
Abstract
Background
Humoral immune responses to infectious agents or vaccination vary substantially among individuals, and many of the factors responsible for this variability remain to be defined. Current evidence suggests that human genetic variation influences (i) serum immunoglobulin levels, (ii) seroconversion rates, and (iii) intensity of antigen-specific immune responses. Here, we evaluated the impact of intrinsic (age and sex), environmental, and genetic factors on the variability of humoral response to common pathogens and vaccines.
Methods
We characterized the serological response to 15 antigens from common human pathogens or vaccines, in an age- and sex-stratified cohort of 1000 healthy individuals (Milieu Intérieur cohort). Using clinical-grade serological assays, we measured total IgA, IgE, IgG, and IgM levels, as well as qualitative (serostatus) and quantitative IgG responses to cytomegalovirus, Epstein-Barr virus, herpes simplex virus 1 and 2, varicella zoster virus, Helicobacter pylori, Toxoplasma gondii, influenza A virus, measles, mumps, rubella, and hepatitis B virus. Following genome-wide genotyping of single nucleotide polymorphisms and imputation, we examined associations between ~ 5 million genetic variants and antibody responses using single marker and gene burden tests.
Results
We identified age and sex as important determinants of humoral immunity, with older individuals and women having higher rates of seropositivity for most antigens. Genome-wide association studies revealed significant associations between variants in the human leukocyte antigen (HLA) class II region on chromosome 6 and anti-EBV and anti-rubella IgG levels. We used HLA imputation to fine map these associations to amino acid variants in the peptide-binding groove of HLA-DRβ1 and HLA-DPβ1, respectively. We also observed significant associations for total IgA levels with two loci on chromosome 2 and with specific KIR-HLA combinations.
Conclusions
Using extensive serological testing and genome-wide association analyses in a well-characterized cohort of healthy individuals, we demonstrated that age, sex, and specific human genetic variants contribute to inter-individual variability in humoral immunity. By highlighting genes and pathways implicated in the normal antibody response to frequently encountered antigens, these findings provide a basis to better understand disease pathogenesis.
Trials registration
ClinicalTrials.gov, NCT01699893
Similar content being viewed by others


Background
Humans are regularly exposed to infectious agents, including common viruses such as cytomegalovirus (CMV), Epstein-Barr virus (EBV), or herpes simplex virus-1 (HSV-1) that have the ability to persist as latent infections throughout life—with possible reactivation events depending on extrinsic and intrinsic factors [1]. Humans also receive multiple vaccinations, which in many cases are expected to achieve lifelong immunity in the form of neutralizing antibodies. In response to each of these stimulations, the immune system mounts a humoral response, triggering the production of specific antibodies that play an essential role in limiting infection and providing long-term protection. Although the intensity of the humoral response to a given stimulation has been shown to be highly variable [2,3,4], the genetic and non-genetic determinants of this variability are still largely unknown. The identification of such factors may lead to improved vaccination strategies by optimizing vaccine-induced immunoglobulin G (IgG) protection, or to new understanding of autoimmune diseases, where immunoglobulin levels can correlate with disease severity [5].
Several genetic variants have been identified that account for inter-individual differences in susceptibility to pathogens [6,7,8,9] and in infectious [10] or therapeutic [11] phenotypes. By contrast, relatively few studies have investigated the variability of humoral responses in healthy humans [12,13,14]. In particular, Hammer et al. examined the contribution of genetics to variability in human antibody responses to common viral antigens, and fine-mapped variants at the HLA class II locus that associated with IgG responses. To replicate and extend these findings, we measured IgG responses to 15 antigens from common infectious agents or vaccines as well as total IgG, IgM, IgE, and IgA levels in 1000 well-characterized healthy donors. We used an integrative approach to study the impact of age, sex, non-genetic, and genetic factors on humoral immunity in healthy humans.
Methods
Study participants
The Milieu Intérieur cohort consists of 1000 healthy individuals that were recruited by BioTrial (Rennes, France). The cohort is stratified by sex (500 men, 500 women) and age (200 individuals from each decade of life, between 20 and 70 years of age). Donors were selected based on stringent inclusion and exclusion criteria, previously described [15]. Briefly, recruited individuals had no evidence of any severe/chronic/recurrent medical conditions. The main exclusion criteria were seropositivity for human immunodeficiency virus (HIV) or hepatitis C virus (HCV); ongoing infection with the hepatitis B virus (HBV)—as evidenced by detectable HBs antigen levels; travel to (sub-) tropical countries within the previous 6 months; recent vaccine administration; and alcohol abuse. To avoid the influence of hormonal fluctuations in women during the peri-menopausal phase, only pre- or post-menopausal women were included. To minimize the importance of population substructure on genomic analyses, the study was restricted to self-reported Metropolitan French origin for three generations (i.e., with parents and grandparents born in continental France). Whole blood samples were collected from the 1000 fasting healthy donors on lithium heparin tubes, from September 2012 to August 2013. The clinical study was approved by the Comité de Protection des Personnes - Ouest 6 on June 13, 2012, and by the French Agence Nationale de Sécurité du Médicament on June 22nd, 2012. The study is sponsored by Institut Pasteur (Pasteur ID-RCB Number: 2012-A00238-35) and was conducted as a single-center study without any investigational product. The protocol is registered under ClinicalTrials.gov (study# NCT01699893).
Serologies
Total IgG, IgM, IgE, and IgA levels were measured using clinical grade turbidimetric test on AU 400 Olympus at the BioTrial (Rennes, France). Antigen-specific serological tests were performed using clinical-grade assays measuring IgG levels, according to the manufacturer’s instructions. A list and description of the assays is provided in Additional file 1: Table S1. Briefly, anti-HBs and anti-HBc IgGs were measured on the Architect automate (CMIA assay, Abbott). Anti-CMV IgGs were measured by CMIA using the CMV IgG kit from Beckman Coulter on the Unicel Dxl 800 Access automate (Beckman Coulter). Anti-measles, anti-mumps, and anti-rubella IgGs were measured using the BioPlex 2200 MMRV IgG kit on the BioPlex 2200 analyzer (Bio-Rad). Anti-Toxoplasma gondi, and anti-CMV IgGs were measured using the BioPlex 2200 ToRC IgG kit on the BioPlex 2200 analyzer (Bio-Rad). Anti-HSV1 and anti-HSV2 IgGs were measured using the BioPlex 2200 HSV-1 and HSV-2 IgG kit on the BioPlex 2200 analyzer (Bio-Rad). IgGs against Helicobacter Pylori were measured by EIA using the PLATELIA H. pylori IgG kit (BioRad) on the VIDAS automate (Biomérieux). Anti-influenza A IgGs were measured by ELISA using the NovaLisa IgG kit from NovaTec (Biomérieux) that explores responses to grade 2 H3N2 Texas 1/77 strain. In all cases, the criteria for serostatus definition (positive, negative, or indeterminate) were established by the manufacturer and are indicated in Additional file 1: Table S2. Donors with an unclear result were retested and assigned a negative result if borderline levels were confirmed with repeat testing.
Non-genetic variables
A large number of demographical and clinical variables are available in the Milieu Intérieur cohort as a description of the environment of the healthy donors [15]. These include infection and vaccination history, childhood diseases, health-related habits, and socio-demographical variables. Of these, 53 where chosen for subsequent analysis of their impact on serostatus. This selection is based on the one done in [16], with a few variables added, such as measures of lipids and C-reactive protein (CRP).
Testing of non-genetic variables
Using serostatus variables as the response, and non-genetic variables as treatment variables, we fitted a logistic regression model for each response and treatment variable pair. A total of 14 × 52 = 742 models where therefore fitted. Age and sex where included as controls for all models, except if that variable was the treatment variable. We tested the impact of the clinical and demographical variables using a likelihood ratio test. All 742 tests where considered a multiple testing family with the false discovery rate (FDR) as error rate.
Age and sex testing
To examine the impact of age and sex, we performed logistic and linear regression analyses for serostatus and IgG levels, respectively. For logistic regression, we included both scaled linear and quadratic terms for the age variable (model = glm(y~Age + I(Age^2) + Sex, family = binomial)). Scaling was achieved by centering age variable at the mean age. When indicated, we used a second model that includes age, sex as well as an interaction term for age and sex (model = glm(y~Age + Sex + Age × Sex, family = binomial)). All continuous traits (i.e., quantitative measurements of antibody levels) were log10-transformed in donors assigned as positive using the clinical cutoff suggested by the manufacturer. We used false discovery rate (FDR) correction for the number of serologies tested (associations with P < 0.05 were considered significant).
DNA genotyping
Blood was collected in 5-mL sodium EDTA tubes and was kept at room temperature (18°–25°) until processing. DNA was extracted from human whole blood and genotyped at 719,665 single nucleotide polymorphisms (SNPs) using the HumanOmniExpress-24 BeadChip (Illumina). The SNP call rate was higher than 97% in all donors. To increase coverage of rare and potentially functional variation, 966 of the 1000 donors were also genotyped at 245,766 exonic variants using the HumanExome-12 BeadChip. The HumanExome variant call rate was lower than 97% in 11 donors, which were thus removed from this dataset. We filtered out from both datasets genetic variants that (i) were unmapped on dbSNP138, (ii) were duplicated, (iii) had a low genotype clustering quality (GenTrain score < 0.35), (iv) had a call rate < 99%, (v) were monomorphic, (vi) were on sex chromosomes, or (vii) diverged significantly from Hardy-Weinberg equilibrium (HWE P < 10−7). These quality-control filters yielded a total of 661,332 and 87,960 variants for the HumanOmniExpress and HumanExome BeadChips, respectively. Average concordance rate for the 16,753 SNPs shared between the two genotyping platforms was 99.9925%, and individual concordance rates ranged from 99.8 to 100%.
Genetic relatedness and structure
As detailed elsewhere [16], relatedness was detected using KING [17]. Six pairs of related participants (parent-child, first and second-degree siblings) were detected, and one individual from each pair, randomly selected, was removed from the genetic analyses. The genetic structure of the study population was estimated using principal component analysis (PCA), implemented in EIGENSTRAT (v6.1.3) [18]. The PCA plot of the study population is shown in Additional file 2: Figure S1.
Genotype imputation
We used positional Burrows-Wheeler transform for genotype imputation, starting with the 661,332 quality-controlled SNPs genotyped on the HumanOmniExpress array. Phasing was performed using EAGLE2 (v2.0.5) [19]. As reference panel, we used the haplotypes from the Haplotype Reference Consortium (release 1.1) [20]. After removing SNPs that had an imputation info score < 0.8, we obtained 22,235,661 variants. We then merged the imputed dataset with 87,960 variants directly genotyped on the HumanExome BeadChips array and removed variants that were monomorphic or diverged significantly from Hardy-Weinberg equilibrium (P < 10−7). We obtained a total of 12,058,650 genetic variants to be used in association analyses.
We used SNP2HLA (v1.03) [21] to impute 104 four-digit HLA alleles and 738 amino acid residues (at 315 variable amino acid positions of the HLA class I and II proteins) with a minor allele frequency (MAF) of > 1%.
We used KIR*IMP [22] to impute KIR alleles, after haplotype inference on chromosome 19 with SHAPEIT2 (v2.r790) [23]. A total of 19 KIR types were imputed: 17 loci plus two extended haplotype classifications (A vs. B and KIR haplotype). A MAF threshold of 1% was applied, leaving 16 KIR alleles for association analysis.
Genetic association analyses
For single-variant association analyses, we only considered SNPs with a MAF of > 5% (N = 5,699,237). We used PLINK (v1.9) [24] to perform logistic regression for binary phenotypes (serostatus: antibody positive versus negative) and linear regression for continuous traits (log10-transformed quantitative measurements of antibody levels in seropositive donors). The first two principal components of a PCA based on genetic data, age and sex, were used as covariates in all tests. In order to correct for baseline difference in IgG production in individuals, total IgG levels were included as covariates when examining associations with antigen-specific antibody levels, total IgM, IgE, and IgA levels. From a total of 53 additional variables additional co-variates, selected by using elastic net [25] and stability selection [26] as detailed elsewhere [16], were included in some analyses (Additional file 1: Table S3). For all genome-wide association studies, we used a genome-wide significant threshold (Pthreshold < 2.6 × 10−9) corrected for the number of antigens and immunoglobulin classes tested (N = 19). For specific HLA analyses, we used PLINK (v1.07) [27] to perform conditional haplotype-based association tests and multivariate omnibus tests at multi-allelic amino acid positions.
Variant annotation and gene burden testing
We used SnpEff (v4.3g) [28] to annotate all 12,058,650 variants. A total of 84,748 variants were annotated as having (potentially) moderate (e.g., missense variant, inframe deletion) or high impact (e.g., stop gained, frameshift variant) and were included in the analysis. We used bedtools v2.26.0 [29] to intersect variant genomic location with gene boundaries, thus obtaining sets of variants per gene. By performing kernel-regression-based association tests with SKAT_CommonRare (testing the combined effect of common and rare variants) and SKATBinary implemented in the SKAT v1.2.1 [30], we tested 16,628 gene sets for association with continuous and binary phenotypes, respectively. By SKAT default parameters, variants with MAF ≤ \( \frac{1}{\sqrt{2n}} \) are considered rare, whereas variants with MAF ≥ \( \frac{1}{\sqrt{2n}} \) were considered common, where N is the sample size. We used genome-wide Bonferroni correction for multiple testing, accounting for the number of phenotypes tested (Pthreshold < 2.6 × 10−9).
Results
Characterization of humoral immune responses in the 1000 study participants
To characterize the variability in humoral immune responses between healthy individuals, we measured total IgG, IgM, IgA, and IgE levels in the plasma of the 1000 donors of the Milieu Interieur (MI) cohort. After log10 transformation, total IgG, IgM, IgA, and IgE levels showed normal distributions, with a median ± sd of 1.02 ± 0.08 g/l, 0.01 ± 0.2 g/l, 0.31 ± 0.18 g/l, and 1.51 ± 0.62 UI/ml, respectively (Additional file 2: Figure S2A).
We then evaluated specific IgG responses to multiple antigens from the following infections and vaccines: (i) seven common persistent pathogens, including five viruses: CMV, EBV (EA, EBNA, and VCA antigens), herpes simplex virus 1 and 2 (HSV-1 & 2), varicella zoster virus (VZV), one bacterium: Helicobacter pylori (H. pylori), and one parasite: Toxoplasma gondii (T. gondii); (ii) one recurrent virus: influenza A virus (IAV); and (iii) four viruses for which most donors received vaccination: measles, mumps, rubella, and HBV (HBs and HBc antigens). The distributions of log10-transformed antigen-specific IgG levels in the 1000 donors for the 15 serologies are shown in Additional file 2: Figure S2B. Donors were classified as seropositive or seronegative using the thresholds recommended by the manufacturer (Additional file 1: Table S2).
The vast majority of the 1000 healthy donors were chronically infected with EBV (seropositivity rates of 96% for EBV VCA, 91% for EBV EBNA, and 9% for EBV EA) and VZV (93%). Many also showed high-titer antibodies specific for IAV (77%), HSV-1 (65%), and T. gondii (56%). By contrast, fewer individuals were seropositive for CMV (35%), HSV-2 (21%), and H. pylori (18%) (Additional file 2: Figure S3A). The majority of healthy donors carried antibodies against five or more persistent/recurrent infections of the eight infectious agents tested (Additional file 2: Figure S3B). Fifty-one percent of MI donors were positive for anti-HBs IgG—a large majority of them as a result of vaccination, as only 15 study participants (3% of the anti-HBs-positive group) were positive for anti-HBc IgG, indicative of previous HBV infection (spontaneously cured, as all donors were negative for HBs antigen, criteria for inclusion in the study). For rubella, measles, and mumps, seropositivity rates were 94, 91, and 89%, respectively. For the majority of the donors, this likely reflects vaccination with a trivalent vaccine, which was integrated in 1984 as part of national recommendations in France, but for some, in particular the > 40-year-old individuals of the cohort, it may reflect acquired immunity due to natural infection.
Associations of age, sex, and non-genetic variables with serostatus
Subjects included in the Milieu Interieur cohort were surveyed for a large number of variables related to infection and vaccination history, childhood diseases, health-related habits, and socio-demographical variables (http://www.milieuinterieur.fr/en/research-activities/cohort/crf-data). Of these, 53 where chosen for subsequent analysis of their impact on serostatus. This selection is based on the one done in [16], with a few variables added, such as measures of lipids and CRP. Applying a mixed model analysis that controls for potential confounders and batch effects, we found expected associations of HBs seropositivity with previous administration of HBV vaccine, as well as of influenza seropositivity with previous administration of flu vaccine. We also found associations of HBs seropositivity with previous administration of typhoid and hepatitis A vaccines—which likely reflects co-immunization, as well as with income, employment, and owning a house—which likely reflects confounding epidemiological factors (Additional file 2: Figure S4). Full results of the association of non-genetic variables with serostatus are available in Additional file 1: Table S4.
We observed a significant impact of age on the probability of being seropositive for antigens from persistent or recurrent infectious agents and/or vaccines. For 14 out of the 15 examined serologies, older people (> 45 years old) were more likely to have detectable specific IgG, with a mean beta estimate of 0.04 for linear associations (Fig. 1a). Additionally, we found a significant quadratic term for five out of the 15 serologies, highlighting that the rate of change in probability of seropositivity with respect to age is higher for rubella and lower for HSV-1, HP, HBs, and EBV EBNA in older people as compared to younger donors (Additional file 2: Figure S5A). We identified four different profiles of age-dependent evolution of seropositivity rates (Fig. 1b). Profile 1 is typical of childhood-acquired infection, i.e., microbes that most donors had encountered by age 20 (EBV, VZV, and influenza). We observed in this case either (i) a limited increase in seropositivity rate after age 20 for EBV; (ii) stability for VZV; or (iii) a small decrease in seropositivity rate with age for IAV (Additional file 2: Figure S5B-F). Profile 2 concerns prevalent infectious agents that are acquired throughout life, with steadily increasing prevalence (observed for CMV, HSV-1, and T. gondii). We observed in this case either (i) a linear increase in seropositivity rates over the five decades of age for CMV (seropositivity rate 24% in 20–29 years old, 44% in 60–69 years old, slope = 0.02) and T. gondii (seropositivity rate 21% in 20–29 years old, 88% in 60–69, slope = 0.08); or (ii) a non-linear increase in seropositivity rates for HSV-1, with a steeper slope before age 40 (seropositivity rate 36% in 20–29 years old, 85% in 60–69, slope = 0.05) (Additional file 2: Figure S5G-I). Profile 3 showed microbial agents with limited seroprevalence—in our cohort, HSV-2, HBV (anti-HBs and anti-HBc positive individuals, indicating prior infection rather than vaccination), and H. pylori. We observed a modest increase of seropositivity rates throughout life, likely reflecting continuous low-grade exposure (Additional file 2: Figure S5J-L). Profile 4 is negatively correlated with increasing age and is unique to HBV anti-HBs serology (Additional file 2: Figure S5M). This reflects the introduction of the HBV vaccine in 1982 and the higher vaccination coverage of younger populations. Profiles for measles, mumps and rubella are provided in Additional file 2: Figure S5N-P.

Age and sex impact on serostatus. a Effect sizes of significant linear associations (adjusted P values (adj. P < 0.05)) between age and serostatus as determined based on clinical-grade serologies in the 1000 healthy individuals from the Milieu Intérieur cohort. Effect sizes were estimated in a generalized linear mixed model, with serostatus as response variable, and age and sex as treatment variables. This model includes both scaled linear and quadratic terms for the age variable. Scaling was achieved by centering age variable at the mean age. All results from this analysis are provided in Additional file 1: Table S5. Dots represent the mean of the beta. Lines represent the 95% confidence intervals. b Odds of being seropositive towards EBV EBNA (Profile 1; upper left), Toxoplasma gondii (Profile 2; upper right), Helicobacter Pylori (Profile 3; bottom left), and HBs antigen of HBV (Profile 4; bottom right), as a function of age in men (blue) and women (red) in the 1000 healthy donors. Indicated P values were obtained using a logistic regression with Wald test, with serostatus binary variables (seropositive versus seronegative) as the response, and age and sex as treatments. Similar plots from all examined serologies are provided in Additional file 2: Figure S5. c Effect sizes of significant associations (adjusted P values (adj. P < 0.05) between sex (men = reference vs. women) and serostatus. Effect sizes were estimated in a generalized linear mixed model, with serostatus as response variable, and age and sex as treatment variables. All results from this analysis are provided in Additional file 1: Table S5. Dots represent the mean of the beta. Lines represent the 95% confidence intervals
We also observed a significant association between sex and serostatus for 8 of the 15 antigens, with a mean beta estimate of 0.07 (Fig. 1c). For six serological phenotypes, women had a higher rate of positivity, IAV being the notable exception. These associations were confirmed when considering “Sharing house with partner” and “Sharing house with children” as covariates. Full results of associations of age and sex with serostatus are present in Additional file 1: Table S5. Finally, we found a significant interaction of age and sex for odds of being seropositive for EBV EBNA, reflecting a decrease in seropositivity rate in older women (beta − 0.0414814; P = 0.02, Additional file 2 Figure S5Q).
Impact of age and sex on total and antigen-specific antibody levels
We further examined the impact of age and sex on the levels of total IgG, IgM, IgA, and IgE detected in the serum of the patients, as well as on the levels of antigen-specific IgGs in seropositive individuals. We observed a low impact of age and sex with total immunoglobulin levels (Fig. 2a). Age also had a strong impact on specific IgG levels in seropositive individuals, affecting 9 out of the 15 examined serologies (Fig. 2b). Correlations between age and pathogen-specific IgG levels were mostly positive, i.e., older donors had more specific IgG than younger donors, as for example in the case of rubella (Additional file 2: Figure S6A). The notable exception was T. gondii, where we observed lower amounts of specific IgG in older individuals (b = − 0.013(− 0.019, − 0.007), P = 3.7 × 10−6, Additional file 2: Figure S6B). On the other hand, sex was significantly correlated with IgG levels specific to mumps and VZV (Fig. 2c). Full results of associations of age and sex with total immunoglobulin and antigen-specific antibody levels are presented in Additional file 1: Table S5.

Age and sex impact on total and antigen-specific antibody levels. a Relationships between Log10-transformed IgG (upper left), IgA (upper right), IgM (bottom left), and IgE (bottom right) levels and age. Regression lines were fitted using linear regression, with Log10-transformed total antibody levels as response variable, and age and sex as treatment variables. Indicated adj. P were obtained using the mixed model and corrected for multiple testing using the FDR method. b, c Effect sizes of significant associations (adjusted P values (adj. P < 0.05) between age (b) and sex (c) on Log10-transformed antigen-specific IgG levels in the 1000 healthy individuals from the Milieu Intérieur cohort. Because of low number of seropositive donors (n = 15), HBc serology was removed from this analysis. Effect sizes were estimated in a linear mixed model, with Log10-transformed antigen-specific IgG levels as response variables, and age and sex as treatment variables. All results from this analysis are provided in Additional file 1: Table S5. Dots represent the mean of the beta. Lines represent the 95% confidence intervals
Genome-wide association study of serostatus
To test if human genetic factors influence the rate of seroconversion upon exposure, we performed genome-wide association studies. Specifically, we searched for associations between 5.7 million common polymorphisms (MAF > 5%) and the 15 serostatus in the 1000 healthy donors. Based on our results regarding age and sex, we included both as covariates in all models. After correcting for the number of antibodies considered, the threshold for genome-wide significance was Pthreshold = 2.6 × 10−9, for which we did not observe any significant association. In particular, we did not replicate the previously reported associations with H. pylori serostatus on chromosomes 1 (rs368433, P = 0.56, OR = 1.08) and 4 (rs10004195, P = 0.83, OD = 0.97) [31]. We verified this result by performing an additional analysis that matched the design of the previous study, i.e., a case-control association study comparing individuals in the upper quartile of the anti-H. pylori antibody distribution to the rest of the study population: no association was found (P = 0.42 and P = 0.48 for rs368433 and rs10004195, respectively). The quantile-quantile (QQ) plots and lambda values of all genome-wide logistic regressions are available in Additional file 2: Figure S7.
We then focused on the HLA region and confirmed the previously published association of influenza A serostatus with specific amino acid variants of HLA class II molecules [12]. The strongest association in the MI cohort was found with residues at position 31 of the HLA-DRβ1 subunit (omnibus P = 0.009, Additional file 1: Table S6). Residues found at that position, isoleucine (P = 0.2, OD (95% CI) = 0.8 (0.56, 1.13)) and phenylalanine (P = 0.2, OR (95% CI) = 0.81 (0.56, 1.13)), are consistent in direction and in almost perfect linkage disequilibrium (LD) with the glutamic acid residue at position 96 in HLA-DRβ1 that was identified in the previous study (Additional file 1: Table S7). As such, our result independently validates the previous observation.
Genome-wide association study of total and antigen-specific antibody levels
To test whether human genetic factors also influence the intensity of antigen-specific immune response, we performed genome-wide association studies of total IgG, IgM, IgA and IgE levels, as well as antigen-specific IgG levels.
We found no SNPs associated with total IgG, IgM, IgE, and IgA levels. Additional file 2: Figure S8 shows QQ plots and lambda values of these studies. However, we observed nominal significance and the same direction of the effect for 3 out of 11 loci previously published for total IgA [13, 32,33,34,35], 1 out of 6 loci for total IgG [13, 32, 36], and 4 out of 11 loci for total IgM [13, 37] (Additional file 1: Table S8). Finally, we also report a suggestive association (genome-wide significant, P < 5.0 × 10−8, but not significant when correcting for the number of antibody levels tested in the study) of a SNP rs11186609 on chromosome 10 with total IgA levels (P = 2.0 × 10−8, beta = − 0.07 for the C allele). The closest gene for this signal is SH2D4B.
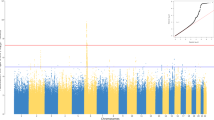
We next explored associations between human genetic variants and antigen-specific IgG levels in seropositive donors. Information on possible inflation of false positive rates of these linear regressions is available in Additional file 2: Figure S9. We detected significant associations for anti-EBV (EBNA antigen) and anti-rubella IgGs. Associated variants were in both cases located in the HLA region on chromosome 6. For EBV, the top SNP was rs74951723 (P = 3 × 10−14, beta = 0.29 for the A allele) (Fig. 3a). For rubella, the top SNP was rs115118356 (P = 7.7 × 10−10, beta = − 0.11 for the G allele) (Fig. 3b). rs115118356 is in LD with rs2064479, which has been previously reported as associated with titers of anti-rubella IgGs (r2 = 0.53 and D′ = 0.76) [38].

Association between host genetic variants and serological phenotypes. Manhattan plots of association results for a EBV anti-EBNA IgG and b rubella IgG levels. The dashed horizontal line denotes genome-wide significance (P = 2.6 × 10−9)
To fine map the associations observed in the HLA region, we tested four-digit HLA alleles and variable amino positions in HLA proteins. At the level of HLA alleles, HLA-DQB1*03:01 showed the lowest P value for association with EBV EBNA (P = 1.3 × 10−7), and HLA-DPB1*03:01 was the top signal for rubella (P = 3.8 × 10−6). At the level of amino acid positions, position 58 of the HLA-DRβ1 protein associated with anti-EBV (EBNA antigen) IgG levels (P = 2.5 × 10−11). This is consistent with the results of previous studies linking genetic variations in HLA-DRβ1 with levels of anti-EBV EBNA-specific IgGs [12, 39, 40] (Additional file 1: Table S9). In addition, position 8 of the HLA-DPβ1 protein associated with anti-rubella IgG levels (P = 1.1 × 10−9, Table 1). Conditional analyses on these amino-acid positions did not reveal any additional independent signals.
KIR associations
To test whether specific KIR genotypes, and their interaction with HLA molecules, are associated with humoral immune responses, we imputed KIR alleles from SNP genotypes using KIR*IMP [22]. First, we searched for potential associations with serostatus or IgG levels for 16 KIR alleles that had a MAF > 1%. We did not find any significant association after Bonferroni correction for multiple testing. Second, we tested specific KIR-HLA combinations. We filtered out rare combinations by removing pairs that were observed less than four times in the cohort. After correction for the number of tests performed and phenotypes considered (Pthreshold < 5.4 × 10−7), we observed significant associations between total IgA levels and the two following HLA-KIR combinations: HLA-B*14:02/KIR3DL1 and HLA-C*08:02/KIR2DS4 (P = 3.9 × 10−9 and P = 4.9 × 10−9 respectively, Table 2).
Burden testing for rare variants
Finally, to search for potential associations between the burden of low-frequency variants and the serological phenotypes, we conducted a rare variant association study. This analysis only included variants annotated as missense or putative loss-of-function (nonsense, essential splice-site, and frame-shift, N = 84,748), which we collapsed by gene and tested together using the kernel-regression-based association test SKAT [30]. We restricted our analysis to genes that contained at least five variants. Two genes were identified as significantly associated with total IgA levels using this approach: ACADL (P = 3.4 × 10−11) and TMEM131 (P = 7.8 × 10−11) (Table 3). By contrast, we did not observe any significant associations between rare variant burden and antigen-specific IgG levels or serostatus. All the QQ plots and lambda values of analysis of binary, total Ig levels, and pathogen-specific quantitative phenotypes are shown in Additional file 2: Figure S10, S11, and S12.
Discussion
We performed genome-wide association studies for a number of serological phenotypes in a well-characterized age- and sex-stratified cohort and included a unique examination of genetic variation at HLA and KIR loci, as well as KIR-HLA associations. As such, our study provides a broad resource for exploring the variability in humoral immune responses across different isotypes and different antigens in humans.
Using a fine-mapping approach, we replicated the previously reported associations of variation in the HLA-DRβ1 protein with influenza A serostatus and anti-EBV IgG titers [4, 12], implicating amino acid residues in strong LD with the ones previously reported by Hammer et al. In accordance with the same study, we did not observe any significant association with another measure of EBV serostatus, the presence of anti-EBNA antibodies, suggesting that a larger sample size will be required to uncover potentially associated variants. We replicated an association between HLA class II variation and anti-rubella IgG titers [38] and further fine-mapped it to position 8 of the HLA-DPβ1 protein. Interestingly, position 8 of HLA-DPβ1 and positions 58 and 31 of HLA-DRβ1 are all part of the extracellular domain of the respective proteins. Our findings confirm these proteins as critical elements for the presentation of processed peptide to CD4+ T cells and as such may reveal important clues in the fine regulation of class II antigen presentation. We also identified specific HLA/KIR combinations, namely HLA-B*14:02/KIR3DL1 and HLA-C*08:02/KIR2DS4, which associate with higher levels of circulating IgA. Combinations of HLA and killer cell immunoglobulin-like receptor (KIR) genes have been associated with diseases as diverse as autoimmunity, viral infections, reproductive failure, and cancer [41]. To date, the molecular basis for these associations is mostly unknown. One could speculate that the association identified between IgA levels and specific KIR-HLA combinations may reflect different levels of tolerance to commensal microbes. However, formal testing of this hypothesis will require additional studies. Also, given the novelty of KIR imputation method and the lack of possibility of benchmarking its reliability in the MI cohort, further replication of these results will be needed. Yet these findings support the concept that variations in the sequence of HLA class II molecules, or specific KIRs/HLA class I interactions play a critical role in shaping humoral immune responses in humans. In particular, our findings confirm that small differences in the capacity of HLA class II molecules to bind specific viral peptides can have a measurable impact on downstream antibody production. As such, our study emphasizes the importance of considering HLA diversity in disease association studies where associations between IgG levels and autoimmune diseases are being explored.
We identified nominal significance for some but not all of the previously reported associations with levels of total IgG, IgM, and IgA, as well as a suggestive association of total IgA levels with an intergenic region on chromosome 10—closest gene being SH2D4B. By collapsing the rare variants present in our dataset into gene sets and testing them for association with the immunoglobulin phenotypes, we identified two additional loci that participate to natural variation in IgA levels. These associations mapped to the genes ACADL and TMEM131. ACADL encodes an enzyme with long-chain acyl-CoA dehydrogenase activity, and polymorphisms have been associated with pulmonary surfactant dysfunction [42]. As the same gene is associated with levels of circulating IgA in our cohort, we speculate that ACADL could play a role in regulating the balance between mucosal and circulating IgA. Further studies will be needed to test this hypothesis, as well as the potential impact of our findings in other IgA-related diseases.
We were not able to replicate previous associations of TLR1 and FCGR2A locus with serostatus for H. pylori [31]. We believe this may be a result of (i) different analytical methods or (ii) notable differences in previous exposure among the different cohorts as illustrated by the different levels of seropositivity—17% in the Milieu Interieur cohort, versus 56% in the previous ones, reducing the likelihood of replication due to decreased statistical power.
In addition to genetics findings, our study re-examined the impact of age and sex, as well as non-genetic variables, on humoral immune responses. Although this question has been previously addressed, our well-stratified cohort brings interesting additional insights. One interesting finding is the high rate of seroconversion for CMV, HSV-1, and T. gondii during adulthood. In our cohort, the likelihood of being seropositive for one of these infections is comparable at age 20 and 40. This observation raises interesting questions about the factors that could prevent some individuals from becoming seropositive upon late-life exposure, considering the high likelihood of being in contact with the pathogens because of their high prevalence in humans (CMV and HSV-1) or because of frequent interactions with an animal reservoir (toxoplasmosis). Second, both age and sex have a strong correlation with serostatus, i.e., older and female donors were more likely to be seropositive. Although increased seropositivity with age probably reflects continuous exposure, the sex effect is intriguing. Indeed, our study considered humoral immunity to microbial agents that differ significantly in terms of physiopathology and that do not necessarily have a childhood reservoir. Also, our analysis shows that associations persist after removal of potential confounding factors such as marital status and/or number of kids. As such, we believe that our results may highlight a general impact of sex on humoral immune response variability, i.e., a tendency for women to be more likely to seroconvert after exposure, as compared to men of same age. Gender-specific differences in humoral responses have been previously observed for a large number of viral and bacterial vaccines including influenza, hepatitis A and B, rubella, measles, rabies, yellow fever, meningococcus, pneumococcus, diphtheria, tetanus, and Brucella [43, 44]. Along the same line, women often respond to lower vaccine doses than men [43, 45], and higher levels of antibodies have been found in female schoolchildren after rubella and mumps vaccination [46] as well as in adult women after smallpox vaccination [47]. This could be explained, at least partially, by a shift towards Th2 immunity in women as compared to men [48]. Finally, we observed an age-related increase in antigen-specific IgG levels in seropositive individuals for most serologies, with the notable exception of toxoplasmosis. This may indicate that aging plays a general role in IgG production. An alternative explanation that requires further study is that this could be the consequence of reactivation or recurrent exposure.
Conclusions
In sum, our study provides evidence that age, sex, and host genetics contribute to natural variation in humoral immunity in humans. The identified associations have the potential to help improve vaccination strategies and/or dissect pathogenic mechanisms implicated in human diseases related to immunoglobulin production such as autoimmunity.
Abbreviations
- CMV:
-
Cytomegalovirus
- CRP:
-
C-reactive protein
- EBV:
-
Epstein-Barr virus
- FDR:
-
False discovery rate
- H. pylori :
-
Helicobacter pylori
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- HLA:
-
Human leukocyte antigen
- HSV1:
-
Herpes simplex virus 1
- HSV2:
-
Herpes simplex virus 2
- IAV:
-
Influenza A virus
- Ig:
-
Immunoglobulin
- LD:
-
Linkage disequilibrium
- MAF:
-
Minor allele frequency
- MI:
-
Milieu Interieur
- QQ:
-
Quantile-quantile
- SNP:
-
Single nucleotide polymorphism
- T. gondii :
-
Toxoplasma gondii
- VZV:
-
Varicella zoster virus
References
Traylen CM, Patel HR, Fondaw W, Mahatme S, Williams JF, Walker LR, Dyson OF, Arce S, Akula SM. Virus reactivation: a panoramic view in human infections. Future Virol. 2011;6:451–63.
Grundbacher FJ. Heritability estimates and genetic and environmental correlations for the human immunoglobulins G, M, and A. Am J Hum Genet. 1974;26:1–12.
Tsang JS, Schwartzberg PL, Kotliarov Y, Biancotto A, Xie Z, Germain RN, Wang E, Olnes MJ, Narayanan M, Golding H, Moir S, Dickler HB, Perl S, Cheung F, Baylor HIPC Center; CHI Consortium. Global analyses of human immune variation reveal baseline predictors of postvaccination responses. Cell. 2014;157:499–513.
Rubicz R, Leach CT, Kraig E, Dhurandhar NV, Duggirala R, Blangero J, Yolken R, Göring HH. Genetic factors influence serological measures of common infections. Hum Hered. 2011;72:133–41.
Almohmeed YH, Avenell A, Aucott L, Vickers MA. Systematic review and meta-analysis of the sero-epidemiological association between Epstein Barr virus and multiple sclerosis. PLoS One. 2013;8:e61110.
Timmann C, Thye T, Vens M, Evans J, May J, Ehmen C, Sievertsen J, Muntau B, Ruge G, Loag W, Ansong D, Antwi S, Asafo-Adjei E, Nguah SB, Kwakye KO, Akoto AO, Sylverken J, Brendel M, Schuldt K, Loley C, Franke A, Meyer CG, Agbenyega T, Ziegler A, Horstmann RD. Genome-wide association study indicates two novel resistance loci for severe malaria. Nature. 2012;489:443–6.
McLaren PJ, Coulonges C, Ripke S, van den Berg L, Buchbinder S, Carrington M, Cossarizza A, Dalmau J, Deeks SG, Delaneau O, De Luca A, Goedert JJ, Haas D, Herbeck JT, Kathiresan S, Kirk GD, Lambotte O, Luo M, Mallal S, van Manen D, Martinez-Picado J, Meyer L, Miro JM, Mullins JI, Obel N, O'Brien SJ, Pereyra F, Plummer FA, Poli G, Qi Y, Rucart P, Sandhu MS, Shea PR, Schuitemaker H, Theodorou I, Vannberg F, Veldink J, Walker BD, Weintrob A, Winkler CA, Wolinsky S, Telenti A, Goldstein DB, de Bakker PI, Zagury JF, Fellay J. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog. 2013;9:e1003515.
Casanova JL, Abel L. The genetic theory of infectious diseases: a brief history and selected illustrations. Annu Rev Genomics Hum Genet. 2013;14:215–43.
Tian C, Hromatka BS, Kiefer AK, Eriksson N, Noble SM, Tung JY, Hinds DA. Genome-wide association and HLA region fine-mapping studies identify susceptibility loci for multiple common infections. Nat Commun. 2017;8:599.
McLaren PJ, Coulonges C, Bartha I, Lenz TL, Deutsch AJ, Bashirova A, Buchbinder S, Carrington MN, Cossarizza A, Dalmau J, De Luca A, Goedert JJ, Gurdasani D, Haas DW, Herbeck JT, Johnson EO, Kirk GD, Lambotte O, Luo M, Mallal S, van Manen D, Martinez-Picado J, Meyer L, Miro JM, Mullins JI, Obel N, Poli G, Sandhu MS, Schuitemaker H, Shea PR, Theodorou I, Walker BD, Weintrob AC, Winkler CA, Wolinsky SM, Raychaudhuri S, Goldstein DB, Telenti A, de Bakker PI, Zagury JF, Fellay J. Polymorphisms of large effect explain the majority of the host genetic contribution to variation of HIV-1 virus load. Proc Natl Acad Sci U S A. 2015;112:14658–63.
Ge D, Fellay J, Thompson AJ, Simon JS, Shianna KV, Urban TJ, Heinzen EL, Qiu P, Bertelsen AH, Muir AJ, Sulkowski M, McHutchison JG, Goldstein DB. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461:399–401.
Hammer C, Begemann M, McLaren PJ, Bartha I, Michel A, Klose B, Schmitt C, Waterboer T, Pawlita M, Schulz TF, Ehrenreich H, Fellay J. Amino acid variation in HLA class II proteins is a major determinant of humoral response to common viruses. Am J Hum Genet. 2015;97:738–43.
Jonsson S, Sveinbjornsson G, de Lapuente Portilla AL, Swaminathan B, Plomp R, Dekkers G, Ajore R, Ali M, Bentlage AEH, Elmér E, Eyjolfsson GI, Gudjonsson SA, Gullberg U, Gylfason A, Halldorsson BV, Hansson M, Holm H, Johansson Å, Johnsson E, Jonasdottir A, Ludviksson BR, Oddsson A, Olafsson I, Olafsson S, Sigurdardottir O, Sigurdsson A, Stefansdottir L, Masson G, Sulem P, Wuhrer M, Wihlborg AK, Thorleifsson G, Gudbjartsson DF, Thorsteinsdottir U, Vidarsson G, Jonsdottir I, Nilsson B, Stefansson K. Identification of sequence variants influencing immunoglobulin levels. Nat Genet. 2017;49:1182–91.
Rubicz R, Yolken R, Drigalenko E, Carless MA, Dyer TD, Kent J Jr, Curran JE, Johnson MP, Cole SA, Fowler SP, Arya R, Puppala S, Almasy L, Moses EK, Kraig E, Duggirala R, Blangero J, Leach CT, Göring HH. Genome-wide genetic investigation of serological measures of common infections. Eur J Hum Genet. 2015;23:1544–8.
Thomas S, Rouilly V, Patin E, Alanio C, Dubois A, Delval C, Marquier LG, Fauchoux N, Sayegrih S, Vray M, Duffy D, Quintana-Murci L, Albert ML. Milieu Intérieur Consortium. The Milieu Intérieur study—an integrative approach for study of human immunological variance. Clin Immunol. 2015;157:277–93.
Patin E, Hasan M, Bergstedt J, Rouilly V, Libri V, Urrutia A, Alanio C, Scepanovic P, Hammer C, Jönsson F, Beitz B, Quach H, Lim YW, Hunkapiller J, Zepeda M, Green C, Piasecka B, Leloup L, Rogge L, Huetz F, Peguillet I, Lantz O, Fontes M, Di Santo JP, Thomas S, Fellay J, Duffy D, Quintana-Murci L, Albert ML, for The Milieu Intérieur Consortium. Natural variation in innate immune cell parameters is preferentially driven by genetic factors. Nat Immunol 2018;19:302–314.
Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen WM. Robust relationship inference in genome-wide association studies. Bioinformatics. 2010;26:2867–73.
Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190.
Loh PR, Danecek P, Palamara PF, Fuchsberger C, A Reshef Y, K Finucane H, Schoenherr S, Forer L, McCarthy S, Abecasis GR, Durbin R, L Price A. Reference-based phasing using the Haplotype Reference Consortium panel. Nat Genet. 2016;48:1443–8.
McCarthy S, et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet. 2016;48:1279–83.
Jia X, Han B, Onengut-Gumuscu S, Chen WM, Concannon PJ, Rich SS, Raychaudhuri S, de Bakker PI. Imputing amino acid polymorphisms in human leukocyte antigens. PLoS One. 2013;8:e64683.
Vukcevic D, Traherne JA, Næss S, Ellinghaus E, Kamatani Y, Dilthey A, Lathrop M, Karlsen TH, Franke A, Moffatt M, Cookson W, Trowsdale J, McVean G, Sawcer S, Leslie S. Imputation of KIR types from SNP variation data. Am J Hum Genet. 2015;97:593–607.
O'Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, Traglia M, Huang J, Huffman JE, Rudan I, McQuillan R, Fraser RM, Campbell H, Polasek O, Asiki G, Ekoru K, Hayward C, Wright AF, Vitart V, Navarro P, Zagury JF, Wilson JF, Toniolo D, Gasparini P, Soranzo N, Sandhu MS, Marchini J. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 2014;10:e1004234.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Zhou X, Stephens M. Efficient multivariate linear mixed model algorithms for genome-wide association studies. Nat Methods. 2014;11:407–9.
Meinshausen N, Bühlmann P. Stability selection. J R Stat Soc Ser B: Stat Methodol. 2010;72:417–73.
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75.
Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, Land SJ, Lu X, Ruden DM. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin). 2012;6:80–92.
Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–2.
Ionita-Laza I, Lee S, Makarov V, Buxbaum JD, Lin X. Sequence kernel association tests for the combined effect of rare and common variants. Am J Hum Genet. 2013;92:841–53.
Mayerle J, den Hoed CM, Schurmann C, Stolk L, Homuth G, Peters MJ, Capelle LG, Zimmermann K, Rivadeneira F, Gruska S, Völzke H, de Vries AC, Völker U, Teumer A, van Meurs JB, Steinmetz I, Nauck M, Ernst F, Weiss FU, Hofman A, Zenker M, Kroemer HK, Prokisch H, Uitterlinden AG, Lerch MM, Kuipers EJ. Identification of genetic loci associated with Helicobacter pylori serologic status. JAMA. 2013;309:1912–20.
Swaminathan B, Thorleifsson G, Jöud M, Ali M, Johnsson E, Ajore R, Sulem P, Halvarsson BM, Eyjolfsson G, Haraldsdottir V, Hultman C, Ingelsson E, Kristinsson SY, Kähler AK, Lenhoff S, Masson G, Mellqvist UH, Månsson R, Nelander S, Olafsson I, Sigurðardottir O, Steingrimsdóttir H, Vangsted A, Vogel U, Waage A, Nahi H, Gudbjartsson DF, Rafnar T, Turesson I, Gullberg U, Stefánsson K, Hansson M, Thorsteinsdóttir U, Nilsson B. Variants in ELL2 influencing immunoglobulin levels associate with multiple myeloma. Nat Commun. 2015;6:7213.
Viktorin A, Frankowiack M, Padyukov L, Chang Z, Melén E, Sääf A, Kull I, Klareskog L, Hammarström L, Magnusson PK. IgA measurements in over 12 000 Swedish twins reveal sex differential heritability and regulatory locus near CD30L. Hum Mol Genet. 2014;23:4177–84.
Frankowiack M, Kovanen RM, Repasky GA, Lim CK, Song C, Pedersen NL, Hammarström L. The higher frequency of IgA deficiency among Swedish twins is not explained by HLA haplotypes. Genes Immun. 2015;16:199–205.
Yang C, Jie W, Yanlong Y, Xuefeng G, Aihua T, Yong G, Zheng L, Youjie Z, Haiying Z, Xue Q, Min Q, Linjian M, Xiaobo Y, Yanling H, Zengnan M. Genome-wide association study identifies TNFSF13 as a susceptibility gene for IgA in a South Chinese population in smokers. Immunogenetics. 2012;64:747–53.
Liao M, Ye F, Zhang B, Huang L, Xiao Q, Qin M, Mo L, Tan A, Gao Y, Lu Z, Wu C, Zhang Y, Zhang H, Qin X, Hu Y, Yang X, Mo Z. Genome-wide association study identifies common variants at TNFRSF13B associated with IgG level in a healthy Chinese male population. Genes Immun. 2012;13:509–13.
Yang M, Wu Y, Lu Y, Liu C, Sun J, Liao M, Qin M, Mo L, Gao Y, Lu Z, Wu C, Zhang Y, Zhang H, Qin X, Hu Y, Zhang S, Li J, Dong M, Zheng SL, Xu J, Yang X, Tan A, Mo Z. Genome-wide scan identifies variant in TNFSF13 associated with serum IgM in a healthy Chinese male population. PLoS One. 2012;7:e47990.
Lambert ND, Haralambieva IH, Kennedy RB, Ovsyannikova IG, Pankratz VS, Poland GA. Polymorphisms in HLA-DPB1 are associated with differences in rubella virus-specific humoral immunity after vaccination. J Infect Dis. 2015;211:898–905.
Rubicz R, Yolken R, Drigalenko E, Carless MA, Dyer TD, Bauman L, Melton PE, Kent JW, Jr HJB, Curran JE, Johnson MP, Cole SA, Almasy L, Moses EK, Dhurandhar NV, Kraig E, Blangero J, Leach CT, Göring HH. A genome-wide integrative genomic study localizes genetic factors influencing antibodies against Epstein-Barr virus nuclear antigen 1 (EBNA-1). PLoS Genet. 2013;9:e1003147.
Pedergnana V, Syx L, Cobat A, Guergnon J, Brice P, Fermé C, Carde P, Hermine O, Le-Pendeven C, Amiel C, Taoufik Y, Alcaïs A, Theodorou I, Besson C, Abel L. Combined linkage and association studies show that HLA class II variants control levels of antibodies against Epstein-Barr virus antigens. PLoS One. 2014;9:e102501.
Rajagopalan S, Long EO. Understanding how combinations of HLA and KIR genes influence disease. J Exp Med. 2005;201:1025–9.
Goetzman ES, Alcorn JF, Bharathi SS, Uppala R, McHugh KJ, Kosmider B, Chen R, Zuo YY, Beck ME, McKinney RW, Skilling H, Suhrie KR, Karunanidhi A, Yeasted R, Otsubo C, Ellis B, Tyurina YY, Kagan VE, Mallampalli RK, Vockley J. Long-chain acyl-CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J Biol Chem. 2014;289:10668–79.
Giefing-Kröll C, Berger P, Lepperdinger G, Grubeck-Loebenstein B. How sex and age affect immune responses, susceptibility to infections, and response to vaccination. Aging Cell. 2015;14:309–21.
Cook IF. Sexual dimorphism of humoral immunity with human vaccines. Vaccine. 2008;26:3551–5.
Klein SL, Jedlicka A, Pekosz A. The Xs and Y of immune responses to viral vaccines. Lancet Infect Dis. 2010;10:338–49.
Ovsyannikova IG, Jacobson RM, Dhiman N, Vierkant RA, Pankratz VS, Poland GA. Human leukocyte antigen and cytokine receptor gene polymorphisms associated with heterogeneous immune responses to mumps viral vaccine. Pediatrics. 2008;121:e1091–9.
Kennedy RB, Ovsyannikova IG, Pankratz VS, Vierkant RA, Jacobson RM, Ryan MA, Poland GA. Gender effects on humoral immune responses to smallpox vaccine. Vaccine. 2009;27:3319–23.
Girón-González JA, Moral FJ, Elvira J, García-Gil D, Guerrero F, Gavilán I, Escobar L. Consistent production of a higher TH1:TH2 cytokine ratio by stimulated T cells in men compared with women. Eur J Endocrinol. 2000;143:31–6.
Acknowledgements
We would like to thank all the donors for their contribution to the study. We also thank the members of the The Milieu Intérieur Consortium for their insightful comments. The Milieu Intérieur Consortium is composed of the following team leaders: Laurent Abel (Hôpital Necker, Paris, France), Andres Alcover (Institut Pasteur, Paris, France), Hugues Aschard (Institut Pasteur, Paris, France), Kalla Astrom (Lund University, Lund, Sweden), Philippe Bousso (Institut Pasteur, Paris, France), Pierre Bruhns (Institut Pasteur, Paris, France), Ana Cumano (Institut Pasteur, Paris, France), Caroline Demangel (Institut Pasteur, Paris, France), Ludovic Deriano (Institut Pasteur, Paris, France), James Di Santo (Institut Pasteur, Paris, France), Françoise Dromer (Institut Pasteur, Paris, France), Darragh Duffy (Institut Pasteur, Paris, France), Gérard Eberl (Institut Pasteur, Paris, France), Jost Enninga (Institut Pasteur, Paris, France), Jacques Fellay (EPFL, Lausanne, Switzerland) Odile Gelpi (Institut Pasteur, Paris, France), Ivo Gomperts-Boneca (Institut Pasteur, Paris, France), Milena Hasan (Institut Pasteur, Paris, France), Serge Hercberg (Université Paris 13, Paris, France), Olivier Lantz (Institut Curie, Paris, France), Claude Leclerc (Institut Pasteur, Paris, France), Hugo Mouquet (Institut Pasteur, Paris, France), Sandra Pellegrini (Institut Pasteur, Paris, France), Stanislas Pol (Hôpital Côchin, Paris, France), Antonio Rausell (INSERM UMR 1163 – Institut Imagine, Paris, France), Lars Rogge (Institut Pasteur, Paris, France), Anavaj Sakuntabhai (Institut Pasteur, Paris, France), Olivier Schwartz (Institut Pasteur, Paris, France), Benno Schwikowski (Institut Pasteur, Paris, France), Spencer Shorte (Institut Pasteur, Paris, France), Vassili Soumelis (Institut Curie, Paris, France), Frédéric Tangy (Institut Pasteur, Paris, France), Eric Tartour (Hôpital Européen George Pompidou, Paris, France), Antoine Toubert (Hôpital Saint-Louis, Paris, France), Mathilde Touvier (Université Paris 13, Paris, France), Marie-Noëlle Ungeheuer (Institut Pasteur, Paris, France), Matthew L. Albert (Roche Genentech, South San Francisco, CA, USA), Lluis Quintana-Murci (Institut Pasteur, Paris, France).
Funding
This work benefited from support of the French government’s Invest in the Future Program, managed by the Agence Nationale de la Recherche (ANR, reference 10-LABX-69-01). It was also supported by a grant from the Swiss National Science Foundation (31003A_175603, to JF). C.A. received a PostDoctoral Fellowship from Institut National de la Recherche Médicale.
Availability of data and materials
Genotype data supporting the conclusions of this article are available in the European Genome-Phenome Archive under the accession code EGAS00001002460. Full summary association results are available for download from Zenodo (https://doi.org/10.5281/zenodo.1217136).
Author information
Authors and Affiliations
Consortia
Contributions
CA, LA, LQ-M, DD, MLA, and JF contributed to the conception of the study. PS, CA, CH, DD, MLA, and JF contributed to the design of the study. PS, CA, CH, FH, EP, and BC contributed to the acquisition of the data. PS, CA, CH, FH, JB, CWT, and NC contributed to the analysis of the data. PS, CA, CH, DD, MLA, and JF contributed to the drafting of the manuscript. PS, CA, CH, FH, JB, EP, CWT, LA, DD, MLA, and JF contributed to the revising of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The clinical study was approved by the Comité de Protection des Personnes - Ouest 6 on June 13, 2012 and by the French Agence Nationale de Sécurité du Médicament on June 22, 2012, and has been performed in accordance with the Declaration of Helsinki. The study is sponsored by the Institut Pasteur (Pasteur ID-RCB Number: 2012-A00238-35) and was conducted as a single-center study without any investigational product. The protocol is registered under ClinicalTrials.gov (study# NCT01699893). Informed consent was obtained from participants after the nature and possible consequences of the studies were explained.
Consent for publication
Not applicable.
Competing interests
C.H. and M.L.A. are employees of Genentech Inc., a member of The Roche Group. The remaining authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Assay details for serologies. Table S2. Cutoffs and seroprevalence for serologies. Table S3. List of covariates used for each phenotype. Table S4. Associations of environmental variables with serostatus. Table S5. Association of serologies with age and sex. Table S6. Associations of amino acid positions in HLA proteins with Influenza A serology. Table S7. LD between residues in HLA-DRβ1 at position 13 and 96. Table S8. Replication of SNPs associated with levels of total IgM, IgA and IgG. Table S9. LD between residues in HLA-DRβ1 at position 15 and 11. (XLSX 65 kb)
Additional file 2:
Figure S1. Principal Component Analysis. Figure S2. Distribution of serological variables, and clinical thresholds. Figure S3. Seroprevalence data. Figure S4. Impact of non-genetic factors on serostatus. Figure S5. Evolution of serostatus with age and sex. Figure S6. Correlations between age and IgG specific to Rubella and T. gondii. Figure S7. QQ plots for logistic regressions preformed in the study. Figure S8. QQ plots for linear regressions preformed on total Ig levels. Figure S9. QQ plots for linear regressions preformed for pathogen-specific IgG levels. Figure S10. QQ plots for burden testing analyses preformed for all binary phenotypes. Figure S11. QQ plots for burden testing analyses preformed for total Ig levels. Figure S12. QQ plots for burden testing analyses preformed for pathogen-specific IgG levels. (DOCX 89996 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Scepanovic, P., Alanio, C., Hammer, C. et al. Human genetic variants and age are the strongest predictors of humoral immune responses to common pathogens and vaccines. Genome Med 10, 59 (2018). https://doi.org/10.1186/s13073-018-0568-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13073-018-0568-8