing loss. Autosomal dominant deafness accounts for approximately 20 % of cases of hereditary hearing loss. Diseases with autosomal dominant inheritance often show pleiotropy, different degrees of penetrance, and variable expressivity.
Methods
A three-generation Chinese family with autosomal dominant nonsyndromic hearing impairment (ADNSHI) was enrolled in this study. Audiometric data and blood samples were collected from the family. In total, 129 known human deafness genes were sequenced using next-generation sequencing (NGS) to identify the responsible gene mutation in the family. Whole Exome Sequencing (WES) was performed to exclude any other variant that cosegregated with the phenotype.
Results
The age of onset of the affected family members was the second decade of life. The condition began with high-frequency hearing impairment in all family members excluding III:2. The novel ACTG1 c.638A > G (p.K213R) mutation was found in all affected family members and was not found in the unaffected family members. A heterozygous c.638A > G mutation in ACTG1 and homozygous c.109G > A (p.V37I) mutation in GJB2 were found in III:2, who was born with hearing loss. The WES result concurred with that of targeted sequencing of known deafness genes.
Conclusions
The novel mutation p.K213R in ACTG1 was found to be co-segregated with hearing loss and the genetic cause of ADNSHI in this family. A homozygous mutation associated with recessive inheritance only rarely co-acts with a dominant mutation to result in hearing loss in a dominant family. In such cases, the mutations in the two genes, as in ACTG1 and GJB2 in the present study, may result in a more severe phenotype. Targeted sequencing of known deafness genes is one of the best choices to identify the genetic cause in hereditary hearing loss families.
Similar content being viewed by others

Background
Nonsyndromic hearing impairment (NSHI) is a common sensory defect in humans, and most cases of NSHI are attributable to genetic factors [1]. The inheritance patterns of NSHI include autosomal dominant, autosomal recessive, X-linked, and mitochondrial inheritances. Autosomal dominant deafness accounts for approximately 20 % of cases of hereditary hearing loss [2]. To date, more than 30 genes and 50 genetic loci have been implicated in autosomal dominant NSHI (ADNSHI) (http://hereditaryhearingloss.org/). These deafness genes encode a multiplicity of proteins that function in various cell types, structures, and processes in the cochlea [3].
Diseases with autosomal dominant inheritance exhibit pleiotropy, different degrees of penetrance, and variable expressivity. ADNSHI is difficult to distinguish phenotypically [2]. In contrast to autosomal recessive nonsyndromic hearing loss, most patients with ADNSHI show large variation in age of onset; hearing impairment often begins before the age of 20 years and progresses gradually. However, deafness, autosomal dominant nonsyndromic sensorineural 4 (DFNA4,OMIM:600652), DFNA9 (OMIM:601369), and DFNA10 (OMIM:601316) are associated with hearing impairment starting in the third and fourth decades of life [2]. Additionally, the ADNSHI phenotypes vary and are divided into low-frequency, mid-frequency, high-frequency, and all-frequency hearing impairment [4]. Thus, in clinical molecular diagnosis, the hearing loss phenotype in a patient can help to select a limited number of genes for mutational analysis.
Digenic mutations leading to hearing loss have been reported in previous studies and include GJB2/GJB6 [5], GJB2/GJB3 [6], and KCNJ10/SLC26A4 [7]. However, all exhibit recessive/recessive inheritance, while dominant/dominant and dominant/recessive inheritances are rare.
We herein report a family with eight individuals affected by sensorineural hearing loss. We used next-generation sequencing (NGS) to analyse 129 known deafness genes and identify the responsible gene mutation in the family. Whole exome sequencing (WES) was performed to exclude any other variant that cosegregated with the phenotype. The results identified one novel mutation, c.638A > G [p.K213R],in ACTG1 in this family. A dominant mutation co-acting with a recessive mutation (heterozygous c.638A > G in ACTG1 and homozygous c.109G > A in GJB2) was found in one sibling.
Results
Clinical presentation of the family
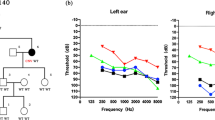
The family evaluated in this study included eight affected individuals and five unaffected members (Fig. 1a). Audiograms of the affected individuals revealed progressive, bilateral, sensorineural hearing loss that began at high frequencies. The degree of hearing loss increased with age, with threshold shifts ultimately seen at all frequencies. After the third decade of life, the progression of hearing loss led to profound hearing loss across all frequencies in the affected family members. The age of onset of hearing loss was during the second decade of life (Table 1). However, the proband, III:2, a 15-year-old boy, was born with hearing loss with a mean hearing threshold of approximately 60 dB (Fig. 1e).

Combined figure. a Pedigree of the family with ADNSHL. Affected family members are denoted in black. The arrow indicates the proband. b–h udiogram of the family members
A CT scan of the temporal bone in the proband excluded inner ear malformation. Physical examination of all family members revealed no signs of systemic illness or dysmorphic features. None of the affected individuals displayed tinnitus.
Mutation detection and analysis
First, mutations of the common deafness genes GJB2, SLC26A4, and mtDNA 12SrRNA were investigated in the affected family members by sequencing. For GJB2, the proband (III:2) was homozygous for the c.109G > A (p.V37I) mutation, while his parents were heterozygous for c.109G > A (Fig. 2a, Table 1). The c.109G > A mutation in GJB2 did not cosegregate with the phenotype in this family. Then we performed the targeted sequencing of 129 known deafness genes in individuals I:1,I:2,II:1,II:2,II:3,II:6 and III:6.

Mutation detection and conservation analysis. a GJB2 mutation analysis. Sequencing results show that the homozygous c.109G > A was found in III:2 and that the parents exhibited heterozygous c.109G > A. b ACTG1 mutation analysis. Sequencing results show that heterozygous c.638A > G was found in all affected family members and that wild-type ACTG1 was found in the unaffected members. c Protein alignment shows conservation of the K213 residue ofACTG1 across 15 species
We identified a novel mutation (c.638A > G [p.K213R]) in exon 4 of ACTG1 in the affected family members. This mutation results in a lysine to arginine substitution at position 213 in ACTG1. Sanger sequencing revealed that all of the affected family members were heterozygous for this mutation, while the mutation was not observed in the unaffected family members (Fig. 2b, Table 1). The ACTG1 c.638A > G mutation was not detected in the normal hearing controls.
The lysine at position 213 in ACTG1 is conserved across 15 species, as depicted in Fig. 2c. Both PolyPhen-2 and MutationTaster predicted that ACTG1 c.638A > G (p.K213R) was a damaging mutation.
To exclude any other variant that cosegregated with the phenotype, whole exome sequencingwas performed. The proband and his parents (III:2, II:1, and II:2) were examined. For each sample, we obtained approximately 4.0–5.3 Gb of data after whole exome sequencing. The data mapped to the targeted region had a mean depth of 135.12 fold, and 99.62 % at the depth of 4X, 98.57 % at the depth of 10X, and 96.50 % at the depth of 20X of targeted bases were covered. For bioinformatic analysis, we focused on variants in coding regions. Variants in individuals and their parents with a MAF < 0.005 were filtered using ten databases: the 1000 Genomes Project, HapMap, EVS, Wellderly, and ExAC_v0.3 databases; and five in-house databases from BGI. After completing this filtering process, we identified twelve genes with variants shared by the two cases (Table 2). Among the twelve genes, the hearing loss-related gene ACTG1 was identified. Considering the WES results, the prediction results using SIFT, PolyPhen, MutationTaster, MutationAssessor, LRT, FATHMM, GERP+, PhyloP, SiPhy, GERP, and phastCons (Table 3); the gene pathways; and their expression in the human fetal cochlear EST database, the variants in COL6A1 were then tested using Sanger sequencing. The variant in COL6A1 did not cosegregate in the affected and unaffected individuals. Therefore, ACTG1 was identified as the gene associated with the family. The whole exome sequencing result concurred with that oftargeted sequencing of known deafness genes.
Structural modelling of p.K213R
A molecular model of γ-actin was constructed based on the crystal structure of the heterodimer (PDB ID: 3ub5A). The constructed model covered the target sequence of ACTG1 (residues 6–375). The sequence identity between the target and template was 99.73 %, higher than the average 25.00 %. Quality of the model were evaluated and fixed by Verify 3D and the results showed 99.46 % of the residues had an averaged 3D-1D score > =0.2 (pass). Using Swiss-PdbViewer 4.1, the mutation was predicted to lose two hydrogen bonds (2.68A, 3.24A) and influence the interaction with ATP due to the substitution of lysine by arginine (Fig. 3). MUpro software predicted that this mutation decreased the stability of the protein structure.

Structure of wild-type and mutant ACTG1. a K213 in the wild-type protein has two hydrogen bonds that interact with ATP. b Mutant R213 has lost its hydrogen bonds and does not interact with ATP
These data together with the clinical presentation of the affected individuals clearly indicate that the ACTG1 c.638A > G (p.K213R) mutation was the genetic cause of ADNSHI in this family.
Discussion
The ACTG1 gene encodes γ-actin, which is a cytoskeletal protein abundantly expressed in the sensory hair cells of the cochlea [8, 9]. ACTG1 is linked to the DFNA20/26 locus on chromosome 17q25.3. ACTG1 is predicted to be essential for the shape and function of the stereocilia of the hair cells [10]. Recently, several exome sequencing studies were carried out and ACTG1 mutations were identified as causes for either deafness or Baraitser-Winter syndrome. Park G et al. designed a multiphasic analysis of copy number variation, linkage, and single nucleotide variation of whole exome sequencing data for the efficient discovery of mutations causing nonsyndromic hearing loss, and selected a novel variant p.M305T in ACTG1 as a disease-causing variant in a three generation pedigree [11]. Using whole-exome sequencing, Rivière JB et al. identified de novo missense mutations in the cytoplasmic actin-encoding genes ACTB and ACTG1 in Baraitser-Winter syndrome (a disorder characterized by distinct craniofacial features, ocular colobomata and neuronal migration defect) patients [12]. Mutations in γ-actin cause hearing loss mainly by impairing the function and/or viability of hair cells [10, 13, 14]. To date, the following 12 missense mutations in ACTG1 have been reported to cause ADNSHI: p.D51N, p.T89I(rs28999111), p.K118N(rs267606630), p.K118M(rs104894544), p.I122V(rs281875330), p.D187H, p.E241K(rs267606631), p.P264L(rs104894546), p.T278I(rs28999112), p.M305T, p.P332A(rs104894545), and p.V370A(rs104894547) [3, 10, 11, 13–17]. All are located in exons 3, 4, 5, and 6 of ACTG1 (Table 4).
γ-Actin comprises four subdomains (subdomains 1–4). The novel missense mutation p.K213R, which is caused by a c.638A > G transversion, results in the substitution of lysine for arginine in subdomain 4, which also causes the mutations p.D187H, p.E241K, and p.P264L. Even minor changes in this domain may lead to major effects on the structural stability of the actin polymer [18]. For p.D187H, it has been suggested that the mutation could collapse the polymerisation–depolymerisation balance of microtubules, leading to the destruction of cellular homeostasis in normal hair cells [19]. The p.E241K mutation was shown to lead to abnormal formation of thick randomly oriented action filament bundles [16]. Mice with the p.P264L mutation develop hearing loss concomitant with loss of the shortest row of actin stererocilia in hair cells [20]. These findings suggest that mutations in subdomain 4 affect normal hair cells.
Mutations in ACTG1 have been associated with DFNA20 and DFNA26. Patients with DFNA20 and DFNA26 disorders display sensorineural hearing loss, initially only at high frequencies and steadily progressing to include all frequencies [21, 22]. Distortion product otoacoustic emission data show a cochlear lesion [21]. Phenotypic differences have been shown between previously reported DFNA20/26 families, in which tinnitus was not a feature, and the p.D51N mutation family, which is associated with tinnitus [15]. Tinnitus was not found in the present study.
The age of onset of hearing loss, which ranges from the first to third decade of life, differs among the different mutations in ACTG1. The age of onset of hearing loss caused by mutation p.D51N located in subdomain 2 of γ-actin is during the first decade of life [15]. And that associated with mutations p.T89I, p.K118M, p.K118N, and p.I122V located in subdomain 1 of γ-actin [3, 13, 16] as well as mutations p.T278I, p.M305T, and p.P332A located in subdomain 3 [11, 13, 14] ranges from the first to third decade. The average age of onset of hearing loss associated with mutations p.D187H, p.E241K, and p.P264L located in subdomain 4 is from the first to second decade of life [13, 16, 17] (Table 4).
In the present study, the age of onset of hearing loss for the affected family members excluding III:2 was during the second decade of life. III:2 was a 13-year-old boy who was born with hearing loss; his pure-tone average threshold for four frequencies (0.5, 1.0, 2.0, and 4.0 KHz) were 61.25 and 56.25 dB HL for the right and left, respectively. Interestingly, homozygous c.109G > A (p.V37I) in GJB2 and heterozygous c.638A > G (p.K213R) in ACTG1 were found in III:2, which differed from the other affected family members (Table 1). The c.109G > A (rs72474224) mutation is very common, although its pathogenicity remains controversial. However, it was previously reported that GJB2 homozygous c.109G > A was considered to be a pathogenic mutation causing moderate to profound deafness [23]. In one study, c.109G > A was the second most frequent mutation among 126 south Chinese patients with NSHI carrying a homozygous or compound heterozygous GJB2 pathogenic mutation (18.0 %, 126/701) [24]. In another study, the c.109G > A mutation had an allelic frequency of 6.7 % (185/2744) in Chinese Han patients, and this frequency was significantly higher than that in the control population (2.8 %, 17/602; P = 0.0003) [25]. These results support the notion that the GJB2 recessive mutation c.109G > A should be considered a pathogenic mutation. Digenic mutations resulting in hearing loss have been reported in previous studies [5–7], but all exhibited recessive/recessive inheritance; a homozygous mutation associated with DFNB co-acting with a heterozygous mutation associated with DFNA is rare. In this study, III:2 carried homozygous c.109G > A in GJB2 and heterozygous c.638A > G in ACTG1. These digenic mutations in III:2 showed an earlier age of onset than in the other affected family members. The audiogram of III:2 showed hearing loss at all frequencies, which was a more severe phenotype than that in III:4, although both of these family members were of the same age (Fig. 1e, g). We reviewed literatures on the phenotype of GJB2 c.109G > A homozygous mutation and found that the onset age of hearing loss in those cases carrying the c.109G > A homozygous mutation varies from born to 13 years old. And the extent of hearing impairment is mainly mild to moderate except 4 cases. For the exception, one case showed progressive hearing loss reaching bilateral severe sensorineural hearing loss (85–90 dB) at 4 years old, another case progressed to severe hearing loss at the age of 2 years old, and the other two showed moderately severe sensorineural hearing loss (Table 5). These observations suggest that 1) additive effect of the GJB2 and ACTG1 gene defects might play a role in the phenotype in III:2; 2) GJB2 might have synergized with ACTG1 to result in a more severe phenotype in III:2.
Conclusions
In this study, by using targeted sequencing of known deafness genes, we identified a novel mutation, c.638A > G (p.K213R), in ACTG1 in a Chinese autosomal dominant deafness family. We found one affected family member with a heterozygous c.638A > G mutation in ACTG1 and a homozygous c.109G > A mutation in GJB2. To our knowledge, this is a rare DFNA family in whichone individual was affected by both a dominant mutation and a recessive mutation in two different genes. The more severe hearing phenotype was suggested to be resulted from these digenic mutations. This rare hereditary mode should be considered in clinical genetic diagnosis and counselling. Our results not only add to the theoretical basis of hereditary hearing loss, but will promote the translation of deafness gene capture and NGS in otology clinics.
Methods
Ethics statement
This study was approved by the Chinese PLA General Hospital Research Ethics Committee. Fully informed written consent for participation and publication of clinical data was attained from each subject or their guardians when the age of subjects <18 years old.
Clinical data
A three-generation Chinese family with eight affected members and five unaffected members from Sichuan Province was evaluated. The medical history of each family member was obtained using a questionnaire (Additional file 1) that included the degree of hearing loss, age of onset of hearing loss, progression of hearing loss, symmetry of hearing loss, use of hearing aids, presence of tinnitus, pathological changes in the ear, infection, ototoxicity, noise exposure, and other relevant clinical manifestations to understand the otologic manifestation and exclude any history of other diseases and environmental factors. The proband underwent a number of clinical tests including general physical examinations, chest X-rays, brain MRI, and temporal bone CT. No abnormalities were detected in these tests, thus excluding the possibility that the hearing loss in this family was syndromic.
All genomic DNA (gDNA) was extracted from peripheral blood using a blood DNA extraction kit according to the protocol provided by the manufacturer (TianGen, Beijing, China).
Audiometric analysis
Pure-tone audiometry with air and bone conduction was performed according to standard protocols in a sound-controlled room at frequencies ranging from 250 to 8000 Hz. Audiograms were available for six of the eight affected family members and for the one unaffected member.
Deafness gene capture and Illumina library preparation
Among the affected family members, mutations in the common deafness genes GJB2, SLC26A4, and mtDNA 12SrRNA mutations were excluded, with the exception of homozygous c.109G > A in GJB2, which was found in III:2. Targeted NGS was then used to sequence 129 known deafness genes (http://www.otogenetics.com/forms/Deafness_v3_gene_list.pdf).DNA specimens from five patients and two normal hearing members of the family were sequenced by Otogenetics Corporation (Atlanta, GA, USA) using next-generation sequencingwith the Illumina platform. The quality of gDNA was examined by checking the optical density ratio (260/280 ratio) and performing gel electrophoresis imaging. High-molecular weight gDNA (approximately 3 μg) was fragmented ultrasonically using a Covaris E210 DNA shearing instrument (Covaris, Inc., Woburn, MA, USA) to an average size of 300 base pairs (bp). The Covaris protocol was a 3-min total duration, duty cycle of 10 %, intensity of 5, and 200 cycles per burst.
Exons and their flanking 50 bp from 129 known human deafness genes were selected for capture and NGS sequencing using an Illumina HiSeq2000. Hybridisation probes of 0.5 to 1.6 kilobase pairs (kb) were generated for these genes from either cDNA clones of the genes or by polymerase chain reaction (PCR) amplification from targeted gDNA regions. To ensure reliable capture of shorter exons, we specifically generated longer hybridisation probes from gDNA for those exons that were shorter than 50 bp by including approximately 100 bp genomic DNA flanking the exons on both sides. All PCR products (10 ng each) were purified using the QIAquick PCR Purification Kit (Qiagen, Valencia, CA, USA) before use. Further details of the capture probe validation and preparation can be found in a previous report [26].
Fragmented gDNA libraries for Illumina GAII sequencing were prepared using the NEBNextTM DNA Sample Prep Master Mix set (E6040; NEB Biolab, Ipswich, MA, USA). End repair of DNA fragments, addition of a 3′ adenine (A), adaptor ligation, and reaction clean-up were performed according to the manufacturer’s protocol. The libraries were purified and size-selected using the AMPure DNA Purification kit (Beckman Agencourt, Danvers, MA, USA). The ligated product (20 ng) was amplified over 14 PCR cycles using the Illumina PCR primers InPE1.0 and InPE2.0 and indexing primers according to the manufacturer’s instructions.
For targeted enrichment of deafness genes, the Illumina library DNA was purified using a QIAquickMinElute column and eluted into 50 μL hybridisation buffer (Roche NimbleGen, Madison, WI, USA). The barcoded Illumina gDNA libraries (5 μg) were incubated in 16 μL hybridisation buffer on the surface of hybridisation glass slides on a hybridisation station (BioMicro Systems, Inc., Salt Lake City, UT, USA) at 42 °C for 72 h. Nonspecific DNA fragments were removed after a series of six washing steps in washing buffer (Roche NimbleGen, Madison, WI, USA). The DNA bound to the probes was eluted by a 10-min incubation with NaOH (425 mL, 125 mM). The eluted solution was transferred to a 1.5-mL Eppendorf tube containing 500 μL neutralisation buffer (Qiagen’s PBI buffer). The neutralised DNA was desalted and concentrated on a QIAquickMinElute column and eluted into 30 μL EB buffer. To increase the yield, we typically amplified 5 μL eluted solution by 12 cycles of PCR using the Illumina PCR primers InpE1.0 and 2.0. Enrichment of the targeted deafness genes was examined by comparing the growth curves of captured and noncaptured samples during quantitative PCR [26]. Twelve barcoded libraries of captured samples were pooled, and paired-end Illumina sequencing was performed using the Illumina HiSeq system (Illumina, San Diego, CA, USA). Details of the bioinformatic analysis methods used have been published previously [26].
The sequence data were mapped with BWA (0.7.4) against the human reference genome index (hg19), and then analyzed with Picard to remove duplicates from the mapped reads. Variants in the data (SNPs/indels) were called with SAMtools (0.1.19) across the genome and exported in VCF format; 516.7 ± 28.5 variants were obtained per sample. All of the variants in the target regions were selected based on the bed file provided by Otogenetics, and then annotated with ANNOVAR and the internal mutation database to get information on the impact of each variant, predicted functional changes, 1000 Genome Project population allele frequency, and associated diseases, if applicable. Variants with known disease associations, a deleterious functional impact, or aMAF(Minor Allele Frequency) <0.04 were selected as candidate mutations for analysis and validation; 20.4 ± 5.4 variants were obtained per sample. To identify the pathogenic mutation, a cosegregation analysis of the family members and an in-house database of 481 Chinese normal hearing controls from Otogeneticswas applied.
Whole exome sequencing
Exome capture was performed in the proband and his parents by BGI–Shenzhen using NimbleGen SeqCap EZ Human Exome Library v3.0 (Roche NimbleGen, Inc., Madison, WI, USA) according to the manufacturer’s protocols, and sequencing was performed using a HiSeq2000 platform (Illumina, San Diego, CA, USA). Illumina base calling Software 1.7 was used with default parameters to process the raw image files and to sequence the individual products as 90-bp paired-end reads. The sequenced reads were aligned to the human genome reference (UCSC hg19 version, build37.1) using SOAP aligner/SOAP2[27]. SNP or indels were called using Soapsnp [28] software and BWA [29], respectively. The alignment results were identified using GATK [30] to identify the breakpoints.
Sanger sequencing
After filtering against multiple databases, Sanger sequencing was used to determine whether any of the potential mutations in known genes causing ADNSHI co-segregated with the phenotype in this family. Direct PCR products were sequenced using Bigdyeterminator v3.1 cycle sequencing kits (Applied Biosystems, Foster City, CA, USA) and analysed using an ABI 3700XL Genetic Analyzer.
Mutational analysis
Segregation of the mutations was evaluated in all family members. Genotyping for c.638A > G was performed by PCR and detected by bidirectional sequencing of the amplified fragments using an automated DNA sequencer (ABI3100); the primers were 5′-CAGAGCCCTCCCTTAGTGAT-3′ and 5′-CGAGGCTACAGCTTCACCAC-3′. Nucleotide alterations were identified by sequence alignment with the ACTG1 GenBank sequence (NG_011433) using Genetool software.
Multiple sequence alignment
Multiple sequence alignment was performed across 15 species using ClustalW2 online (http://www.ebi.ac.uk/Tools/msa/clustalw2/).
Model building and structure-based analysis
Three-dimensional modelling of the human wild-type and p.K213R mutation was performed using SWISS-MODEL [31], an automated homology modelling program (http://swissmodel.expasy.org/workspace/). We used the automatic modelling approach to model the complete human ACTG1 protein, including its 375 amino acids (NP_001186883.1) with or without the mutations. Data obtained from the homology models were visualised using Swiss-PdbViewer 4.1. Quality of the structure model were assessed by Verify 3D.
Availability of supporting data
Sequence read data of the affected subject (II:3) has been deposited into Sequence Read Archive ([32]; accession number SRP064631).
References
Morton CC, Nance WE. Newborn hearing screening--a silent revolution. N Engl J Med. 2006;354(20):2151–64.
Bitner-Glindzicz M. Hereditary deafness and phenotyping in humans. Br Med Bull. 2002;63:73–94.
Liu P, Li H, Ren X, Mao H, Zhu Q, Zhu Z, et al. Novel ACTG1 mutation causing autosomal dominant non-syndromic hearing impairment in a Chinese family. J Genet Genomics. 2008;35(9):553–8.
Kunst H, Huybrechts C, Marres H, Huygen P, Van Camp G, Cremers C. The phenotype of DFNA13/COL11A2: nonsyndromic autosomal dominant mid-frequency and high-frequency sensorineural hearing impairment. Am J Otol. 2000;21(2):181–7.
Bolz H, Schade G, Ehmer S, Kothe C, Hess M, Gal A. Phenotypic variability of non-syndromic hearing loss in patients heterozygous for both c.35delG of GJB2 and the 342-kb deletion involving GJB6. Hear Res. 2004;188(1–2):42–6.
Liu XZ, Yuan Y, Yan D, Ding EH, Ouyang XM, Fei Y, et al. Digenic inheritance of non-syndromic deafness caused by mutations at the gap junction proteins Cx26 and Cx31. Hum Genet. 2009;125(1):53–62.
Yang T, Gurrola 2nd JG, Wu H, Chiu SM, Wangemann P, Snyder PM, et al. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84(5):651–7.
Hofer D, Ness W, Drenckhahn D. Sorting of actin isoforms in chicken auditory hair cells. J Cell Sci. 1997;110(Pt 6):765–70.
Vandekerckhove J, Leavitt J, Kakunaga T, Weber K. Coexpression of a mutant beta-actin and the two normal beta- and gamma-cytoplasmic actins in a stably transformed human cell line. Cell. 1980;22(3):893–9.
Rendtorff ND, Zhu M, Fagerheim T, Antal TL, Jones M, Teslovich TM, et al. A novel missense mutation in ACTG1 causes dominant deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are not frequent among families with hereditary hearing impairment. Eur J Hum Genet. 2006;14(10):1097–105.
Park G, Gim J, Kim AR, Han KH, Kim HS, Oh SH, et al. Multiphasic analysis of whole exome sequencing data identifies a novel mutation of ACTG1 in a nonsyndromic hearing loss family. BMC Genomics. 2003;14:191.
Riviere JB, van Bon BW, Hoischen A, Kholmanskikh SS, O'Roak BJ, Gilissen C, et al. De novo mutations in the actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nature genetics. 2012;44(4):440–4. S441-442.
Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, et al. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet. 2003;73(5):1082–91.
van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, Cremers CW, et al. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J Med Genet. 2003;40(12):879–84.
de Heer AM, Huygen PL, Collin RW, Oostrik J, Kremer H, Cremers CW. Audiometric and vestibular features in a second Dutch DFNA20/26 family with a novel mutation in ACTG1. Ann Otol Rhinol Laryngol. 2009;118(5):382–90.
Morin M, Bryan KE, Mayo-Merino F, Goodyear R, Mencia A, Modamio-Hoybjor S, et al. In vivo and in vitro effects of two novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing impairment. Hum Mol Genet. 2009;18(16):3075–89.
Baek JI, Oh SK, Kim DB, Choi SY, Kim UK, Lee KY, et al. Targeted massive parallel sequencing: the effective detection of novel causative mutations associated with hearing loss in small families. Orphanet J Rare Dis. 2012;7:60.
Otterbein LR, Graceffa P, Dominguez R. The crystal structure of uncomplexed actin in the ADP state. Science. 2001;293(5530):708–11.
Verrills NM, Po'uha ST, Liu ML, Liaw TY, Larsen MR, Ivery MT, et al. Alterations in gamma-actin and tubulin-targeted drug resistance in childhood leukemia. J Natl Cancer Inst. 2006;98(19):1363–74.
Drummond MC, Belyantseva IA, Friderici KH, Friedman TB. Actin in hair cells and hearing loss. Hear Res. 2012;288(1–2):89–99.
Elfenbein JL, Fisher RA, Wei S, Morell RJ, Stewart C, Friedman TB, et al. Audiologic aspects of the search for DFNA20: a gene causing late-onset, progressive, sensorineural hearing loss. Ear Hear. 2001;22(4):279–88.
Yang T, Smith A. A novel locus DFNA 26 maps to chromosome 17q25 in two unrelated families with progressive autosomal dominant hearing loss. Am J Hum Genet. 2000, Suppl(67):300
Pollak A, Skorka A, Mueller-Malesinska M, Kostrzewa G, Kisiel B, Waligora J, et al. M34T and V37I mutations in GJB2 associated hearing impairment: evidence for pathogenicity and reduced penetrance. Am J Med Genet A. 2007;143A(21):2534–43.
Chen K, Zong L, Liu M, Wang X, Zhou W, Zhan Y, et al. Developing regional genetic counseling for southern Chinese with nonsyndromic hearing impairment: a unique mutational spectrum. J Transl Med. 2014;12:64.
Wu BL, Lindeman N, Lip V, Adams A, Amato RS, Cox G, et al. Effectiveness of sequencing connexin 26 (GJB2) in cases of familial or sporadic childhood deafness referred for molecular diagnostic testing. Genet Med. 2002;4(4):279–88.
Tang W, Qian D, Ahmad S, Mattox D, Todd NW, Han H, et al. A low-cost exon capture method suitable for large-scale screening of genetic deafness by the massively-parallel sequencing approach. Genet Test Mol Biomarkers. 2012;16(6):536–42.
Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7.
Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, et al. SNP detection for massively parallel whole-genome resequencing. Genome research. 2009;19(6):1124–32.
Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95.
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research. 2010;20(9):1297–303.
Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006;22(2):195–201.
Snoeckx RL, Huygen PL, Feldmann D, Marlin S, Denoyelle F, Waligora J, et al. GJB2 mutations and degree of hearing loss: a multicenter study. Am J Hum Genet. 2005;77(6):945-957.
Kenna MA, Wu BL, Cotanche DA, Korf BR, Rehm HL. Connexin 26 studies in patients with sensorineural hearing loss. Archives of otolaryngology--head and neck surgery. 2001; 127(9):1037-1042.
Marlin S, Garabedian EN, Roger G, Moatti L, Matha N, Lewin P, Petit C, Denoyelle F: Connexin 26 gene mutations in congenitally deaf children: pitfalls for genetic counseling. Archives of otolaryngology--head and neck surgery. 2001; 127(8):927-933.
Dahl HH, Tobin SE, Poulakis Z, Rickards FW, Xu X, Gillam L, et al. The contribution of GJB2 mutations to slight or mild hearing loss in Australian elementary school children. J Med Genet. 2006;43(11):850-855.
Bason L, Dudley T, Lewis K, Shah U, Potsic W, Ferraris A, et al. Homozygosity for the V37I Connexin 26 mutation in three unrelated children with sensorineural hearing loss. Clinical genetics. 2002; 61(6):459-464.
Huculak C, Bruyere H, Nelson TN, Kozak FK, Langlois S. V37I connexin 26 allele in patients with sensorineural hearing loss: evidence of its pathogenicity. Am J Med Genet A. 2006; 140(22):2394-2400.
Gallant E, Francey L, Tsai EA, Berman M, Zhao Y, Fetting H, et al: Homozygosity for the V37I GJB2 mutation in fifteen probands with mild to moderate sensorineural earing impairment: further confirmation of pathogenicity and haplotype analysis in Asian populations. Am J Med Genet A. 2013; 161A(9):2148-2157.
Acknowledgements
These investigations were supported by National Natural Science Foundation of China (81230020) to P.D. and grant from National Natural Science Foundation of China (81371098), Beijing Natural Science Foundation (7132177), Beijing Nova programme (2009B34) to Y.Y.Y. Grants from the U.S. National Institute on Deafness and other Communication Disorders (NIDCD R01DC010204, RO1 DC006483 and 4R33DC010476) to X.L. Grants from China Postdoctoral Science Foundation (No. 2012 M521878, No. 2013 T60947) and National Natural Science Foundation of China (81570929)to X.G. Grants from The National High Technology Research and Development Program of China 863 Program (2012AA020101), National Natural Science Foundation of China (81360159) and National Basic Research Program of China (2014CB541706) to G.J.W. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We sincerely thank all the family members for their participation and cooperation in this study.
Web Resources
dbSNP: http://www.ncbi.nlm.nih.gov/projects/SNP/
1000 genomes: http://www.1000genomes.org
Hapmap: http://hapmap.ncbi.nlm.nih.gov/
EVS: http://evs.gs.washington.edu/EVS/
Wellderly: http://www.stsiweb.org/wellderly/
ExAC_v0.3: http://exac.broadinstitute.org/
SIFT: http://sift.jcvi.org/
Polyphen2: http://genetics.bwh.harvard.edu/pph2/index.shtml
MutationTaster: http://www.mutationtaster.org/
MutationAssessor: http://mutationassessor.org
LRT: http://www.unc.edu/~yunmli/UNCcombo/
FATHMM: http://fathmm.biocompute.org.uk/
PhyloP: http://compgen.bscb.cornell.edu/phast/
SiPhy: http://www.broadinstitute.org/genome_bio/siphy/index.html
Gerp/GERP+: http://mendel.stanford.edu/sidowlab/downloads/gerp/index.html
Phastcons: http://compgen.bscb.cornell.edu/phast/
Morton Cochlear ESTDatabase:
http://www.brighamandwomens.org/bwh_hearing/human-cochlear-ests.aspx
Verify3D: http://nihserver.mbi.ucla.edu/Verify_3D
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests. Xi Lin owns stocks in Otogenetics Corporation, which may be a source of potential conflict of interest for materials presented in this paper. This does not alter our adherence to BMC Genetics policies on sharing data and materials.
Authors’ contributions
YY and XG carried out the molecular genetic studies and sequence alignment. JL, BH, GW and XL carried out the bioinformatic analysis of the sequencing data. YY and BH drafted the manuscript. YQ and PD conceived the study, participated in its design and coordination. All authors have read and approved the final manuscript.
Additional file
Additional file 1:
Questionnaire for deafness. (DOCX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Yuan, Y., Gao, X., Huang, B. et al. Phenotypic Heterogeneity in a DFNA20/26 family segregating a novel ACTG1 mutation. BMC Genet 17, 33 (2016). https://doi.org/10.1186/s12863-016-0333-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12863-016-0333-1