
Abstract
Background
Highly pathogenic influenza A (H5N8) viruses have caused several worldwide outbreaks in birds and are of potential risk to humans. Thus, a specific, rapid and sensitive method for detection is urgently needed.
Methods
In the present study, TaqMan minor groove binder probes and multiplex real-time RT-PCR primers were designed to target the H5 hemagglutinin and N8 neuraminidase genes. A total of 38 strains of avian influenza viruses and other viruses were selected to test the performance of the assay.
Results
The results showed that only H5 and N8 avian influenza viruses yielded a positive signal, while all other subtypes avian influenza viruses and other viruses were negative. High specificity, repeatability, and sensitivity were achieved, with a detection limit of 10 copies per reaction.
Conclusions
The developed assay could be a powerful tool for rapid detection of H5N8 influenza viruses in the future.
Similar content being viewed by others


Background
Highly pathogenic avian influenza (HPAI) viruses are a threat to humans and animals, and cause considerable economic damage. The first H5N1 HPAI virus was detected in 1996 in a domestic goose in Guangdong, China (Gs/GD lineage), and caused deaths in wild birds, poultry and humans, and has since spread to over 80 countries in Asia, Europe, Africa and North America [1]. Since 2008, HPAI subtypes H5N2, H5N6 and H5N8 carrying the genetic backbone of the Gs/GD lineage H5 clade 2.3.4 have been identified in poultry worldwide, especially in domestic ducks and other birds in live poultry markets, and these subtypes have subsequently evolved into different subclades including 2.3.4.4 [2,3,4].
In early 2014, reassortant clade 2.3.4.4 H5N8 HPAI virus caused outbreaks in poultry in South Korea [4], and by late 2014, it had spread to Japan, Russia and Europe, with multiple cases reported from wild birds, including apparently healthy birds [5,6,7]. Subsequently, HPAIV H5N8 virus spread from Asia to North America and caused an outbreak leading to heavy losses of poultry in commercial farms in 2014–15 [8, 9]. The reassortant HPAIV H5N2 was composed of Eurasian HPAIV H5N8 and North American lineage AIVs, causing several outbreaks in Canada and North America [10], and affecting 232 farms in 15 states and more than 50 million birds in 2015 in the US [11]. In 2016 and 2019, the HPAI H5N8 virus caused successive epidemics in Nigeria, Cameroon, Egypt, Saudi Arabia and Namibia [12,13,14,15,16,17]. An increasing number of reports indicate that HPAI H5N8 viruses continuously cause deaths in wild migratory birds and birds in live poultry markets [18,19,20].
For more effective prevention and control of H5N8 infection, the development of a rapid, sensitive and specific diagnostic assay is critical. Currently, viral culture is the most traditional method for influenza diagnosis, and is considered the gold standard. However, it is time-consuming and complicated, and requires a laboratory with bio-safety level 3 practices [21]. Reverse-transcription PCR (RT-PCR) is the most well-established molecular detection technology currently available to detect and/or type influenza viruses [22]. Real-time RT-PCR (RRT-PCR), developed from RT-PCR, can monitor the progress of reactions by detecting the fluorescence signal in real time, resulting in higher sensitivity, specificity and simplicity [23]. In the present study, we developed a TaqMan minor groove binder (MGB) RRT-PCR assay to detect H5N8 subtype avian influenza viruses (AIVs) rapidly and specifically.
Method
Three pairs each of specific primers and corresponding probes targeting H5 hemagglutinin (HA) and N8 neuraminidase (NA) genes were designed based on the nucleotide sequences of H5-HA (H5N1, H5N2, H5N6 and H5N8) (2.3.2 and 2.3.4.4) and N8-NA (H2N8, H3N8, H5N8, H6N8 and H10N8) genes from 1998 to 2018, obtained from the GenBank database, using Primer Express software as described previously [24]. Finally, two optimal sets of primers and probes for H5-HA and N8-NA (Table 1 and Fig. S1) were chosen after numerous comparison experiments as described previously [25]. A total of 34 strains of AIVs (Table 2) were selected to test the performance of the assay. Newcastle disease virus (NDV), infectious bronchitis (IBV) and infectious bursal disease virus (IBDV) were also used to assess specificity. And a reference real-time RT-PCR was performed using an Influenza A Virus Real Time RT-PCR Kit (Liferiver, Shanghai, China) according to the manufacturer’s instructions [26].
By combining the sequences of H5 and N8, we developed a duplex RRT-PCR assay with two sets of primers and probes. Optimal concentrations of the two probes and primers were determined using the matrix method. H5 and N8 plasmids (pHW2000-H5 and pHW2000-N8 [27]) were serially diluted in 10-fold, with DNA ranging from 1 copy/mL to 1 × 105 copies/mL and was detected with different amounts of forward primer, reverse primer and probe (Table S1 and Table S2). The optimal primer and probe concentration for the H5-HA primer pairs, N8-NA primer pairs, H5-HA probe, and N8-NA probe in the 20 μL RRT-PCR system was 250 nM in all cases. The RRT-PCR assay was performed in a 20 μL reaction mixture consisting of 10 μL 2 × One Step PCR Mix (Vazyme, China), 1 μL Enzyme Mix containing reverse transcription enzyme and DNA polymerase, 0.5 μL H5 forward primer (10 μM), 0.5 μL H5 reverse primer (10 μM), 0.5 μL H5 probe (10 μM), 0.5 μL N8 forward primer (10 μM), 0.5 μL N8 reverse primer (10 μM), 0.5 μL N8 probe (10 μM), 5 μL RNA sample, and 1 μL RNase-free water, as described previously [24]. Reactions were carried out in a C1000 Thermal Cycler Real-time RT-PCR instrument (Bio-Rad) and cycling parameters were 15 min at 55 °C, 5 min at 95 °C, 40 cycles of 5 s at 95 °C, and 34 s at 60 °C. No template control (NTC), positive control (H5N8 RNA) and negative control (water) reactions were also included, and data were analysed using a CFX96 Real-Time System.
The sensitivity of the RRT-PCR assay was determined for each reaction using 10-fold serial dilutions of H5 and N8 plasmids, with DNA ranging from 1 to 109 copies per reaction [28]. To evaluate the clinical sensitivity and specificity of the RRT-PCR assay, six-week-old female BALB/c mice (n = 24) were anesthetized by isoflurane and inoculated intranasally with H5N8 virus in 0.05 mL phosphate buffered saline. Respiratory specimens and cloacal swab samples were harvested from mice at 3 days post-inoculation, and the mice were sacrificed with 5% isoflurane.
Results
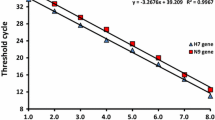
In this study, DNA plasmids were used for analytical sensitivity testing instead of RNA run-off transcripts as described previously [28]. The detection limit of the assay was 10 copies per reaction for both H5 and N8 genes. Standard curves for the two plasmids were generated by plotting their cycle threshold (Ct) values against DNA copy number, and both followed a linear correlation between 10 and 109 copies of target DNA in each multiplex detection reaction (Fig. 1). Linear correlations of the standard curves of H5 and N8 were y = − 3.407x + 40.688 (efficiency = 96.6%, R2 = 0.991), and y = − 3.325x + 40.016 (efficiency = 99.9%, R2 = 0.991), respectively.

Amplification plots and standard curves of H5 (A and B) and N8 (C and D) assays. HA and NA gene sequences of pHW2000-H5 and pHW2000-N8 are from A/duck/Zhejiang/W24/ 2013(H5N8), an H5N8 AIV of clade 2.3.4.4 isolated in 2013
The diagnosis specificity of the assay was evaluated using the viruses listed in Table 2. The results showed that only H5 and N8 AIVs yielded a positive signal, while all other AIV subtypes and other viruses were negative.
Regarding reproducibility, inter-assays and intra-assays were analysed using different concentrations of plasmids as described previously [29]. The results of intra-assays (Table 3) and inter-assays (Table 4) revealed that the coefficients of variation (CV%) were all < 2%, suggesting our RRT-PCR method is highly reproducible [30].
Respiratory specimens and cloacal swab samples (n = 24) from mice infected with H5N8 [27] were collected and tested to evaluate the clinical sensitivity and specificity of the RRT-PCR assay. Concurrently, samples were also tested using an Influenza A Virus Real-Time RT-PCR Kit (Liferiver, China), and the results were used as a reference as described previously [24, 26]. Positive signals were obtained for all H5 and N8 samples, and the results were consistent with those obtained with the Influenza A Virus Real Time RT-PCR Kit (Fig. 2).

Detection of 24 laboratory-confirmed samples of H5N8 viruses by the multiplex real-time RT-PCR developed in this study. The Influenza Virus A&B Real-Time RT-PCR Kit was used in parallel as a reference. Ct values obtained from H5 and N8 assays for each sample are presented. Samples 1–12 are from respiratory specimens, and samples 13–24 are from cloacal swabs
Additionally, a total of 148 cloacal swabs were collected from poultry in Zhejiang from 2013 to 2018 [31,32,33] and tested using both the RRT-PCR assay and virus isolation. The results of the RRT-PCR assay showed that there were 12 positive samples of H5N8 subtype AIVs, six positive samples of H5Nx subtype, and eight positive samples of HxN8 subtype, consistent with the results of virus isolation (Table 5).
Discussion
Increasing evidence suggests that many subtypes of AIVs, such as H7N9, H10N8, H6N1, H9N2 and H7N7, are not only pathogenic for poultry, but they can also infect humans, and even cause death [34,35,36]. Historically, H5N1 and H7N9 AIVs have caused great economic losses and numerous deaths in humans [37, 38]. H5N8 HPAI has caused multiple disease outbreaks in poultry and wild birds, and has the potential to be transmitted from birds to humans. In view of the global threat posed by the H5N8 virus, an appropriate technology for timely detection and surveillance of this virus is required. A multiplex RRT-PCR assay for detecting H5N8 has been developed previously with a detection limit of 99.9 copies per reaction for the H5 gene and 15.9 copies per reaction for the N8 gene [39]. A riems influenza a typing array (RITA) was developed by duplex TaqMan reactions for detection and identification of 14 HA and 9 NA subtypes of AIVs, including H5 and N8 subtype AIVs. But no H5N8 AIVs were included in this study to verify the specificity of the RITA [40]. Additionally, a real-time PCR assay was developed to sensitively detect H5N8 of clade 2.3.4.4b HPAIVs, originating from European poultry and wild bird cases during 2016–2018 [41]. In the current study, the RRT-PCR was developed to detect the currently circulating H5N8 (including Eurasian lineage and North American lineage) by in silico analysis of published H5N8 sequence data. However, a total of 44 sequences (44/781) were incompletely covered by the primers and probe of H5, such as A/duck/Quang Ninh/19c511/2013 (H5N8), A/chicken/South Africa/499723/2018 (H5N8), and A/Duck/Egypt/F131/2019 (H5N8). In addition, a total of 56 sequences (56/781) were incompletely covered by the primers and probe of N8, such as A/common teal/Shanghai/PD1108–13/2013 (H5N8), A/duck/Taiwan/A3400/2015 (H5N8), and A/chicken/Belgium/807/2017 (H5N8). In silico mismatches do not necessarily translate into failure of detection in the wet assay. The capacity of the RRT-PCR developed in this study to cover the above strains should be further verified. In the present work, an efficient RRT-PCR assay was developed with a detection limit of 10 copies per reaction for both H5 and N8 genes via careful design and optimisation of primers and probes. Additionally, this assay performed well in the analysis of clinical samples.
Conclusions
These results indicate that the duplex assay designed in this study is sufficiently sensitive and specific to be used for the detection of the H5N8 virus.
Availability of data and materials
The datasets supporting the conclusions of this article are contained within the article and its supporting files.
Abbreviations
- AIVs:
-
Avian influenza viruses
- Ct:
-
Cycle threshold
- CV%:
-
Coefficients of variation
- HA:
-
Hemagglutinin
- HPAI:
-
Highly pathogenic avian influenza
- IBDV:
-
Infectious bursal disease virus
- IBV:
-
Infectious bronchitis virus
- MGB:
-
Minor groove binder
- NA:
-
Neuraminidase
- NDV:
-
Newcastle disease virus
- NTC:
-
No template control
- RITA:
-
Riems influenza a typing array
- RRT-PCR:
-
Real-time RT-PCR
- RT-PCR:
-
Reverse-transcription PCR
References
Lee DH, Bertran K, Kwon JH, et al. Evolution, global spread, and pathogenicity of highly pathogenic avian influenza H5Nx clade 2.3.4.4. J Vet Sci. 2017;18(S1):269–80.
Lee CW, Swayne DE, Linares JA, et al. H5N2 avian influenza outbreak in Texas in 2004: the first highly pathogenic strain in the United States in 20 years? J Virol. 2005;79(17):11412–21.
Wu H, Peng X, Xu L, et al. Novel reassortant influenza A(H5N8) viruses in domestic ducks, eastern China. Emerg Infect Dis. 2014;20(8):1315–8.
Lee YJ, Kang HM, Lee EK, et al. Novel reassortant influenza A(H5N8) viruses, South Korea, 2014. Emerg Infect Dis. 2014;20(6):1087–9.
Bae YJ, Lee SB, Min KC, et al. Pathological evaluation of natural cases of a highly pathogenic avian influenza virus, subtype H5N8, in broiler breeders and commercial layers in South Korea. Avian Dis. 2015;59(1):175–82.
Fan ST, Zhou LC, Wu D, et al. A novel highly pathogenic H5N8 avian influenza virus isolated from a wild duck in China. Influenza Other Resp. 2014;8(6):646–53.
Wu HB, Peng XR, Xu LH, et al. Novel reassortant influenza A(H5N8) viruses in domestic ducks, Eastern China. Emerg Infect Dis. 2014;20(8):1315–8.
Lee DH, Torchetti MK, Winker K, et al. Intercontinental spread of Asian-origin H5N8 to North America through Beringia by migratory birds. J Virol. 2015;89(12):6521–4.
Lee DH, Torchetti MK, Hicks J, et al. Transmission dynamics of highly pathogenic avian influenza virus A(H5Nx) clade 2.3.4.4, North America, 2014-2015. Emerg Infect Dis. 2018;24(10):1840–8.
Pasick J, Berhane Y, Joseph T, et al. Reassortant highly pathogenic influenza A H5N2 virus containing gene segments related to Eurasian H5N8 in British Columbia, Canada, 2014. Sci Rep. 2015;5:9484.
Lee DH, Torchetti MK, Killian ML, et al. Reoccurrence of avian influenza A(H5N2) virus clade 2.3.4.4 in wild birds, Alaska, USA, 2016. Emerg Infect Dis. 2017;23(2):365–7.
Selim AA, Erfan AM, Hagag N, et al. Highly pathogenic avian influenza virus (H5N8) clade 2.3.4.4 infection in migratory birds, Egypt. Emerg Infect Dis. 2017;23(6):1048–51.
Wade A, Jumbo SD, Zecchin B, et al. Highly pathogenic avian influenza A(H5N8) virus, Cameroon, 2017. Emerg Infect Dis. 2018;24(7):1367–70.
Abolnik C. Outbreaks of Clade 2.3.4.4 H5N8 highly pathogenic avian influenza in 2018 in the northern regions of South Africa were unrelated to those of 2017. Transbound Emerg Dis. 2020;67(3):1371–81.
Hassan KE, El-Kady MF, EL-Sawah AAA, et al. Respiratory disease due to mixed viral infections in poultry flocks in Egypt between 2017 and 2018: Upsurge of highly pathogenic avian influenza virus subtype H5N8 since 2018. Transbound Emerg Dis. 2019. https://doi.org/10.1111/tbed.13281.
Hemida MG, Chu D, Abdelaziz A, et al. Molecular characterisation of an avian influenza (H5N8) outbreak in backyard flocks in Al Ahsa, eastern Saudi Arabia, 2017-2018. Vet Rec Open. 2019;6(1):e000362.
Molini U, Aikukutu G, Roux JP, et al. Avian influenza H5N8 outbreak in African penguins (Spheniscus demersus), Namibia, 2019. J Wildl Dis. 2020;56(1):214–8.
Li MX, Liu HZ, Bi YH, et al. Highly pathogenic avian influenza A(H5N8) virus in wild migratory birds, Qinghai Lake, China. Emerg Infect Dis. 2017;23(4):637–41.
Nagarajan S, Kumar M, Murugkar HV, et al. Novel reassortant highly pathogenic avian influenza (H5N8) virus in zoos, India. Emerg Infect Dis. 2017;23(4):717–9.
Al-Ghadeer H, Chu DKW, Rihan EA, et al. Circulation of influenza A(H5N8) virus, Saudi Arabia. Emerg Infect Dis. 2018;17:24(10).
Stephenson I, Heath A, Major D, et al. Reproducibility of serologic assays for influenza virus A (H5N1). Emerg Infect Dis. 2009;15(8):1250–9.
Espy MJ, Uhl JR, Sloan LM, et al. Real-time PCR in clinical microbiology: Applications for a routine laboratory testing. Clin Microbiol Rev. 2006;19(1):165−+.
Spackman E, Senne DA, Myers TJ, et al. Development of a real-time reverse transcriptase PCR assay for type A influenza virus and the avian H5 and H7 hemagglutinin subtypes. J Clin Microbiol. 2002;40(9):3256–60.
Yang F, Chen B, Liu F, et al. Development of a TaqMan MGB RT-PCR assay for the detection of type A and subtype H10 avian influenza viruses. Arch Virol. 2018;163:2497.
Peng Y, Xie ZX, Liu JB, et al. Visual detection of H3 subtype avian influenza viruses by reverse transcription loop-mediated isothermal amplification assay. Virol J. 2011;8:337.
Yang F, Wu H, Liu F, et al. Establishment of a multiplex real-time RT-PCR assay for rapid identification of H6 subtype avian influenza viruses. Arch Virol. 2018;163:1671.
Wu HB, Peng XM, Lu RF, et al. Virulence of an H5N8 highly pathogenic avian influenza is enhanced by the amino acid substitutions PB2 E627K and HA A149V. Infect Genet Evol. 2017;54:347–54.
Zhang ZJ, Liu D, Sun WQ, et al. Multiplex one-step real-time PCR by Taqman-MGB method for rapid detection of pan and H5 subtype avian influenza viruses. PLoS One. 2017;12(6):e0178634.
Zhang Z, Liu D, Sun W, et al. Multiplex one-step real-time PCR by Taqman-MGB method for rapid detection of pan and H5 subtype avian influenza viruses. PLoS One. 2017;12(6):e0178634.
Chang CW, Wu YC. Evaluation of DNA extraction methods and dilution treatment for detection and quantification of Acanthamoeba in water and biofilm by real-time PCR. Water Sci Technol. 2010;62(9):2141–9.
Wu H, Wu N, Peng X, et al. Molecular characterization and phylogenetic analysis of H3 subtype avian influenza viruses isolated from domestic ducks in Zhejiang Province in China. Virus Genes. 2014;49(1):80–8.
Wu HB, Guo CT, Lu RF, et al. Genetic characterization of subtype H1 avian influenza viruses isolated from live poultry markets in Zhejiang Province, China, in 2011. Virus Genes. 2012;44(3):441–9.
Hai-bo W, Ru-feng L, En-kang W, et al. Sequence and phylogenetic analysis of H7N3 avian influenza viruses isolated from poultry in China in 2011. Arch Virol. 2012;157(10):2017–21.
Chen Y, Liang W, Yang S, et al. Human infections with the emerging avian influenza A H7N9 virus from wet market poultry: clinical analysis and characterisation of viral genome. Lancet. 2013;381(9881):1916–25.
Wei SH, Yang JR, Wu HS, et al. Human infection with avian influenza A H6N1 virus: an epidemiological analysis. Lancet Respir Med. 2013;1(10):771–8.
Montomoli E, Trombetta CM. Is influenza A/H10N8 a potential candidate for the next pandemic? (vol 108, pg 213, 2014). Pathog Glob Health. 2014;108(6):302.
Gao R, Cao B, Hu Y, et al. Human infection with a novel avian-origin influenza A (H7N9) virus. N Engl J Med. 2013;368(20):1888–97.
To KKW, Ng KHL, Que TL, et al. Avian influenza A H5N1 virus: a continuous threat to humans. Emerg Microbes Infect. 2012;1:e25.
Park YR, Kim EM, Lee YJ, et al. Multiplex real-time reverse transcription polymerase chain reaction for diff erential detection of H5, N1, and N8 genes of highly pathogenic avian infl uenza viruses. Vet Med-Czech. 2017;62(4):211–20.
Hoffmann B, Hoffmann D, Henritzi D, et al. Riems influenza a typing array (RITA): an RT-qPCR-based low density array for subtyping avian and mammalian influenza a viruses. Sci Rep. 2016;6:27211.
James J, Slomka MJ, Reid SM, et al. Proceedings paper-avian diseases 10th AI symposium issue development and application of real-time PCR assays for specific detection of contemporary avian influenza virus subtypes N5, N6, N7, N8, and N9. Avian Dis. 2019;63(sp1):209–18.
Acknowledgements
We would like to thank the native English speaking scientists of Elixigen Company (Huntington Beach, California) for editing our manuscript.
Funding
This work was supported by grants from the National Science and Technology Major Project for the Control and Prevention of Major Infectious Diseases in China (2018ZX10711001, 2018ZX10102001, and 2020ZX09001016–004-002), Zhejiang Provincial Natural Science Foundation of China (LY19H260006).
Author information
Authors and Affiliations
Contributions
FY, NW and HW conceived and designed the assays. FY, XL and FL conducted experimental work. HY, NW and HW data analysed the data and wrote the paper. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The animal experiment was approved by the Institutional Animal Care Use Committee (IACUC) of Zhejiang University (No. 2017–015).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1:
Table S1. The optimal concentrations of H5 primers and probe. a. The most optimal concentrations of H5 primers and probe.
Additional file 2:
Table S2. The optimal concentrations of N8 primers and probe. a. The most optimal concentrations of N8 primers and probe.
Additional file 3:
Figure S1. Phylogenetic analysis (A and B) and sequence alignments (C and D) of the H5 and N8 genes of H5N8 influenza viruses. The tree was created by the maximum likelihood method and bootstrapped with 1000 replicates using the MEGA6 software version 6.0. The scale bar represents the distance unit between sequence pairs.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
Cite this article
Yang, F., Xu, L., Liu, F. et al. Development and evaluation of a TaqMan MGB RT-PCR assay for detection of H5 and N8 subtype influenza virus. BMC Infect Dis 20, 550 (2020). https://doi.org/10.1186/s12879-020-05277-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-020-05277-z