
Abstract
Background
Maternal cytomegalovirus (CMV) infection and/or reactivation in pregnancy is associated with a myriad of adverse infant outcomes. However, the role of host genetic polymorphisms in modulating maternal CMV status is inconclusive. This study investigated the possible association of single nucleotide polymorphisms in toll-like receptor (TLR) and cytokine genes with maternal plasma CMV DNA status in black Zimbabweans.
Methods
In a cross-sectional study, 110 women in late gestation who included 36 CMV infected cases and 74 CMV uninfected, age and HIV status matched controls were enrolled. Twenty single nucleotide polymorphisms in 10 genes which code for proteins involved in immunity against CMV were genotyped using Iplex GOLD SNP genotyping protocol on the Agena MassARRAY® system. Statistical analyses were performed using Stata SE and the ‘Genetics’ and ‘SNPassoc’ packages of the statistical package R.
Results
The TLR7 rs179008A > T (p < 0.001) polymorphism was associated while the TLR9 rs352139T > C (p = 0.049) polymorphism was on the borderline for association with CMV positive (CMV+) status. In contrast, the interleukin (IL)-6 rs10499563T > C (p < 0.001) and TLR2 rs1816702C > T (p = 0.001) polymorphisms were associated with CMV negative (CMV-) status. Furthermore, allele frequencies of SNPs in TLR2, TLR4, TLR9, TLR7, IL-6, IL-10, IL-28B, IL-1A and interferon AR1 (IFNAR1) genes are being reported here for the first time in a Zimbabwean population. The allele frequencies in the Zimbabwean population are generally comparable to other African populations but different when compared to European and Asian populations.
Conclusions
Toll-like receptor and interleukin genetic polymorphisms influence CMV status in late gestation among black Zimbabweans. This is attributable to possible modulation of immune responses to CMV reactivation in a population previously exposed to CMV infection.
Similar content being viewed by others


Background
Seroprevalence of cytomegalovirus (CMV) amongst women of reproductive age ranges from 40 to 65% in the developed world and can reach 100% in developing countries [1, 2]. CMV infection in pregnancy, in the setting of both primary infection and reinfection, can be potentially transmitted to the foetus and or neonate, resulting in congenital CMV (cCMV). The consequences of CMV range from asymptomatic viraemia to potentially life changing conditions which include mental retardation and congenital sensorineural hearing loss. Studies have implicated maternal demographics, socioeconomics and HIV status among the strongest determinants of the biased occurrence and vertical transmission of CMV [3,4,5,6,7]. Furthermore, maternal immune responses to CMV infection and/or reactivation actively modulate CMV related disease outcomes [8]. Thus, variation in genes that encode components of the immune system that are directly or indirectly involved in the pathogenesis of CMV have been implicated in CMV infection outcomes [9]. However, the genetic variants, like seroprevalences and the factors influencing CMV epidemiology are heterogenous among populations hence research findings are equivocal.
Toll-like receptors (TLR) are crucial in the detection of viruses in circulation and the subsequent elicitation of an antiviral response [10, 11]. TLRs act as pattern recognition receptors of non-methylated viral CpG-containing DNA which signals the presence of CMV infection [12]. TLR2 and TLR4 are cell surface receptors while TLR3, − 7 and − 9 are endosomal receptors [13, 14]. TLRs facilitate viral attachment and entry resulting in CMV-elicited signalling antiviral responses such as type 1 interferon activation of nuclear factor kappa β (NF-k β) and pro-inflammatory cytokine gene expression [12, 15]. Activation of the type 1 interferon producing cascade and production of cytokines form the major cellular antiviral mechanisms against CMV [16,17,18]. Single nucleotide polymorphisms (SNPs) in the TLR2, TLR4, TLR7 and TLR9 genes were inconclusively reported to be associated with CMV infection [19,20,21,22,23].
In response to TLR activation, chemokine (interleukin and interferon) genes signal immediate secretion of ILs from cells such as macrophages and T-helper cells. Chemokines that trigger an immune cascade by signalling direct growth, development, maturation, activation and increased life-span of immune cells. In the case of CMV infection, chemokines signal: maturation of B-lymphocytes into plasma cells which produce anti-CMV antibodies, and activation of cytotoxic T cells for destruction of CMV infected cells [24, 25]. The differential response to CMV exposure with some but not all exposed individuals developing CMV-related diseases suggests a possible role of host genetic variation in immune response. A study by Sezgin et al. [26] showed that human interleukin-10 receptor variants potentially interfere with IL-10 binding and signal transduction influencing susceptibility to CMV retinitis. In a large Swiss HIV Cohort Study, the effect of IFNL3 TT/−G substitution, the variant allele was associated with occurrence of CMV retinitis [27]. The same allele was also associated with susceptibility to CMV replication in transplant patients [28].
Detection of host genetic variants which may confer resistance to CMV infection and reactivation could reveal potential therapeutic targets against pregnancy related CMV disease. Furthermore, host genetic determinants of CMV disease outcomes could be used as predictors of adverse outcomes of maternal CMV. While the host genetics of CMV have been studied in other populations, a glaring gap in knowledge exists among Africans. The differences in genomic variation between Africans and other populations cannot be over-emphasised, hence findings from other populations may not be an accurate reflection in Africans.
The aim of the present study was to determine if single nucleotide polymorphisms in genes that encode components of the immune system are associated reactivation of CMV in late pregnancy.
Methods
Study participants
This study was carried out among pregnant women in late gestation, seeking antenatal care at three polyclinics in Harare’s Kuwadzana, Dzivarasekwa and Glenview high density suburbs who were recruited in the University of Zimbabwe College of Health Sciences Birth Cohort (MRCZ/A/1968). The general study design, setting and participants characteristics for the main cohort are described elsewhere [29]. In summary, this cross-sectional nested sub-study enrolled 110 women aged 18 to 42 years, including 36 CMV infected cases and 74 CMV uninfected, age- and HIV status matched controls. All participants previously tested positive for CMV IgG antibodies hence, cases were presumed to have reinfection/reactivation. Whole blood and plasma specimens archived at enrolment were retrieved for host genotyping and CMV DNA detection, respectively. CMV status of participants was determined by detection of CMV DNA in plasma using the real time polymerase chain reaction (PCR) kit (RealStar CMV Kit v1.0, Altona Diagnostics, Hamburg, Germany), following manufacturer’s instructions.
Genotyping of candidate genes
Using candidate gene approach, 20 SNPs in 10 genes were selected for genotyping (Table 1). Selection of SNPS was based on the following criteria: previously reported association or plausible association with CMV infection and/or other viral infection, a minor allele frequency (MAF) ≥10% in African populations reported in the dbSNP database (Available from: http://www.ncbi.nlm.nih.gov/SNP/), except for the rs113181057 SNP whose MAF in African populations was not previously reported. Host genomic DNA was extracted from 200 μl of whole blood using the Quick-DNA™ MiniPrep Plus Kit (Zymo Research, Irvine, CA, USA), according to manufacturer’s instructions. All DNA samples were diluted to a concentration of approximately 50 ng/ul in preparation of genotyping. SNPs were genotyped using Iplex GOLD SNP genotyping protocol on the Agena MassARRAY® system (Agena BioscienceTM, San Diego, CA, USA).
Statistical analysis
Data were compiled and managed in Research Electronic Data Capture (REDCap) [30]. Statistical analyses were performed using Stata SE, version 15 (StataCorp, College Station, Texas, USA) and the ‘Genetics’ and ‘SNPassoc’ packages of the statistical package R (version 3.4.3 [2017-11-30], The R Foundation for Statistical Computing, Vienna, Austria). Numerical variables are described as either median and 25th to 75th percentiles for skewed variables or mean and standard deviation for normally distributed variables, with groups comparisons via Mann-Witney U-test and Student’s t-test respectively. Categorical variables are described as frequencies and compared across groups using Chi squared test. p-value < 0.05 was considered statistically significant. Genotype and allele frequencies were calculated using ShesisPlus [31]. SNPs were tested for departure from Hardy-Weinberg Equilibrium (HWE) expectation using a Chi square goodness of fit test. Association between SNPs and CMV status was determined using univariable logistic regression analysis. Bonferroni correction was used to account for simultaneous comparison of multiple SNPs. Dominant, log-additive, codominant, recessive and overdominant inheritance models were interrogated for association of SNPs with CMV infection. Furthermore, multivariate logistic regression analysis of SNPs that were associated with CMV infection in the univariate analysis was carried out to adjust for their effect on each other in a model that also contained BMI as covariate.
Results
Study participants’ demographic and clinical characteristics
The demographic and clinical characteristics of the 110 participants are summarised in Table 2. All participants were of child bearing age (median 28 years, 25th–75th percentile: 23–34). The group of women with a positive CMV DNA (CMV+, n = 36) status (median 24 kg/m2, 25th–75th percentile: 22–27) had a significantly lower body max index (BMI) than the group who tested negative for CMV DNA (CMV-, n = 74) (median 26 kg/m2, 25th–75th percentile: 24–29); p = 0.006. CMV+ participants also had significantly lower systolic blood pressure when compared with the CMV- participants. Age, gestational age, parity, gravidity, diastolic blood pressure, pulse rate, income, level of education and HIV status were comparable between CMV+ cases and CMV- controls.
Association between SNPs and CMV infection
Genotype data for the 20 SNPs genotyped was available for all 110 participants and the SNP rs113181057 on the IFNAR1 gene was monomorphic in the study population. There was a departure from Hardy-Weinberg equilibrium (HWE) for four of the 20 SNPs: TLR7 rs179008 in cases, TLR2 rs1816702 and IL-6 rs10499563 in the controls and IFNAR1 rs2843710 in both groups. An additional table shows genotype frequencies in the CMV+ and CMV- negative groups and the univariate logistic regression analyses of SNPs and CMV status (see Additional Table 1). Using the univariate logistic regression analysis of codominant and log additive inheritance models, 4 SNPs (rs10499563 (p < 0.001), rs179008 (p < 0.001), rs1816702 (p = 0.002) and rs352139 (p = 0.003) were significantly associated with CMV DNA status (Additional Table 1). The IL-6 rs10499563T > C polymorphism was significantly associated with lower risk of CMV infection. When compared to the IL-6 rs10499563T/T genotype, the rs10499563T/C was associated with a lower risk of CMV infection as the genotype was significantly (p < 0.001) less frequent in the CMV+ group (14%) than the CMV- group (70%). Likewise, the TLR2 rs1816702C > T SNP was significantly associated with lower risk of CMV infection. Genotype rs1816702C/C genotype was significantly (p = 0.002) higher in the CMV+ (47%) than the CMV- women (11%).
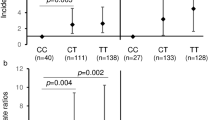
In contrast, TLR7 (rs179008A > T) and TLR9 (rs352139T > C) polymorphisms were associated with an increased risk of CMV infection. The TLR7 rs179008C/C genotype was significantly higher in the CMV+ group than the CMV- group (31% vs. 3%; p < 0.001. With reference to the TLR9 rs352139T/T genotype, both the rs352139T/C and rs352139C/C genotypes were significantly (p = 0.005) higher in the CMV+ women (28 and 58% respectively) than in the CMV- women (11 and 47% respectively). These associations remained significant after correction for multiple comparisons (Fig. 1). When other models of genetic inheritance were considered, the association of IL-6 rs10499563 maintained significant association with CMV status after Bonferonni correction (BC) in dominant, and overdominant models. SNPs rs1816702 and rs179008 also maintained significance with CMV status after BC in the dominant and recessive models (Fig. 1).

Plot of log10p-values for the association of gene with CMV DNA across models of genetic associations. For each figure panel, the lower dotted horizontal line is for the nominal p-value threshold for significance (0.05), while the upper dotted blue line for the Bonferroni corrected threshold p-value for significance
Table 3 shows multivariable logistic regression analysis of SNPs (rs10499563, rs179008, rs1816702 and rs352139) that were associated with CMV status in the univariable analyses. BMI was also included in the model. All SNPs maintained significant association with CMV infection status in at least one of the models. However significant association of rs352139 with CMV status was borderline (p = 0.049) in the log additive model while it was not significant in the other models. BMI’s association with CMV status also substantially attenuated in the multivariable logistic regression model (p = 0.068).
The IL-6 rs10499563T > C SNP was associated with low likelihood of CMV positivity in codominant (OR = 0.05; 95%CI = 0.01–0.25, p = 0.001) as well as in the log additive, dominant and overdominant models. The result shows the association of the C allele with lower odds of CMV infection even in heterozygous state (rs10499563 T/C). For the TLR7 rs179008A > T SNP, the T allele was significantly associated with higher odds of CMV infection in the codominant model (OR = 3.67; 95%CI = 0.79–116.99; p < 0.011) which, was maintained in the log additive, recessive and dominant models. Hence T allele will likely be associated with CMV+ status in both homozygous and heterozygous states (rs179008T/T and rs179008A/T). The TLR2 rs1816702T > C was significantly associated with decreased risk of CMV positivity both in the codominant (OR = 0.05; 95%CI = 0.01–0.25, p = 0.001) as well as in the log additive, dominant and overdominant models. Hence, risk of CMV infection will be decreased in the homozygous state, rs1816702C/C. The TLR9 rs352139 was significantly associated with likelihood of CMV infection, only in the log additive model (OR = 2.85: 95%CI = 0.95–8.58; p = 0.049).
Comparison of variant allele frequencies from this study with other populations
The variant allele frequencies of the genotyped SNPs were compared with data from two other populations: Asians and Europeans. Table 4 gives variant allele frequencies for the genotyped SNPs in this study as well as for Asians and Europeans as reported on dbSNP.
Discussion
The outcome of an infection is determined, in part, by the intensity of the inflammatory response [32], which varies between individuals and can be regulated at the genetic level [33]. In this study, we hypothesised the possible contribution of genetic variation to the biased occurrence of CMV infection among pregnant women. SNPs may influence the rate and regulatory dynamics of gene transcription, stability of mRNA as well as production and biological activity of resultant protein. We therefore investigated possible association between CMV infection and SNPs in 19 genes which encode proteins that are or may be involved in the immune reaction cascade against CMV. The departure from HWE in polymorphic SNPs is due to their association with CMV infection mainly because the departure is being observed when cases and controls are separated but HWE is maintained when the two groups are combined. We report a significant association between each of; rs10499563, rs179008, rs1816702 and rs352139 SNPs and CMV DNA status. To our knowledge, this is the first report on SNPs and CMV infection in an African setting.
To minimise the confounding effects of age and HIV status, which are directly related to immune function, enrolled participants were age and HIV status matched. The observation that overweight women were less likely to be CMV+ contradicts findings from previous studies where CMV infection was associated with metabolic syndrome, higher BMI and or obesity [34, 35]. Our findings could be due to none of the participants having any form or history of metabolic syndrome. Hence, we were unlikely to observe any significant associations. The observation that CMV positivity is significantly associated with low systolic blood pressure contrasts with previous findings which have shown increasing systolic blood pressure with CMV positivity [36, 37]. It is worth noting that the previous studies were carried out in non-pregnant adults, hence discrepancy in findings could be due to the well documented effects of pregnancy on fluctuations in blood pressure [38, 39] masking the effects of CMV infection.
We found an association between SNP rs10499563 (− 6331 T > C), located within the promoter region of IL-6 gene which regulates the rate of IL-6 gene transcription [40] and CMV DNA status. Individuals carrying the C allele were less likely to be CMV infected, hence likelihood of being CMV DNA positive decreased with genotypes T/T>> > T/C> > C/C. Individuals heterozygous (T/C) and homozygous (C/C) for the variant allele were significantly less likely to be CMV infected than individuals homozygous for the T allele (T/T). The IL-6 gene codes for IL-6, a versatile inflammatory cytokine whose function is related to its expression in the tissue. Smith et al. previously reported higher level of serum IL-6, in individuals with wildtype T/T genotype compared to individuals with C/C genotype, among coronary artery bypass patients (Smith et al. [41]).
Our findings could at least in part, be explained by results from the Smith et al. study. Being a pro-inflammatory cytokine, abundance of IL-6 in circulation could promote CMV activation. In contrast, the low levels of IL-6 associated with the rs10499563C allele would disfavour the occurrence of CMV infection. Serum IL-6 levels were reported to be significantly higher among the CMV infected pregnant women compared to the CMV uninfected in a Chinese cohort [42].
We also report an association between CMV DNA status and rs179008, a non-synonymous A > T (Gln11Leu) polymorphism within exon 3 of the TLR7 gene [43]. The resulting glycine to leucine change has been suggested to code for a functionally impaired TLR7 protein [44, 45]. In the present study, the T allele was associated with significantly lower odds of CMV positivity. Individuals homozygous for the variant allele T/T were significantly less likely to be CMV infected compared to individuals homozygous for the wildtype allele A/A.
Upon recognising pathogen associated molecular patterns (PAMP), TLR7 activate a signalling cascade which activates type I IFN, dendritic cells (DCs) and B lymphocytes [46]. Activated type 1 IFN, DCs and B cells are responsible for pathogen clearance, antigen recognition and antibody production. The induced immune cascade is critical in CMV clearance. In the presence of the T allele which results in a less potent protein, an insufficient signal is mounted by TLR7, hence carriers of the rs179008 T allele are at a greater risk of CMV infection. The rs179008 T allele has been linked with unfavourable outcomes in HIV and other viral infections. The variant was associated with increased susceptibility to HIV-1 and decreased IFNα production in HIV uninfected women [47]. The T allele has also been previously associated with a higher risk of hepatitis C infection and cCMV. Our findings are therefore contrasting with previous reports suggesting that the rs179008A > T SNP could be in linkage disequilibrium with another functional SNP or epistatic gene which masks the effects of rs179008A > T.
CMV DNA status was also associated with rs1816702C > T, a SNP located in intron 2 of the TLR2 gene. The C variant was significantly more prevalent in cases than in controls which means that participants with the rs1816702 C/C genotype were at a higher risk of being CMV+ than those with rs1816702 T/T genotype. TLR2 recognise CMV glycoproteins B (gB) and gH in a process which facilitates entry of CMV into immune cells [15, 48]. The rs1816702T allele is associated with significantly elevated levels of inflammatory monocytes expressing CD14+/TLR2+ receptors than rs1816702C allele [49]. This could explain our findings of a higher risk of CMV among rs1816702C/C carriers because their immune response against CMV is impaired due to lower TLR2 expression compared to the T/T. Homozygosity for the rs1816702C allele has also been associated with increased odds of Mycobacteria leprae infection and inflammatory bowel disease which were attributable to altered NFκB-mediated inflammatory response [50, 51].
The intronic SNP rs352139T > C in the TLR9 gene was also associated with CMV DNA status. Homozygous rs352139C/C individuals were at a significantly higher risk of being CMV+ compared to homozygous T/T carriers. The effect of the C allele on risk of CMV infection was also observed in the dominant and recessive models where the significance of the compound heterozygous (T/C) and homozygous (C/C) genotypes had a greater risk than the homozygous (C/C) alone, relative to the T/T genotype in both cases. The higher risk of CMV positivity in homozygous carriers of the C allele suggest that the polymorphism results in a less potent protein compared to the T allele. Since the polymorphism is intronic, it likely creates an alternative splicing site thus, affecting mRNA transcription and the final protein product. A less potent protein would have decreased ability to form dimers that are required to illicit an immune reaction. Individuals who are homozygous T/T have impaired immune responses against CMV infection, hence are more likely to experience CMV infection or reactivation. The HIV rapid progressor phenotype has been linked to homozygosity for rs352139T allele also due to reduced TRL9 potency [52].
Conflicting findings were reported reduced risk of cCMV associated with the rs352139T/T genotype among infants in Poland [53]. The conflicting effect of rs352139T variant have also been reported in bacterial infection studies in Indonesia and Mexico, perhaps due to ethnic differences [54, 55]. We suggest that rs352139 could be in linkage disequilibrium (LD) with a polymorphic regulatory region that controls TLR9 expression or serves as a functional region SNP. LD patterns differ with level of genetic diversity among different ethnic groups, hence the effects of one SNP may vary from one population to another. Minor allele frequencies for these SNPs which seem to affect CMV infection risk were compared to other populations. TLR2 rs4696480A and TLR4 rs1075993T alleles, respectively, have lower frequencies among Zimbabweans (0.31 and 0.20) and other African populations (0.37 and 0.21) when compared to European (0.52 and 0.77) and Asian (0.57 and 0.86) populations. On the other hand, TLR4 rs7856729T and TLR9 rs5743836G, respectively, are proportionally higher in Zimbabwean (0.38 and 0.36) and other Africans (0.33 and 0.42) when compared to European (0.13 and 0.13) and Asian (0.10 and 0.00) populations. These differences in the distribution of risk alleles of world populations, is likely to lead to differential responses upon exposures to infectious pathogens. Indeed, the adaptive immune responses to the β-coronaviruses, MERS-CoV and SARS-CoV, are that can cause fatal lower respiratory tract infections, are marshalled by T cells, CD4+ T cells, and CD8+ T cells, through among other processes, activate other downstream cytokine and chemokine cascades, such as IL-1, IL-6, IL-8, IL-21 and TNF-β [56]. The molecular patterns displayed by viruses are then sensed by different immune cellular pathogen recognition receptors, including toll-like receptors (TLR:2, 3, 4, 7, 8, and 9) [57]. Whether this genetic heterogeneity among populations plays an active role in the differential prevalence of CMV is unclear and is an area of further research which should also consider the strong influence of environmental factors.
Conclusions
We conclude that TLR2, − 7, − 9 and IL-6 genetic polymorphisms are associated CMV status in late gestation among the black Zimbabweans. TLRs and ILs modulate immune responses to CMV, hence polymorphisms in genes encoding the receptors and cytokines could interfere with the immune mechanisms, hence their association with CMV status. We recommend that future studies consider evaluating the profiles of immune response genes and the polymorphisms in these genes on their possible effects in viral infections. With respect to CMV, we recommend a mother-infant longitudinal approach that will seek to factor in the effect of these immunogenetic profiles in congenital CMV and its possible sequelae.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on request or in the dbSNP repository, http://www.ncbi.nlm.nih.gov/SNP/snp_viewTable.cgi?handle=HUMGEN_PHARMGX
Abbreviations
- CMV:
-
Cytomegalovirus
- TRL:
-
Toll-like receptor
- SNP:
-
Single nucleotide polymorphism
- IL:
-
Interleukin
- IFN:
-
Interferon
- HWE:
-
Hardy-Weinberg Equilibrium
- BMI:
-
Basal metabolic index
- PAMP:
-
Pathogen associated molecular pattern
- DC:
-
Dendritic cell
- NFκB:
-
Nuclear factor kappa β
References
Cannon MJ, Schmid DS, Hyde TB. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev Med Virol. 2010;20:202–13.
Zuhair M, Smit GSA, Wallis G, Jabbar F, Smith C, Devleesschauwer B, et al. Estimation of the worldwide seroprevalence of cytomegalovirus: a systematic review and meta-analysis. Rev Med Virol. 2019;29:e2034.
Chibwe E, Mirambo MM, Kihunrwa A, Mshana SE. Magnitude of the Cytomegalovirus infection among pregnant women attending antenatal clinics in the city of Mwanza, Tanzania. BMC Res Notes. 2017;10:489.
Freeman ML, Lederman MM, Gianella S. Partners in Crime: the role of CMV in immune dysregulation and clinical outcome during HIV infection. Curr HIV/AIDS Rep. 2016;13:10–9.
Goncalves A, Makalo P, Joof H, Burr S, Ramadhani A, Massae P, et al. Differential frequency of NKG2C/KLRC2 deletion in distinct African populations and susceptibility to trachoma: a new method for imputation of KLRC2 genotypes from SNP genotyping data. Hum Genet. 2016;135:939–51.
Hoes J, Boef AGC, Knol MJ, de Melker HE, Mollema L, van der Klis FRM, et al. Socioeconomic status is associated with antibody levels against vaccine preventable diseases in the Netherlands. Front Public Health. 2018;6:209.
Jin Q, Su J, Wu S. Cytomegalovirus infection among pregnant women in Beijing: Seroepidemiological survey and intrauterine transmissions. J Microbiol Biotechnol. 2017;27:1005–9.
Casanova J-L, Abel L. Human genetics of infectious diseases: unique insights into immunological redundancy. Semin Immunol. 2018;36:1–12.
Sezgin E, An P, Winkler CA. Host genetics of cytomegalovirus pathogenesis. Front Genet. 2019;10. https://doi.org/10.3389/fgene.2019.00616.
Botos I, Segal DM, Davies DR. The structural biology of Toll-like receptors. Struct Lond Engl. 2011;19:447–59.
Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nat Immunol. 2010;11:373–84.
Compton T, Kurt-Jones EA, Boehme KW, Belko J, Latz E, Golenbock DT, et al. Human Cytomegalovirus activates inflammatory cytokine responses via CD14 and toll-like receptor 2. J Virol. 2003;77:4588–96.
Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15.
Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7.
Boehme KW, Guerrero M, Compton T. Human cytomegalovirus envelope glycoproteins B and H are necessary for TLR2 activation in permissive cells. J Immunol Baltim Md 1950. 2006;177:7094–102.
Crouse J, Kalinke U, Oxenius A. Regulation of antiviral T cell responses by type I interferons. Nat Rev Immunol. 2015;15:231–42.
González-Navajas JM, Lee J, David M, Raz E. Immunomodulatory functions of type I interferons. Nat Rev Immunol. 2012;12:125–35.
Samuel CE. Antiviral actions of interferons. Clin Microbiol Rev. 2001;14:778–809.
Arav-Boger R, Wojcik GL, Duggal P, Ingersoll RG, Beaty T, Pass RF, et al. Polymorphisms in toll-like receptor genes influence antibody responses to cytomegalovirus glycoprotein B vaccine. BMC Res Notes. 2012;5:140.
Bochud P-Y, Chien JW, Marr KA, Leisenring WM, Upton A, Janer M, et al. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med. 2008;359:1766–77.
Schneider M, Matiqi T, Kundi M, Rieder FJJ, Andreas M, Strassl R, et al. Clinical significance of the single nucleotide polymorphism TLR2 R753Q in heart transplant recipients at risk for cytomegalovirus disease. J Clin Virol. 2016;84:64–9.
Studzińska M, Jabłońska A, Wiśniewska-Ligier M, Nowakowska D, Gaj Z, Leśnikowski ZJ, et al. Association of TLR3 L412F polymorphism with Cytomegalovirus infection in children. PLoS One. 2017;12:e0169420.
Wujcicka W, Paradowska E, Studzińska M, Gaj Z, Wilczyński J, Leśnikowski Z, et al. TLR9 2848 GA heterozygotic status possibly predisposes fetuses and newborns to congenital infection with human Cytomegalovirus. PLoS One. 2015;10. https://doi.org/10.1371/journal.pone.0122831.
Kogut MH, Chiang H-I, Swaggerty CL, Pevzner IY, Zhou H. Gene expression analysis of toll-like receptor pathways in heterophils from genetic chicken lines that differ in their susceptibility to Salmonella enteritidis. Front Genet. 2012;3. https://doi.org/10.3389/fgene.2012.00121.
Smith PD, Shimamura M, Musgrove L, Dennis EA, Bimczok D, Novak L, et al. Cytomegalovirus enhances macrophage TLR expression and MyD88-mediated signal transduction to potentiate inducible inflammatory responses. J Immunol Baltim Md 1950. 2014;193:5604–12.
Sezgin E, Jabs DA, Hendrickson SL, Van Natta M, Zdanov A, Lewis RA, et al. Effect of host genetics on the development of cytomegalovirus retinitis in patients with AIDS. J Infect Dis. 2010;202:606–13.
Bibert S, Wojtowicz A, Taffé P, Manuel O, Bernasconi E, Furrer H, et al. The IFNL3/4 ΔG variant increases susceptibility to cytomegalovirus retinitis among HIV-infected patients. AIDS Lond Engl. 2014;28:1885–9.
Manuel O, Wójtowicz A, Bibert S, Mueller NJ, van Delden C, Hirsch HH, et al. Influence of IFNL3/4 polymorphisms on the incidence of cytomegalovirus infection after solid-organ transplantation. J Infect Dis. 2015;211:906–14.
Mhandire D, Duri K, Kaba M, Mhandire K, Musarurwa C, Chimusa E, et al. Seroprevalence of Cytomegalovirus infection among HIV-infected and HIV-uninfected pregnant women attending antenatal clinic in Harare, Zimbabwe. Viral Immunol. 2019;32(7):289–95.
Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)-a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42:377–81.
Shen J, Li Z, Chen J, Song Z, Zhou Z, Shi Y. SHEsisPlus, a toolset for genetic studies on polyploid species. Sci Rep. 2016;6:24095.
Opie EL. Inflammation and immunity. J Immunol. 1929;17:329–42.
Scepanovic P, Alanio C, Hammer C, Hodel F, Bergstedt J, Patin E, et al. Human genetic variants and age are the strongest predictors of humoral immune responses to common pathogens and vaccines. Genome Med. 2018;10:59.
Genoni G, Prodam F, Marolda A, Giglione E, Demarchi I, Bellone S, et al. Obesity and infection: two sides of one coin. Eur J Pediatr. 2014;173:25–32.
Huttunen R, Syrjänen J. Obesity and the risk and outcome of infection. Int J Obes. 2013;37:333–40.
Firth C, Harrison R, Ritchie S, Wardlaw J, Ferro CJ, Starr JM, et al. Cytomegalovirus infection is associated with an increase in systolic blood pressure in older individuals. QJM Int J Med. 2016;109:595–600.
Hui J, Qu Y, Tang N, Liu Y, Zhong H, Wang L, et al. Association of cytomegalovirus infection with hypertension risk: a meta-analysis. Wien Klin Wochenschr. 2016;128:586–91.
Rebelo F, Farias DR, Mendes RH, Schlüssel MM, Kac G. Blood pressure variation throughout pregnancy according to early gestational BMI: a Brazilian cohort. Arq Bras Cardiol. 2015;104:284–91.
Shen M, Tan H, Zhou S, Smith GN, Walker MC, Wen SW. Trajectory of blood pressure change during pregnancy and the role of pre-gravid blood pressure: a functional data analysis approach. Sci Rep. 2017;7:1–6.
Chen J, Liu R-Y, Yang L, Zhao J, Zhao X, Lu D, et al. A two-SNP IL-6 promoter haplotype is associated with increased lung cancer risk. J Cancer Res Clin Oncol. 2013;139:231–42.
Smith AJP, D’Aiuto F, Palmen J, Cooper JA, Samuel J, Thompson S, et al. Association of serum interleukin-6 concentration with a functional IL6 -6331T>C polymorphism. Clin Chem. 2008;54:841–50.
Zhang JP, Li F, Yu XW, Sheng Q, Shi XW, Zhang XW. Trace elements and cytokine profile in Cytomegalovirus-infected pregnancies: a controlled study. Gynecol Obstet Investig. 2008;65:128–32.
Askar E, Ramadori G, Mihm S. Toll-like receptor 7 rs179008/Gln11Leu gene variants in chronic hepatitis C virus infection. J Med Virol. 2010;82:1859–68.
Buschow SI, Biesta PJ, Groothuismink ZMA, Erler NS, Vanwolleghem T, Ho E, et al. TLR7 polymorphism, sex and chronic HBV infection influence plasmacytoid DC maturation by TLR7 ligands. Antivir Res. 2018;157:27–37.
Mosaad YM, Metwally SS, Farag RE, Lotfy ZF, AbdelTwab HE. Association between toll-like receptor 3 (TLR3) rs3775290, TLR7 rs179008, TLR9 rs352140 and chronic HCV. Immunol Investig. 2019;48:321–32.
Saitoh S-I, Abe F, Kanno A, Tanimura N, Saitoh YM, Fukui R, et al. TLR7 mediated viral recognition results in focal type I interferon secretion by dendritic cells. Nat Commun. 2017;8:1592.
Oh DY, Baumann K, Hamouda O, Eckert JK, Neumann K, Kücherer C, et al. A frequent functional toll-like receptor 7 polymorphism is associated with accelerated HIV-1 disease progression. AIDS Lond Engl. 2009;23:297–307.
Juckem LK, Boehme KW, Feire AL, Compton T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J Immunol Baltim Md 1950. 2008;180:4965–77.
Bielinski SJ, Hall JL, Pankow JS, Boerwinkle E, Matijevic-Aleksic N, He M, et al. Genetic variants in TLR2 and TLR4 are associated with markers of monocyte activation: the atherosclerosis risk in communities MRI study. Hum Genet. 2011;129:655–62.
Bank S, Andersen PS, Burisch J, Pedersen N, Roug S, Galsgaard J, et al. Associations between functional polymorphisms in the NFκB signaling pathway and response to anti-TNF treatment in Danish patients with inflammatory bowel disease. Pharmacogenomics J. 2014;14:526–34.
Mazini PS, Rodrigues-Santos P, Santos-Rosa M, Vicentin HA, Shinzato AH, Shinzato MH, et al. Evaluation of SNPS on the toll-like receptors, NOD-like receptors and CD14 gene in the recognition and in autophagic modulation in response to infection by mycobacterium leprae. Hum Immunol. 2015;76:83.
Bochud PY, Hersberger M, Taffé P, Bochud M, Stein CM, Rodrigues SD, et al. Polymorphisms in toll-like receptor 9 influence the clinical course of HIV-1 infection. AIDS Lond Engl. 2007;21:441–6.
Paradowska E, Jabłońska A, Studzińska M, Skowrońska K, Suski P, Wiśniewska-Ligier M, et al. TLR9 -1486T/C and 2848C/T SNPs are associated with human Cytomegalovirus infection in infants. PLoS One. 2016;11:e0154100.
Kobayashi K, Yuliwulandari R, Yanai H, Naka I, Lien LT, Hang NTL, et al. Association of TLR polymorphisms with development of tuberculosis in Indonesian females. Tissue Antigens. 2012;79:190–7.
Torres-García D, Cruz-Lagunas A, García-Sancho Figueroa MC, Fernández-Plata R, Baez-Saldaña R, Mendoza-Milla C, et al. Variants in toll-like receptor 9 gene influence susceptibility to tuberculosis in a Mexican population. J Transl Med. 2013;11:220.
Velazquez-Salinas L, Verdugo-Rodriguez A, Rodriguez LL, Borca MV. The role of interleukin 6 during viral infections. Front Microbiol. 2019;10. https://doi.org/10.3389/fmicb.2019.01057.
Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92:424–32.
Acknowledgements
We would like to acknowledge the Principle Investigator, employees, and participants of the UZ-CHS Birth Cohort from which we acquired samples for this study.
Funding
The National Research Foundation (NRF) South Africa and the South African Medical Research Council (SAMRC) funded all the laboratory work through grants to Professor Collet Dandara.
Author information
Authors and Affiliations
Contributions
DM conceived the ideas, carried out some laboratory experiments, analysed data and drafted the manuscript; KM reviewed the manuscript draft, assisted with laboratory experiments; AK analysed data and reviewed the manuscript draft; MM and AW assisted with analysis and interpretation of the data and read the manuscript draft; CD conceived the ideas, supervised all components as principal investigator and reviewed the manuscript draft. All authors contributed to the final version of the article. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The current study received ethical clearance from the Medical Research Council of Zimbabwe (MRCZ/A/2177) as well as the University of Cape Town Institutional Review Board (HREC628/2017). The study was carried out in accordance with the guidelines of the Helsinki Declaration of 2008 ethics clearance. All participants signed an informed consent form prior to enrolment into the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table 1.
Genotype frequencies and univariate logistic regression of SNP with CMV infection status.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mhandire, D.Z., Mhandire, K., Magadze, M. et al. Genetic variation in toll like receptors 2, 7, 9 and interleukin-6 is associated with cytomegalovirus infection in late pregnancy. BMC Med Genet 21, 113 (2020). https://doi.org/10.1186/s12881-020-01044-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-020-01044-8