
Abstract
Background
Heterozygous mutations in the inverted formin 2 (INF2) gene are related to secondary focal segmental glomerulosclerosis (FSGS), a rare secondary disease associated with rapidly progressive renal failure.
Case presentation
We report a patient with familial autosomal INF2 mutation manifesting nephritic syndromes and elevated serum creatinine levels. Mutational analysis revealed an autosomal dominant (AD) inheritance pattern and a mutation in exon 4 (p.Arg214Cys) of INF2 as the likely cause, which has not been previously described in an Asian family. The patient progressed to end-stage renal disease (ESRD) and received hemodialysis. His mother had undergone renal transplant 3 years earlier, and his grandmother had carried the p.Arg214Cys mutation for more than 80 years without any sign of renal dysfunction.
Conclusions
This is the first report to identify an association between a familial autosomal dominant INF2 p.Arg214Cys mutation and rapidly progressive renal disease in an Asian family. INF2 mutation analysis should not be restricted to individuals without family history of FSGS, rather it should also be performed on individuals for whom drug-based therapies are not effective. In this case, kidney transplant is an effective alternative.
Similar content being viewed by others


Background
Formins are widely expressed proteins that govern remodeling of the actin cytoskeleton during cytokinesis, cell polarization, and tissue morphogenesis [1].
In the last 10 years, the inverted formin 2 (INF2) has been an important target of mutations responsible for focal segmental glomerulosclerosis (FSGS) [2]. Brown et al. [3] identified an association between heterozygous mutations in INF2 gene and FSGS in 12% of the families studied and recorded the ages of affected individuals at the time of diagnosis and the time of developing ESRD. INF2 encodes an actin regulatory protein of the formin family, which plays an important role in maintaining podocyte plasticity [4, 5].
INF2 mutations have been identified as being responsible for the development of autosomal dominant (AD) FSGS [3, 4, 6, 7]. These mutations are also associated with Charcot-Marie-Tooth disease (CMT) [2, 5, 8,9,10,11], which is characterized by a demyelinating peripheral neuropathy [9]. In CMT patients, an increased prevalence of FSGS has been documented [12]. While the incidence of FSGS is one in a million of the general population, its incidence is one out of four hundred in CMT patients [13]. INF2 mutations are thought to be the link between FSGS and CMT: the frequency of INF2 mutation in patients with both FSGS and CMT is much higher (75%) than that in patients affected by FSGS alone (12–17%) [3, 14, 15]; no INF2 mutations were found in patients with CMT alone. However, it has not been clearly explained why INF2 mutation does not always lead to nerve phenotypic abnormalities. Boyer et al. identified INF2 mutations in exons 2 and 3 in 75% of patients with both CMT and FSGS [5]. De Jonghe et al. reported that mutations in exon 3 of INF2 are responsible for causing CMT-FSGS, whereas mutations in exon 4 and 6 are associated with FSGS alone [10]. However, Caridi et al. reported that CMT patients carried a mutation in exon 4 [9]. Quantitative live-cell imaging may identify distinct subsets of INF2 variants that are linked to FSGS alone or to CMT-FSGS.
In terms of clinical outcomes, intra-familial phenotypes of INF2 mutation are varied. Lee et al. [16] and Caridi et al. [9] observed marked clinical heterogeneity and different severities of disease within families carrying the same INF2 mutation. Due to incomplete penetrance, family members that carry autosomal dominant FSGS may also be asymptomatic [16]. Although more than 70 families and 250 patients have been reported to carry an INF2 mutation in previous reports [4, 9, 17], the affected families are all of European descent. Here, we report a new familial autosomal INF2 mutation in exon 4 (p.Arg214Cys) present in two patients from the same three-generation family of Chinese origin; these patients presented with proteinuria, high blood pressure, and hyperuricemia, and exhibited rapid progression of renal failure. This is the first report of INF2 p.Arg214Cys in Asian families, and one of the few reported cases worldwide.
Case presentation
We report the case of a 23-year-old male patient admitted to our hospital in June 2018. The patient had a 4-month history of foamy urine prior to admission. His medical report revealed proteinuria (+++), mild edema of the lower limbs, elevated serum creatinine (259 μmol/L) levels, and mild hypertension (145/78 mmHg). The patient had no prior history of renal problems.
The patient was hospitalized due to nephrotic syndrome and renal failure. Laboratory testing revealed the following: blood urea nitrogen (BUN), 7.1 mmol/L; serum creatinine (Scr), 282 μmol/L; urine albumin/creatinine (UACR), 2701.53 mg/g; urine protein/creatinine (UPCR), 3360 mg/g; urinary protein quantitative (24 h), 6.678 g; serum albumin, 34.3 g/L; serum uric acid, 561 μmol/L; total cholesterol, 7.63 mmol/L; low density lipoprotein, 5.18 mmol/L; hemoglobin (Hb), 130 g/L; anti-PLA2R, negative; parathyroid hormone (iPTH), 135.79 pg/mL; and homocysteine (HCY), 22.23 μmol/L. Interventricular septal thickness was found to be 12 mm using cardiac ultrasound. As renal ultrasound revealed renal cortex thinning and an unclear boundary of the renal cortex and medulla, renal biopsy was not performed.
After discharge, the patient was prescribed α-keto acid, atorvastatin calcium, and febuxostat tablets to control proteinuria and edema. Unfortunately, the patient’s proteinuria was not improved, and his serum creatinine levels rose rapidly (Table 1). Prednisone (Starting dose 1 mg/kg) was administered for several months but was not effective as renal failure rapidly progressed. In March 2020, the patient started hemodialysis. In June 2020, the patient underwent renal transplantation. The transplantation was successful, and the patient has not shown any sign of proteinuria or hematuria since.
Eight years ago, the patient’s mother had manifested proteinuria, elevated serum creatinine levels (142 μmol/L), high blood pressure, and hyperuricemia. Angiotensin receptor blockers (ARBs) treatment was ineffective, and renal failure rapidly progressed. Peritoneal dialysis was initiated 3 years later (12/31/2014), and kidney transplantation was performed after another 2 years (03/03/2017). After transplantation, proteinuria was completely resolved; blood pressure and uric acid returned to normal; renal function improvement was also recorded (recent serum creatinine, 114 μmol/L) (Table 2).
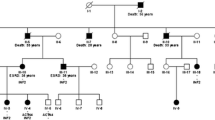
Given the patient’s familial history of renal failure, whole exome sequencing (WES) was performed. Genomic DNA from the patient, his mother, and a control was extracted from buccal swab samples, using a QIAmp DNA Mini Kit (Qiagen China Co., Ltd., Shanghai, China). Target capture sequencing was performed on the extracted DNA. All coding exons were enriched. Genetic analysis, variation annotation, and identification of candidate pathogenic mutations were performed. For our study, a total of 16 members of the patient’s family were recruited. Three individuals were affected in this pedigree, including the patient, his mother, and his maternal grandmother. Sequencing of exon 4 of autosomal INF2 revealed a nonsynonymous missense variant, wherein a C was substituted with T (c.640C > T), resulting in the p.Arg214Cys mutation (Figs. 1 and 2).

Pedigree of a family with the INF2 p.Arg214Cys mutation (I2, II3, III5). The arrow indicates the problem (III5). Black boxes (II3, III5) indicate the affected members of the family related kidney disease. White boxes indicates a genetically tested patient with a normal result

Sequence alterations in members (I2, II3, III5) of families. Sequencing in families INF2 gene was obtained from amplified genomic DNA
We conducted a review of the literature to compare the phenotypes of our patients with other patients reported to have been carrying the INF2 p.Arg214Cys mutation (Table 3).
Discussion and conclusions
We report a familial autosomal mutation in exon 4 of INF2 (p.Arg214Cys) in two patients from the same three-generation family of Chinese origin. Informed consent was provided by the patient. Two patients (son and mother) manifested proteinuria, high blood pressure, and hyperuricemia, and exhibited rapid progression of renal failure, which may be related to FSGS, and ultimately progressed to ESRD. In both cases, kidney transplantation was effective, while drug-based therapies were not.
Several studies have reported a familial autosomal mutation in exon 4 of INF2 (p.Arg214Cys) in patients of Caucasian descent (Table 3). The patients reported were relatively young at the time of diagnosis and at ESRD. Some carriers progressed to ESRD, while others were healthy. In our study, kidney dysfunction progressed rapidly in the patient and the patient’s mother. The patient’s grandmother carried an INF2 mutation without any sign of renal dysfunction. In our two clinical cases—son and mother—the age of symptoms onset was 20 and 40 years old, respectively, and no symptoms of CMT were observed. This may be due to the autosomal mutation located in exon 4 of INF2 [9, 10]. Current literature describing patients with p.Arg214Cys mutations reports the same phenotype but without CMT symptoms. In a study on a family of a father and two sons, Lee et al. reported diseases of very different severity with the same INF2 mutation [16]. In our cases, we also observed that the patient’s grandmother, who carried an INF2 mutation without any sign of renal dysfunction, is consistent with reports of patients of Caucasian descent. However, it is worth noting that previously reported healthy carriers were younger, thus a delayed onset is possible. Therefore, our case confirms the existence of healthy carriers. Some families showed marked clinical heterogeneity, suggesting the involvement of environmental factors or a specific genetic background [5].
In our study, the son and the mother both presented complications such as elevated blood pressure and serum uric acid levels, which may be the prominent phenotype. In general, corticosteroids are used to treat idiopathic FSGS. However, corticosteroid therapy is not effective for genetic FSGS [2, 13]. In our case, before verifying INF2 mutation with WES, corticosteroids in combination with angiotensin receptor blockers (ARBs) were used for treatment, which were ineffective for managing genetic disorders [13].
The patient’s mother had undergone renal transplantation 3 years ago. In June 2020, the patient underwent renal transplantation. After renal transplantation, the patient’s proteinuria was completely resolved; his blood pressure and uric acid returned to normal; his renal function improved as the transplanted kidney functioned properly. This indicates that renal transplantation is an effective treatment for this condition [2].
To our knowledge, this is the first report on INF2 p.Arg214Cys in Asia and one of the few reports worldwide. In addition to confirming previously reported characteristics associated with INF2 mutation, our study also suggests new clinical manifestations and treatment outcomes that are worthy of note. INF2 mutation analysis should not be restricted to individuals without family history of FSGS, rather it should also be performed on individuals for whom drug-based therapies are not effective. For these patients, Kidney transplantation is an effective treatment.
Availability of data and materials
The datasets used and analyzed during the course of the current study are available from the corresponding author on reasonable request.
Abbreviations
- INF2 :
-
Inverted formin 2 gene
- FSGS:
-
Focal segmental glomerulosclerosis
- ESRD:
-
End-stage renal disease
- AD:
-
Autosomal dominant
- CMT:
-
Charcot-Marie-Tooth disease
- UACR:
-
Urine albumin/creatinine
- ARB:
-
Angiotensin receptor blockers
- HCY:
-
Homocysteine
- WES:
-
Whole exome sequencing
References
Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627.
Labat-de-Hoz L, Alonso MA. The formin INF2 in disease: progress from 10 years of research. Cell Mol Life Sci. 2020;77:4581–600.
Brown EJ, Schlöndorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, et al. Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet. 2010;42:72–6.
Boyer O, Benoit G, Gribouval O, Nevo F, Tête MJ, Dantal J, et al. Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol. 2011;22:239–45.
Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, et al. INF2 mutations in Charcot-Marie-tooth disease with glomerulopathy. N Engl J Med. 2011;365:2377–88.
Gbadegesin RA, Lavin PJ, Hall G, Bartkowiak B, Homstad A, Jiang R, et al. Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int. 2012;81:94–9.
Münch J, Grohmann M, Lindner TH, Bergmann C, Halbritter J. Diagnosing FSGS without kidney biopsy - a novel INF2-mutation in a family with ESRD of unknown origin. BMC Med Genet. 2016;17:73.
Echaniz-Laguna A, Latour P. A cryptic splicing mutation in the INF2 gene causing Charcot-Marie-tooth disease with minimal glomerular dysfunction. J Peripher Nerv Syst. 2019;24:120–4.
Caridi G, Lugani F, Dagnino M, Gigante M, Iolascon A, Falco M, et al. Novel INF2 mutations in an Italian cohort of patients with focal segmental glomerulosclerosis, renal failure and Charcot-Marie-tooth neuropathy. Nephrol Dial Transplant. 2014;29(Suppl 4):iv80–6.
Mademan I, Deconinck T, Dinopoulos A, Voit T, Schara U, Devriendt K, et al. De novo INF2 mutations expand the genetic spectrum of hereditary neuropathy with glomerulopathy. Neurology. 2013;81:1953–8.
Bayraktar S, Nehrig J, Menis E, Karli K, Janning A, Struk T, et al. A deregulated stress response underlies distinct INF2-associated disease profiles. J Am Soc Nephrol. 2020;31:1296–313.
Paul MD, Fernandez D, Pryse-Phillips W, Gault MH. Charcot-Marie-tooth disease and nephropathy in a mother and daughter with a review of the literature. Nephron. 1990;54:80–5.
De Rechter S, De Waele L, Levtchenko E, Mekahli D. Charcot-Marie-tooth: are you testing for proteinuria. Eur J Paediatr Neurol. 2015;19:1–5.
Rodriguez PQ, Lohkamp B, Celsi G, Mache CJ, Auer-Grumbach M, Wernerson A, et al. Novel INF2 mutation p. L77P in a family with glomerulopathy and Charcot-Marie-tooth neuropathy. Pediatr Nephrol. 2013;28:339–43.
Chhabra ES, Ramabhadran V, Gerber SA, Higgs HN. INF2 is an endoplasmic reticulum-associated formin protein. J Cell Sci. 2009;122:1430–40.
Lee HK, Han KH, Jung YH, Kang HG, Moon KC, Ha IS, et al. Variable renal phenotype in a family with an INF2 mutation. Pediatr Nephrol. 2011;26:73–6.
Barua M, Brown EJ, Charoonratana VT, Genovese G, Sun H, Pollak MR. Mutations in the INF2 gene account for a significant proportion of familial but not sporadic focal and segmental glomerulosclerosis. Kidney Int. 2013;83:316–22.
Acknowledgements
We thank Qiagen China Co., Ltd. for genetic analyses, Zixin Sha from Carnegie Mellon University and Editage (www.editage.cn) for English language editing.
Funding
The National Natural Science Foundation of Guangdong, China (2017A030313714 to H.P.), and the Science and Technology Program of Guangzhou, China (International Science & Technology Cooperation Program; 201807010037 to H.P.) supported the design of the study, whole exome sequencing analysis, and interpretation of the data.
Author information
Authors and Affiliations
Contributions
WBZ and XXM were the physicians who conducted the renal follow-up; they reviewed the literature and contributed to manuscript drafting; XHZ reviewed the literature and contributed to manuscript drafting; DL and JZ performed the interpretation and contributed to manuscript drafting; ZCY and ML reviewed the literature and drafted the manuscript; HP revised the manuscript with respect to important intellectual content; all authors approved the final version of the submitted manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Written informed consent was obtained from the patient.
Consent for publication
Written informed consent was obtained from the patients for the publication of this Case Report and any accompanying images. Written consent was obtained from all family members who participated in this genetic study and no minors were involved. A copy of the written consent is available for review.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhao, W., Ma, X., Zhang, X. et al. INF2 p.Arg214Cys mutation in a Chinese family with rapidly progressive renal failure and follow-up of renal transplantation: case report and literature review. BMC Nephrol 22, 51 (2021). https://doi.org/10.1186/s12882-021-02254-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12882-021-02254-9