
Abstract
Background
Glycogen Storage Disease Type III (GSD III) is a rare autosomal recessive metabolic disorder caused by AGL gene mutation. There is significant heterogeneity between the clinical manifestations and the gene mutation of AGL among different ethnic groups. However, GSD III is rarely reported in Chinese population.
Case presentation
In this study, we aimed to study the genetic and clinical characteristics of four patients with GSD IIIa from China, especially the neurological manifestations. Meanwhile, we conducted a literature review of GSD IIIa cases reported in Chinese population to investigate the relationship between genotype and phenotype.
Conclusions
Three different AGL gene mutations were identified in our patients: c.206dupA, c.1735 + 1G > T and c.2590 C>T. Moreover, progressive myopathy accompanied by elevated creatine kinase level was the main manifestation of our patients in adolescents. Our results showed that AGL c.206dupA was a novel mutation and caused severe clinical manifestations. AGL c.1735 + 1G > T might be a recurrent mutation in the Chinese population. Genetic analysis of AGL gene mutation combined with muscle magnetic resonance imaging (MRI) might provide greater benefit to the patient in diagnosing GSD IIIa, rather than an invasive diagnostic procedure of biopsy.
Similar content being viewed by others
Background
Glycogen Storage Disease Type III (GSD III) is a rare autosomal recessive metabolic disorder. There are two major clinical types of GSD III. GSD IIIa involves liver and muscle while GSD IIIb only affects the liver. GSD III commonly presents with growth retardation, fasting hypoglycemia, hepatomegaly, and seizures during childhood. The neuromuscular manifestations of GSD IIIa are mainly reported in adults [1,2,3]. GSD III is caused by AGL gene mutation leading a deficiency of glycogen debrancher enzyme activity. The human AGL gene is located on chromosome 1p21 and consists of 35 exons spanning ~ 85 kb of genomic DNA. AGL gene is expressed from the third exon [4]. Genetic analysis of the AGL gene in several ethnic populations has revealed over 150 different AGL gene mutations [5]. However, few AGL mutations have been reported in the Chinese population in mainland China. In this study, we reported four Chinese patients with GSD IIIa and analyzed the relationship between genotype and phenotype of GSD IIIa in the Chinese population.
Case presentation
Our study included three male and one female GSD IIIa Chinese patients from three families. Patients 3 and 4 were siblings from one family. When these patients were admitted to our hospital, their clinical information, such as age, gender, height, weight, liver function and clinical symptoms were collected. Then, routine laboratory test, echocardiography, electromyogram, abdominal ultrasound, muscle MRI and neuropsychological test were conducted. All of this clinical information of the first visit and two years follow-up were shown in Table 1. The guideline for the diagnosis of GSD III had been followed [5].
Our patients mainly complained physical weakness accompanied by elevated creatine kinase level. Although patients had not received high-protein and uncooked cornstarch (UCS) diet therapy in childhood, growth retardation was not observed after adolescence. Treatment with high-protein and UCS diet was implemented in all patients after the first visit, according to preprandial blood glucose and the level of abnormal alanine aminotransferase (ALT) and aspartate aminotransferase (AST). After high-protein and UCS diet therapy, clinical manifestations of weakness of our patients were significantly relieved at the last follow-up. Currently, their therapy followed standard treatment guidelines, with UCS in 1.5 g/kg/time for 1–3 times per day and protein in 2 g/kg/d.
The results of laboratory tests showed the level of creatine kinase (CK) was significantly increased among all patients, but ketonic hypoglycemia was only detected in patient 1. Serum cholesterol and triglyceride were normal in patient 1 but they were elevated in other patients. After a high-protein and UCS diet therapy for two years, the level of CK was still 5–10 times over the upper limit of normal, but preprandial blood glucose, serum cholesterol and triglyceride of all patients had nearly normalized at the last follow-up.
All our patients showed asymptomatic cardiac hypertrophy accompanied by normal ejection fraction and cardiac conduction (Table 1). Meanwhile, hepatomegaly accompanied by abnormal ALT and AST was common during our patients’ childhood. After a high-protein and UCS diet therapy for two years, cardiac hypertrophy of our patients did not deteriorated. Moreover, levels of AST and ALT had decreased and hepatic adenoma was not detected in our patients. We didn’t observe hepatic cirrhosis or hepatocellular carcinoma in our patients by liver ultrasound or laboratory tests. Indeed, we did not perform the liver biopsy which was invasive and potentially detrimental to the patients.
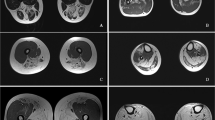
To better identify the neurological manifestations of GSD IIIa in our patients, nerve conduction studies (NCS), electromyogram (EMG), muscle MRI, brain MRI, electroencephalogram and neuropsychological tests were performed. All patients showed normal results in NCS, brain MRI and electroencephalogram, but the results of EMG were abnormal. Myopathic motor unit potential was detected in all patients, but the specificity and sensitivity were not satisfactory. Notably, Muscle MRI transverse sections from the mid-thigh and lower leg muscles of patient 2 presented mild T1 signal intensity in the long head of femoris and peroneus longus muscles (arrows), indicating an increase in adipose-like tissue (Fig. 1).What’s more, lateral heads of gastrocnemius muscles showed atrophy in patient 2 (Fig. 1). In general, the posterior and lateral muscles of the lower limbs were mostly affected (Fig. 1).

Muscle MRI comprising T1-weighted images of the thigh and lower leg from patient 2. a. Transverse cuts from the mid-thigh muscles showed mild increase in signal intensity within the femoris long head (arrows); b, Transverse cuts from the lower leg showed mild increase in signal intensity within the peroneus longus (arrows) and there was atrophy of gastrocnemius lateral heads (asterisk)
DNA sequence analysis of AGL gene
The genomic DNA of all family members was extracted from peripheral blood by using the standard phenol–chloroform extraction method. Polymerase chain reaction using the primers located in the flanking introns was conducted to amplify the AGL gene. AGL gene mutations were screened by direct sequencing using an ABI Prism 3100 Genetic Analyzer (Applied Biosystems, USA). The sample sequences were compared with the genomic DNA sequence of AGL (GenBank accession no.NM_000642.2).
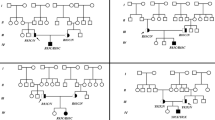
We found three different AGL mutations (Fig. 2) in our four patients with GSD IIIa from three families. Among these three mutations, one mutation was novel and the other two were reported. Patient 1, who still had frequent episodes of hypoglycemia in adolescence, carried a homozygous insert mutation AGL c.206dupA (p. N69Kfs*8). This AGL c.206dupA mutation is a frameshift mutation causing premature termination codons in the exon4 and is predicted to be “disease-causing mutation” by MutationTaster (http://www.mutationtaster. org/).The AGL c.206dupA mutation has not been documented in any public database, nor in our internal exonic database of OMIM genes from 1000 individuals. Patient 2 was homozygous for the AGL c.1735 + 1G > T (IVS14 + 1G > T) splice site mutation, which was predicted to impair normal splicing. For patients 1 and 2, the homozygous mutations were inherited from their parents separately. Patients 3 and 4 had compound heterozygous mutations. One of the mutations is AGL c.2590 C>T which is inherited from their mother. This mutation results in substitution of arginine at codon 864 by termination codon (p.R864X) and causes premature termination. Another mutation is AGL c.1735 + 1G > T which is inherited from their father.

Pedigrees of our patients with GSD IIIa and mutation analysis of AGL gene. Filled circles and squares represent affected females and males, respectively. Proband is indicated with an arrow; a. patient 1 (II:1) from family 1. The DNA sequence chromatogram of the proband indicates AGL c.206dupA homozygous mutation indicated with an arrow; b. patient 2 (II:1) from family 2. The DNA sequence chromatogram of the proband indicates AGL c.1735 + 1G > T homozygous mutation with an arrow; c. patient 3 (II:1) and patient 4 (II:2) from family 3. The DNA sequence chromatogram of the proband indicates AGL C.1735 + 1G > T and AGL c.2590 C>T compound heterozygous mutations indicated with arrows
Literature review
Literature review of GSD IIIa reported in Chinese patients was conducted by searching for studies published from 1996 to 2017, with the keywords “Glycogen Storage Disease Type III”, “glycogen debranching enzyme” and “AGL gene”.The databases included Pub Med, Medline, VIP database and Chinese Biology Medicine. Only studies published in English or Chinese were included. All relevant papers were read carefully.
We had reviewed five papers including eighteen cases of GSD IIIa Chinese patients [6,7,8,9,10]. The detailed clinical information of these patients were shown in Additional file 1: Table S1. There was fifteen male and three female cases. The mean age was 5.7 ± 10.3 years old (median 4 years; range 1–46 years). Hepatomegaly and myopathy were common clinical manifestations in Chinese GSD IIIa patients on different stages of individual development, but hepatic adenoma was rarely reported. CK, ALT, AST, cholesterol, triglyceride and fasting blood-glucose were significantly abnormal in GSD IIIa patients in China. A variety of mutations of the AGL gene were found in Chinese GSD IIIa patients, including deletion, missense, nonsense and splicing mutations. There were five patients with AGL c.1735 + 1G > T mutation. Therefore, AGL c.1735 + 1G > T might be the most recurrent mutation in Chinese patients.
Discussion and conclusions
GSD III is caused by mutations in the AGL gene. Over 150 different mutations in the AGL gene have been identified. However, few AGL mutations have been reported in mainland China [4, 5]. Genetic analysis of our four Chinese GSD IIIa patients revealed three different mutations in AGL gene. One novel frameshift insert mutation c.206dupA (N69Kfs*8) is located in exon4. It is likely be classified as a pathogenic mutation by following the ACMG/AMP sequence variant pathogenicity classification guideline [11]. As to the relationship between genotype and phenotype, we found the patient with AGL c.206dupA mutation showed severe clinical manifestations compared with the other patients. The patient carrying the AGL c.206dupA mutation still suffered severe hypoglycemia symptoms even in adolescence, which were absent in other patients. These manifestations indicated AGL c.206dupA mutation might cause more severe functional deficiency of the glycogen debranching enzyme than other mutations. Therefore, the patient with AGL c.206dupA mutation required a high-protein and UCS diet therapy even in adults. Then, AGL c.1735 + 1G > T mutation, which is first reported in Japan [12], is predicted to impair normal splicing. AGL c.1735 + 1G > T mutation might be a recurrent mutation in Chinese patients and should be given priority when gene detection is conducted in Chinese GSD IIIa patients. Moreover, AGL c.2590 C>T,which is first reported by Caucasians [13], is predicted to lead to premature termination, which completely abolishes the enzyme activity. In summary, the patient carrying AGL c.206dupA homozygous mutation suffered more severe disease, whereas patient 2 carrying the splicing homozygous mutation had milder disease. This observation is in keeping with the notion that some residual functional protein may be produced from the splicing mutation.
We have collected detailed clinical information of four Chinese GSD IIIa patients from three families. We revealed clinic characteristics of Chinese GSD IIIa: Firstly, all of our patients mainly complained of weakness when running and climbing stairs, and a high-protein and UCS diet therapy could improve the symptom of muscle weakness effectively. Secondly, our patients showed growth retardation in childhood, but they can catch up to normal level in adolescence. Mental or physical development disabilities did not appear in most of patients. Thirdly, asymptomatic hepatomegaly is common in our patients. Liver and cardiac dysfunction is not significant in all of our patients. Finally, our study showed muscle MRI combined with genetic testing might be reliable, convenient and less invasive compared with muscle biopsy [14, 15].
In conclusion, our study provides a comprehensive overview of genetic and clinical features of GSD IIIa in China. Our results revealed that muscle weakness accompanied with high level of CK are the main clinical manifestations in adolescence. Cardiomyopathy and hepatic adenoma needs long term follow-up. AGL c.206dupA mutation is a novel mutation leading severe clinical manifestations and AGL C.1735 + 1G > T might be a recurrent mutation in Chinese patients with GSD IIIa. Moreover, clinical history, muscle MRI and genetic testing may provide greater benefit to the patient in diagnosing GSD IIIa.
Abbreviations
- EMG:
-
Electromyogram;
- GSD III:
-
Glycogen storage disease type III
- NCS:
-
Nerve conduction studies
- UCS:
-
Uncooked cornstarch
References
Shen JJ, Chen YT. Molecular characterization of glycogen storage disease type III. Curr Mol Med. 2002;2:167–75.
Shin YS. Glycogen storage disease: clinical, biochemical, and molecular heterogeneity. Semin Pediatr Neurol. 2006;13:115–20.
Kishnani PS, Austin SL, Arn P, Bali DS, Boney A, Case LE, Chung WK, Desai DM, El-Gharbawy A, Haller R, et al. Glycogen storage disease type III diagnosis and management guidelines. Genet Med. 2010;12:446–63.
Yang-Feng TL, Zheng K, Yu J, Yang BZ, Chen YT, Kao FT. Assignment of the human glycogen debranching gene to chromosome 1p21. Genomics. 1992;13:931–4.
Goldstein JL, Austin SL, Boyette K, Kanaly A, Veerapandiyan A, Rehder C, Kishnani PS, Bali DS. Molecular analysis of the AGL gene: identification of 25 novel mutations and evidence of genetic heterogeneity in patients with glycogen storage disease type III. Genet Med. 2010;12:424–30.
Yu X, Pan K, Huang Y, Zhang J. Four cases of the glycogen storage diseases. J Clin Pediatr. 2009;10:987–8.
Qiu Z, Huang W, Wei M, Qiu J, Zhang H, Wu X. Mutation analysis of AGL gene in 7 Chinese patients with glycogen storage disease type III. Basic & Clinical Medicine. 2011;31:471–4.
Wu X, Pan J, Guo Y. AGL gene analysis of a pedigree with glycogen storage disease type III and identification of a novel mutation. Chin J Pediatr. 2013;51:915–20.
Chen X, Niu H, Yan W, He N, Cheng S. Clinical manifestation, pathological feature and treatment of 14 cases of glycogen storage disease. J Development Med. 2014;2:224–7.
Guo L, Lin W, Zhang Z, Zhao X, Zhang S, Cai X, Zhou Q, Song Y. Analysis of clinical features and AGL gene mutations in a family with glycogen storage disease type IIIa. Clin. J Med Genet. 2015;32:502–5.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24.
Horinishi A, Okubo M, Tang NL, Hui J, To KF, Mabuchi T, Okada T, Mabuchi H, Murase T. Mutational and haplotype analysis of AGL in patients with glycogen storage disease type III. J Hum Genet. 2002;47:55–9.
Endo Y, Hofinishi A, Vorgerd M, Aoyama Y, Ebara T, Murase T, Odawara M, Podskarbi T, Shin YS, Okubo M. Molecular analysis of the AGL gene:heterogeneity of mutations in patients with glycogen storage disease type III from Germany, Canada, Afghanistan, Iran, and Turkey. J Hum Genet. 2006;51:958–63.
Preisler N, Laforêt P, Madsen KL, Prahm KP, Hedermann G, Vissing CR, Galbo H, Vissing J. Muscle metabolism is impaired during exercise in glycogen storage disease type III. Neurology. 2015;84:1767–71.
Preisler N, Pradel A, Husu E, Madsen KL, Becquemin MH, Mollet A, Labrune P, Petit F, Hogrel JY, Jardel C, et al. Exercise intolerance in glycogen storage disease type III: weakness or energy deficiency? Mol Genet Metab. 2013;109:14–20.
Acknowledgments
We thank all patients for participating in this study.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81301081,81170811, 30973216), Shanghai Municipal Commission of Health and Family Planning Foundation (20134005),the National key R&D Program of China (2017YFC1310300, 2016YFC0905100).
Availability of data and materials
All data generated or analysed during this study are included in this published article.
Authors’contributions
YZ analyzed the patient data and written the manuscript. MX, XC, GZ and AY are responsible for collecting patient data. ZL and WQ are responsible for research designs. All authors read and approved the final manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was performed with the approval of the Ethics Committee of Xinhua Hospital affiliated to Shanghai Jiao Tong University School of Medicine.
Consent for publication
Consent Form for Publication of personal information in a scientific journal, including clinical data and image and photographs was obtained from all individual participants included in the study or from their parents in the case of minors.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional file
Additional file 1:
Table S1. Clinical and genetic features of GSD IIIa patients reported in China. Literature review of GSD IIIa reported in Chinese patients was conducted by searching for studies published from 1996 to 2017. (DOCX 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y., Xu, M., Chen, X. et al. Genetic analysis and clinical assessment of four patients with Glycogen Storage Disease Type IIIa in China. BMC Med Genet 19, 54 (2018). https://doi.org/10.1186/s12881-018-0560-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12881-018-0560-6