
Abstract
Background
Mutations in INF2 are frequently responsible for focal segmental glomerulosclerosis (FSGS), which is a common cause of end stage renal disease (ESRD); additionally, they are also connected with Charcot-Marie-Tooth neuropathy. INF2 encodes for inverted formin 2. This protein participates in regulation of the dynamics of the actin cytoskeleton, involving not only the polymerisation, but also the depolymerisation of filaments. The present study is the first mutational analysis of INF2 done in the Czech Republic.
Methods
Mutational analysis of INF2 was performed on 109 patients (mean age at onset 41.44 ± 18.91 years) with FSGS or minimal change disease (MCD); and also in 6 patients without renal biopsy who had already developed chronic kidney disease (CKD)/ESRD at the time of diagnosis. We used high resolution melting method (HRM), with subsequent Sanger sequencing, in suspect samples from HRM analysis. The HRM method is an effective method for the screening of large cohorts of patients.
Results
Two pathogenic mutations (p.Arg214His and p.Arg218Gln) were detected in INF2. The first (p.Arg214His) was identified in the FSGS patient with a positive family history. The second mutation (p.Arg218Gln) was found in two brothers with ESRD of unknown etiology. The most frequent sequence change was the substitution p.P35P, the incidence of which corresponded with the frequencies available in the ExAC Browser and gnomAD Browser databases. This analysis also detected different exonic and intronic changes that probably did not influence the phenotype of the included patients.
Conclusions
The INF2 mutational screening is useful in familial FSGS cases as well as in patients with an unknown cause for their ESRD, but with a positive family history. INF2 seems to be not only the cause of FSGS, but also of ESRD of unknown etiology. Our study has confirmed that the HRM analysis is a very useful method for the identification of single nucleotide substitutions.
Similar content being viewed by others

Background
Podocytes are highly specialized cells with a unique and highly differentiated cytoarchitecture. Their main functions include: support of the glomerular capillaries, synthesis of glomerular basement membrane (GBM), and regulation of permselectivity. Any damage to the podocytes is usually characterized by nephrotic syndrome and (if persistent) it may progress into end stage renal disease (ESRD). Minimal change disease (MCD) and focal segmental glomerulosclerosis (FSGS) are common causes of nephrotic syndrome.
The major pathological feature of both MCD and FSGS is increased permeability of the glomerular capillary filter. Under light microscopy the glomeruli of patients with MCD have a normal size and cellularity and show no evidence of segmental sclerosis; whereas electron microscopy demonstrates the same podocyte foot processes effacement as in glomeruli affected by FSGS. Whereas the outcome of MCD in terms of long-term preservation of renal function is good; by comparison, FSGS often leads to ESRD. We still have only limited insight into the pathogenesis of both MCD and FSGS. Moreover, both the etiology and histology of FSGS as well as of MCD may be very variable, including: viral infections, toxic agents, adaptive structural-functional responses, and mutations in the genes encoding proteins specific for podocytes (or highly expressed in them) [1,2,3], such as: ACTN4 [4], INF2 [5], and TRPC6 [6].
Inverted formin 2, encoded by INF2, is a member of the formin family of proteins, which regulates the dynamics of the actin cytoskeleton. It not only accelerates actin polymerisation, but also filament depolymerisation. INF2 is expressed in many tissues such as: kidney, liver, heart, skeletal muscles, and placenta. In renal podocytes, it plays a key role in the function and structure of the actin cytoskeleton. The main regions of INF2 are: the N-terminal diaphanous inhibitory domain (DID), the formin homology FH1 and FH2 domains, and the terminal diaphanous autoregulatory domain (DAD) [7, 8]. Most of the reported mutations are localized in the DID domain, which probably functions as a regulatory domain of the polymerisation and depolymerisation of the actin filaments. The previous experiments demonstrated that substitutions in INF2 caused an abnormal distribution of the slit diaphragm proteins nephrin and podocin, and dysregulation of the podocyte cytoskeleton; suggesting its involvement in the pathogenesis of the autosomal dominant form of FSGS [5, 9]. Mutations in INF2 are not only connected with FSGS, but also with Charcot-Marie-Tooth neuropathy [10].
In this study, we present a mutational analysis of INF2 (whose mutations are frequent causes of FSGS in adults) in patients with FSGS or MCD as well as in a group of patients with a positive family history of ESRD of unknown etiology, using a high resolution melting method (HRM) and Sanger sequencing.
Methods
Patients and study design
Our study, focused upon INF2, was carried out in 109 patients with biopsy-proven FSGS or MCD (see Table 1 for detailed information). The origins of FSGS or MCD of the involved individuals were both familial (inherited) in 19 cases, and idiopathic (sporadic). The study also included a group of 6 patients with a positive family history for ESRD in combination with developed advanced chronic kidney disease (CKD)/ESRD at the time of diagnosis. There were no renal biopsies performed in this group of patients thought to have burnt out diagnostic features because of advanced disease. Healthy controls from anonymous volunteers were used to test for variants in healthy individuals.
Ethical approval
The study was performed with the approval of the Ethics Committee of the General University Hospital in Prague. The blood of patients, their relatives, and the anonymized controls was obtained after written informed consent was given by all in accordance with the protocol approved by the institutional review board at the General University Hospital.
Mutational analysis
Genomic DNA was isolated from the peripheral blood lymphocytes using standard procedures. The complete coding region and intron-exon boundaries of INF2 were amplified, and were subsequently analyzed using the HRM method and LightCycler 480, as well as HRM Master Mix (both by Roche Diagnostics). The data obtained from the HRM analysis were analyzed using Light Cycler Gene Scanning Software (Roche Diagnostics). The suspect samples from the HRM analysis were screened for substitutions by Sanger sequencing performed on an ABI Prism™ 3130 Genetic Analyser using BigDye® Terminator v1.1 Cycle Sequencing Kit (both by Applied Biosystems). All primer sequences and annealing temperatures are available upon request. Targeted Sanger sequencing was also performed in the parents and relatives of probands with causal mutations or novel substitutions in order to determine their carryover and position on homologous chromosomes.
The HRM method is a fast, simple, and cheap method for the analysis of large cohorts of patients. Especially for SNP scanning, this is a highly sensitive and specific method, with some limitations (e.g., GC-rich fragments, or detection of homozygous variants). Sanger sequencing is the technique commonly used in molecular laboratories. On the one hand, it is a simple method in terms of sample preparation, and an effective way to analyze single genes, or to confirm variations identified by next generation sequencing. Then again, the procedure is an expensive procedure with detection limitations (e.g., mosaicism).
Sequence analysis and assessment of sequence changes
Sequence chromatograms were analyzed using a BioEdit Sequence Alignment Editor (http://www.mbio.ncsu.edu/BioEdit). All sequenced samples were compared to the reference coding sequence NM_022489.3 as well as the reference genome sequence NG_027684.1, respectively. Substitutions found in this analysis were checked against the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and/or the Ensembl [11] and NCBI dbSNP database (https://www.ncbi.nlm.nih.gov/projects/SNP/). In the case of novel missense variants, their pathogenicity was assessed computationally using Mutation Taster [12], PolyPhen-2 [13], PROVEAN [14], and SIFT [15]. The possible splice effect of intronic changes was evaluated using Human Splicing Finder [16] and NetGene2 (http://www.cbs.dtu.dk/services/NetGene2/).
Results
INF2 mutational analysis was performed in a total of the 115 patients with FSGS (77 patients) /MCD (32 patients), or the characteristics of a separate group including a positive family history for ESRD in combination with developed advanced CKD/ESRD at the time of diagnosis. The described mutations and both the exonic and intronic polymorphisms had been identified. See Table 2 for the exonic variants identified.
Two mutations in INF2 (p.Arg218Gln and p.Arg214His) already known to be responsible for the familial form of FSGS [5] were detected in two different families with a positive family history for FSGS, or ESRD of unknown etiology. Both of the mutations found are localized in exon 4, which is involved in the highly conserved DID region, and that is situated at the N-terminus of the INF2 protein.
The most frequent substitution in INF2 (p.P35P) located in exon 2, had been found in all patients, mostly in the homozygous state (allele frequency = 97.4%). Exon 21 of INF2 seemed to be a very variable region because of the high number of changes identified. However, these substitutions probably did not influence the phenotype of the patients included in our study.
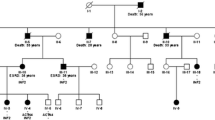
Family no. 133 (INF2, p.Arg218Gln)
This family (see Fig. 1a), with a positive family history of ESRD of unknown etiology, and with mild proteinuria (affecting two brothers, their father, aunt, and cousin), had already been identified as one described by the causal missense mutation p.Arg218Gln (c.653 G > A) in INF2, with a damaging effect on podocyte function [5]. The substitution was first detected in the two brothers who had already developed ESRD. Afterwards, the same mutation was found in their father and cousin. The ages of the brothers at ESRD were 27 and 31 years, respectively; while their father developed ESRD at age 57. For that reason, in the case of these two patients, we presume the co-action of more factors, such as other substitutions in INF2 or some influence from the environment. The younger affected brother as well as his healthy mother also had the SNP p.Pro528Ser (c.1582 C > T) in INF2. Additionally, we found other different intronic and exonic same-sense substitutions; however, there is not any hypothesis of their effects on the phenotype.

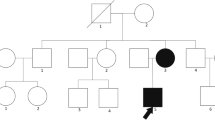
Pedigree diagrams of selected families from our cohorts of patients. The figure shows the pedigrees of the INF2 mutated families; black arrows indicate original probands; black filled individuals were affected by renal disease. All affected individuals whose DNA was available for analysis carried the mutation in INF2
Family no. 50 (INF2, p.Arg214His)
Also identified was the known mutation p.Arg214His (c.641 G > A), with a proven damaging effect [5], in a 51 year old woman with FSGS (from a renal biopsy) and with a positive family history (her father had ESRD at age 33, and an uncle at age 30) (see Fig. 1b). Her two daughters were healthy. At her age, this patient has only mild chronic renal insufficiency with moderate proteinuria (1-3 g/24 h), although she suffers from diabetes mellitus, hypertension, and obesity (BMI = 35.58). The biopsy sample showed a perihilar variant of FSGS, with a mild increase of the mesangial matrix (see Fig. 2). This type of injury represents a common part of the so-called secondary FSGS, which is mediated by adaptive structural-functional responses, and frequently is associated with obesity of all of grades with a BMI ≥ 30.0. Even though the renal biopsy looked like secondary FSGS, a mutational analysis was done because of the positive family history of ESRD of unknown etiology.

Kidney biopsy findings in a proband from family No. 50. Enlarged glomerulus with perihilar segmental sclerotic lesion. Simultaneously, there is showed a small sclerotic lesion (arrow) with a cell bridging between the Bowman capsule and the GBM (PAS, high power field, objective 40×)
Discussion
Several genes highly expressed in podocytes (e.g., ACTN4, INF2, and TRPC6) had previously been reported to be a cause of familial and sporadic autosomal dominant forms of FSGS. Herein, we present the molecular genetic analysis of INF2 (using the HRM method and Sanger sequencing) performed on a large cohort of patients with FSGS/MCD (FSGS = 77 patients, MCD = 32 patients), as well as in a group of patients (6) characterized by a positive family history for ESRD in combination with advanced CKD/ESRD at the time of diagnosis. These types of analyses can lead to the identification of new causal mutations; therefore, they are very important in clinical practice for the diagnosis of patients.
The INF2 protein, belonging to the formin family, plays a key role in the influence of the actin cytoskeleton dependent processes in podocytes because of its capability to accelerate both the polymerisation and depolymerisation of actin. The protein consists of three main parts including: the DID domain, two FH domains, and the DAD domain. Previous studies have demonstrated the abnormal distribution of the detected mutations; all of them having been identified in exons encoding the DID domain, suggesting its crucial role for the protein function. The DID domain appears to be important for actin cytoskeleton regulation [7, 8].
All identified substitutions in the exons were determined to be disease causing candidates if they: (a) segregated with the disease (in the case that other relatives were also affected, and we had samples of their DNA); (b) were predicted to be damaging by the Mutational Taster program [12], PolyPhen-2 [13], PROVEAN [14], and SIFT [15]; (c) were not carried by both healthy family members (whose DNA samples we had) as well as the healthy controls; and (d) were not present in control chromosomes in the db SNP and 1000 Genomes project.
We identified the mutation p.Arg218Gln in INF2 with a damaging effect on the podocyte function in two brothers from a family with the positive history for ESRD. This mutation was first described by Brown et al. in a large family with the familial form of FSGS [5]. Both brothers quickly progressed to ESRD, aged 27 and 31, respectively.
The other detected casual mutation in INF2 was p.Arg214His, also first identified by Brown et al. [5]. The mutation was found in a woman with a perihilar variant of FSGS and simultaneously suffering from diabetes mellitus. Although the family history of the proband was positive for renal diseases (father and uncle), her two healthy daughters had refused the opportunity for genetic testing. The incidences of this variant in our analysis were 0.9% (the whole cohort of patients), 1.3% (only the cohort of FSGS patients), and 5.6% (the cohort of FSGS patients with a positive family history), respectively. Both identified casual mutations are localized in the DID domain of INF2, which is necessary for normal protein function because it is involved in actin cytoskeleton regulation.
The mutational analysis of INF2 also identified other different exonic changes and intronic substitutions that seemed to have no effect on the phenotype of the patients included in this study.
The most frequent substitution in INF2 was p.P35P within exon 2, and with an allele frequency of more than 97%. This incidence corresponds with the results available in the ExAC Browser (http://exac.broadinstitute.org/) and gnomAD Browser (http://gnomad.broadinstitute.org/) databases, confirming the sensitivity of HRM. Other highly frequent changes in INF2 were p.D1022D and p.D880D, with allele frequencies of 68% and 60%, respectively.
The HRM analysis was a very effective method for mutational screening in such a large cohort of patients. It proved to be of sufficient sensitivity in the detection of single nucleotide substitutions.
According to previous studies focused on INF2, it appears that this gene is responsible for a high percentage of patients with familial FSGS [5, 17, 18]. Even though the proportion of mutations in sporadic FSGS is significantly lower [9, 18], we included both familiar and idiopathic patients with FSGS. Although the incidence of pathogenic mutations was lower than our assumptions at the beginning of the study, we established the method for the molecular analysis of INF2 and analyzed a large cohort of patients with FSGS/MCD; confirming that MCD cases are less likely to harbor deleterious genetic variants in those genes implicated in FSGS [4,5,6, 19]. This was the first molecular genetic analysis focused on INF2 in the Czech Republic. A mutational analysis of INF2 should be performed in all patients with a positive family history of FSGS or unknown ESRD with an autosomal dominant inheritance. Proteinuria is frequently mild or moderate, the patients are asymptomatic, and advanced renal insufficiency is already present, so a renal biopsy is not indicated in many cases.
Conclusions
In conclusion, pathogenic mutations of INF2 are not a frequent cause of FSGS nor of MCD in the Czech Republic. The INF2 mutations must be taken into account; not only in patients with FSGS and a positive family history, but also in patients with ESRD of unknown etiology and a positive family history of ESRD in other generations. The HRM method seems to be a reliable and cheaper method than is direct sequencing for an analysis of the concrete gene in large cohorts of patients.
Abbreviations
- ACTN4:
-
Alpha-actinin 4
- BMI:
-
Body mass index
- CKD:
-
Chronic kidney disease
- DAD:
-
Diaphanous autoregulatory domain
- DID:
-
Diaphanoury domain
- ESRD:
-
End stage renal disease
- FH:
-
Formin homology domain
- FSGS:
-
Focal segmental glomerulosclerosis
- GBM:
-
Glomerular basement membrane
- HRM:
-
High resolution melting
- INF2:
-
Inverted formin 2
- MCD:
-
Minimal change disease
- PAS:
-
Periodic acid-Schiff stain
- R:
-
Spectrin repeat
- SNP:
-
Single nucleotide polymorphism
- TRPC6:
-
Transient receptor potential cation channel, subfamily C, member 6.
References
D’Agati VD, Fogo AB, Bruijn JA, Jennette JC. Pathologic classification of focal segmental glomerulosclerosis: a working proposal. Am J Kidney Dis. 2004;43:368–82.
Howie AJ. Pathology of minimal change nephropathy and segmental sclerosing glomerular disorders. Nephrol Dial Transplant. 2003;18:vi33–8.
Becker DJ. Minimal change disease. Nephrology Rounds. 2008;6:304–066.
Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, et al. Mutations in ACTN4, encoding α-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet. 2000;24(3):251–6.
Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42(1):72–6.
Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF, et al. A mutation in the TRPC6 cation channel causes familial focal segmental glomerulosclerosis. Science. 2005;308(5729):1801–4.
Chhabra ES, Higgs HN, et al. INF2 is a WASP homology 2 motif-containing formin that severs actin filaments and accelerates both polymerization and depolymerization. J Biol Chem. 2006;281(36):26754–67.
Schönichen A, Geyer M. Fifteen formins for an actin filament: a molecular view on the regulation of human formins. BBA-Mol Cell Res. 2010;1803(2):152–63.
Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013;83(2):316–22.
Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, et al. INF2 mutations in Charcot-Marie-tooth disease with glomerulopathy. N Engl J Med. 2011;365(25):2377–88.
Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, et al. Ensembl 2016. Nucleic Acids Res. 2016;44(Database issue):D710–6.
Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–2.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–9.
Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the Functional Effect of Amino Acid Substitutions and Indels. PLoS One. 2015;7(10):e46688.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11(5):863–74.
Desmet FO, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C. Human splicing finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37(9):e67.
Caridi G, Lugani F, Dagnino M, Gigante M, Iolascon A, Falco M, et al. Novel INF2 mutations in an Italian cohort of patients with focal segmental glomerulosclerosis, renal failure and Charcot-Marie-Tooth neuropathy. Nephrol Dial Transplant. 2014;29(Suppl 4):iv80–6.
Boyer O, Benoit G, Gribouval O, Nevo F, Tête MJ, Dantal J, et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22(2):239–45.
Gigante M, Caridi G, Montemurno E, Soccio M, d’Apolito M, Cerullo G, Messina G. TRPC6 mutations in children with steroid-resistant nephrotic syndrome and atypical phenotype. Clin J Am Soc Nephrol. 2011;6(7):1626–34.
Acknowledgements
The authors wish to thank all included patients and their relatives, healthy controls, and the staff of the Laboratory of molecular genetics at the Institute of Biology and Medical Genetics, First Faculty of Medicine, Charles University and General University Hospital in Prague, Czech Republic.
Funding
This study was supported by Charles University project grants PROGRES Q25/LF1 and SVV 260373. Charles University had no role in the design of the study and collection, analysis and interpretation of data and in writing the manuscript.
Availability of data and materials
Data supporting the findings are included in the article. Any further information is available from the corresponding author upon reasonable request.
Author information
Authors and Affiliations
Contributions
MS carried out the molecular genetic analysis and the drafting of the manuscript. JS conceived and designed the study, and coordinated the analyses. EH made the histopathologic analyses. VH recruited the participants and collected clinical data. VT participated in the design of the study. JR recruited the participants, collected the clinical data, and helped to draft the manuscript. All of the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was performed in accordance with the Helsinki Declaration, and with the approval of the Ethics Committee of the General University Hospital in Prague, Ref. No. 1443/11 S-IV. The blood of patients, their relatives, as well as anonymized controls was obtained after their written informed consent had been obtained.
Consent for publication
The written consent for publication of case presentations was obtained from each relevant participant.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Safarikova, M., Stekrova, J., Honsova, E. et al. Mutational screening of inverted formin 2 in adult-onset focal segmental glomerulosclerosis or minimal change patients from the Czech Republic. BMC Med Genet 19, 147 (2018). https://doi.org/10.1186/s12881-018-0667-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0667-9