
Abstract
Background
Antibiotic resistance is rising at disturbing rates and contributes to the deaths of millions of people yearly. Antibiotic resistant infections disproportionately affect those with immunocompromising conditions, chronic colonization, and frequent antibiotic use such as transplant patients or those with cystic fibrosis. However, clinicians lack the diagnostic tools to confidently diagnose and treat infections, leading to widespread use of empiric broad spectrum antimicrobials, often for prolonged duration.
Case presentation
A 22 year-old Caucasian female with cystic fibrosis received a bilateral orthotopic lung transplantation 5 months prior to the index hospitalization. She underwent routine surveillance bronchoscopy and was admitted for post-procedure fever. A clear cause of infection was not identified by routine methods. Imaging and bronchoscopic lung biopsy did not identify an infectious agent or rejection. She was treated with a prolonged course of antimicrobials targeting known colonizing organisms from prior bronchoalveolar lavage cultures (Pseudomonas, Staphylococcus aureus, and Aspergillus). However, we identified Stenotrophomonas maltophilia in two independent whole blood samples using direct-pathogen sequencing, which was not identified by other methods.
Conclusions
This case represents a common clinical conundrum: identification of infection in a high-risk, complex patient. Here, direct-pathogen sequencing identified a pathogen that would not otherwise have been identified by common techniques. Had results been clinically available, treatment could have been customized, avoiding a prolonged course of broad spectrum antimicrobials that would only exacerbate resistance. Direct-pathogen sequencing is poised to fill a diagnostic gap for pathogen identification, allowing early identification and customization of treatment in a culture-independent, pathogen-agnostic manner.
Similar content being viewed by others

Background
The last two decades have seen a rapid rise in antibiotic-resistant infections, triggering alarm within global communities [1]. Nearly 10 million additional annual deaths due to antimicrobial resistance are predicted to occur by 2050 at a cost of $100 trillion [2]. Despite antibiotic stewardship programs and increased awareness, clinicians continue to prescribe antibiotics without clear evidence of bacterial infection, especially in high risk populations such as immunocompromised patients, children, and the elderly. It is well recognized that early and appropriate antibiotics reduce septic shock mortality [3]. Although similar data about the timing of antibiotics in less acutely ill patients is lacking, it remains common clinical practice to prescribe broad, empiric antibiotic therapy as a precaution. One of the greatest impediments to more appropriate antibiotic utilization is the lack of diagnostic tools to identify when antibiotics can be safely withheld, delayed, or stopped. Standard clinical diagnostics for detection of bacterial pathogens rely heavily on culture-based methods, which require incubation and relatively large blood volumes for detection. Furthermore, these techniques may not reliably detect some slow growing or fastidious organisms. Whereas antibiotic stewardship is essential to slowing the development and spread of antibiotic resistance, these programs are most effective when coupled with improved diagnostics for infectious disease [4].
Metagenomic sequencing has significant potential as a complimentary routine molecular diagnostic strategy for infectious diseases. Advantages of metagenomic sequencing include its unbiased nature, detection of unculturable organisms, and the potential to obtain strain typing information as well as genotypic susceptibility. Despite these advantages, there are challenges to overcome. Sensitivity is lower for organisms with small genomes or pathogens present at low titers. Also, contaminating sequences can be introduced from the environment, laboratory reagents [5] (Naccache, Hackett, Delwart, & Chiu, 2014), run-to-run carryover, or from bleed-through among multiplexed samples or index cross-contamination [6] (P. E [7].). The resulting massive, complex data require specialized tools and expertise for analysis. Interpreting results is not yet standard in clinical labs or in clinical practice. There are technical challenges and financial considerations as well. Current methods typically require 24 h or more to generate a result. Although this is faster than culture in many cases, it is too long to inform initial treatment decisions. Furthermore, the cost is substantial when compared to existing microbiological diagnostic tools. However, sequencing does not replace existing clinical microbiology costs and thus, in most cases, it represents an added expense. There are, of course, anecdotes where sequencing could have saved time, money, and most importantly lives. However, delineating when to perform this type of test and in which patients has yet to be defined. Notwithstanding these challenges, there are many examples of metagenomic sequencing approaches providing new insights into the etiology of infections (M. R [8].). Perhaps as a starting point, this new diagnostic strategy is best considered in patients at highest risk and for whom conventional microbiological testing is challenging. In this report, we describe the application of such technology to an immunocompromised patient with cystic fibrosis (CF) and lung transplant.
Fiberoptic bronchoscopy (FOB) and bronchoalveolar lavage (BAL) are routinely used to monitor and treat patients with chronic lung disease or lung transplant. Post-bronchoscopy fever develops up to 37% of the time in children and 16% of the time in adults [9,10,11,12,13,14,15]. Fever is more common in patients with focal bronchitis, known infections, and age less than two or over sixty [9, 11, 14, 16]. Case studies have reported transient bacteremia [10, 11, 17] and cytokine release [18, 19] after bronchoscopy, offering an explanation for the fever. However, blood culture data is insensitive and transient bacteremia is rarely reported [10, 11, 17]. Cytokine release is nonspecific and could be related to inflammation from the procedure, transient infection, or the disease process that led to the bronchoscopy in the first place.
We present the case of a 22 year-old female with CF five months post bilateral orthotopic lung transplantation who had a routine surveillance bronchoscopy performed. That procedure was complicated by fever and systemic inflammatory response. We retrospectively identified the cause as Stenotrophomonas maltophilia bacteremia using direct-from-blood RNA sequencing. This case report highlights the application of a metagenomic sequencing technology to a poorly characterized condition and how this information could impact clinical decision making.
The patient was enrolled at Duke University Hospital as part of the Austere environments Consortium for Enhanced Sepsis Outcomes (ACESO) Study to identify early host-based determinants of sepsis. This study was a multi-center clinical trial conducted at Duke University Medical Center and hospitals in Cambodia, Ghana, Liberia, and Uganda in which patients who met two of four systemic inflammatory response syndrome (SIRS) criteria [20] were enrolled. Studies were approved by relevant Institutional Review Boards (IRBs) and in accordance with the Declaration of Helsinki. After providing written informed consent, blood samples were collected in PAXgene Blood RNA tubes (BD Biosciences) and nasopharyngeal swabs were collected for respiratory pathogen testing. All other laboratory analysis and culture results were obtained through routine clinical care and obtained from the medical record.
Case presentation
A 22 year-old female with end stage lung disease secondary to CF underwent bilateral orthotopic lung transplant (BOLT) five months prior to enrollment in our study. The patient’s pre-transplant history was notable for airway colonization with mucoid Pseudomonas and Stenotrophomonas maltophilia, Methicillin Resistant Staphylococcus aureus (MRSA), and Aspergillus in addition to severe chronic sinusitis. Her post-transplant history was remarkable for multidrug resistant pseudomonal infection of her surgical incision, C. tropicalis bloodstream infection, and mild acute cellular rejection (ACR stage A1Bx) although none of these were active issues at the time of presentation.
Six weeks prior to presentation to the emergency department (ED), the patient began having persistent low-grade fevers of 99-101 °F. She was treated for a possible urinary tract infection with a course of ciprofloxacin due to an abnormal urinalysis but urine culture only grew mixed flora without a predominant pathogen. She continued to have low grade fevers and was treated with a course of levofloxacin for nonspecific pulmonary complaints but without clear evidence of infection on chest CT. The patient returned to clinic two weeks prior to enrollment with continued low grade fevers and was started on tobramycin nasal washes for mild sinus symptoms. The trimethoprim/sulfamethoxazole she used for Pneumocystis jirovecii prophylaxis was changed to pentamidine due to concerns about drug-induced fever. She was scheduled for outpatient bronchoscopy to monitor for infection and rejection as a possible cause of her persistent fevers.
At the time of bronchoscopy, her fevers had completely resolved and she reported feeling well without new symptoms. The patient underwent the scheduled bronchoscopy with bronchoalveolar lavage (BAL) and biopsies. Approximately 12 h later, the patient began having fevers and chills at home, which led her to come to the ED. She demonstrated a temperature of 103.1 °F, heart rate of 124 beats/minute, white blood cell count of 13.8 × 109 cells/uL (Ref 3.2–9.8 × 109 cells/uL) and lactate of 4.2 mmol/L (Ref 0.5–2.2 mmol/L). All other vital signs and laboratory analysis were within normal limits (Table 1). Two sets of blood cultures and urine culture showed no growth. Cytomegalovirus (CMV) and Epstein Barr Virus (EBV) quantitative PCR testing were negative. Cultures from the bronchoscopy performed one day prior to presentation grew rare mucoid Pseudomonas, rare MRSA, and Aspergillus. Respiratory viral pathogen PCR panel did not demonstrate viral pathogens on either routine clinical testing or supplemental study testing. Pathology did not show evidence of acute cellular rejection or infection. The patient was started on broad spectrum antibiotics with vancomycin and piperacillin/tazobactam at the time of enrollment. All of her presenting signs and symptoms, vital signs and laboratory testing were within normal limits within 24 h of admission. Biopsy from BAL and chest imaging did not reveal evidence of invasive infection. CT chest revealed ground glass opacities in the region where the BAL and biopsies were performed, interpreted by treating clinicians to be procedure related. The clinical team discharged the patient with two weeks of IV doxycycline, three weeks of IV piperacillin/tazobactam, and three months of voriconazole to cover bacterial and fungal elements noted on BAL, although no clear sources of infection were discovered.
At the time of her initial evaluation in the ED, the patient was identified for enrollment in a research study on host-based biomarkers for sepsis: The Austere environments Consortium for Enhanced Sepsis Outcomes (ACESO). As part of study-related activities, we obtained a blood sample for RNA analysis.
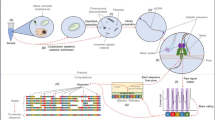
Metagenomic sequencing was performed on RNA isolated from peripheral blood collected at the time of this visit, which was five months after lung transplant. Blood samples were stabilized in two independent PAXgene Blood RNA tubes yielding Replicates 1 and 2, respectively; replicates were sequenced on different sequencing runs. EDGE Bioinformatics Software version 1.5 (P. E [7].) was used to classify the composition of microbes in the subject’s peripheral blood sample. All EDGE parameters for taxonomy tools were set to default as described (Philipson, 2017). Reads that were classified by BWA-mem mapping to RefSeq were used to calculate a sepsis indicating quantifier (SIQ) score as previously described [21]. All non-human reads were assigned a taxonomic classification by three separate algorithms and five separate databases (Fig. 1A). Stenotrophomonas maltophilia was the dominant microbe detected by all tools screening against bacterial databases for Replicate 2. Results were reproduced in Replicate 1 except the MetaPh1An algorithm did not detect the pathogen in Replicate 1, which was due to different sequencing depths for the independent runs. When detected, the top hit for all tools agreed at the strain level for S. maltophilia strain D457. The SIQ score [21] was calculated using classification results from the BWA-mem algorithm to ascertain the significance of each microbe as an etiological agent of bacteremia versus a false positive result. S. maltophilia taxonomy assignments resulted in the highest SIQ score for both replicates (Fig. 1B). Additionally, the pattern of SIQ scores indicates S. maltophilia was the singular dominant microbe in the patient’s blood samples.

High-throughput sequencing based microbial detection in replicate samples from blood. a Quality controlled reads from two replicates were profiled by four different taxonomic classifiers: BWA-mem against RefSeq; GOTTCHA against a bacterial database (GOTTCHA.b); GOTTCHA against a viral database (GOTTCHA.v); Kraken-mini; and MetaPhlAn. Normalized read counts mapping to the top 20 organisms by one or more tools are presented at the species level (heatmap). b The SIQ score was calculated for each replicate using reads that mapped to RefSeq using BWA-mem. Coordinates on SIQ plots are relative abundance of all species (x-axis), by significance (1 – p value, y-axis), and the size of the dot represents the SIQ score. The dashed red line indicates a p value of 0.05. Microbes detected at significant levels with a normalized abundance level > 4 are labeled: S, Stenotrophomonas maltophilia; P, Propionibacterium acnes; E, Escherichia coli. Plots were generated using R statistical programming
Discussion
We report a case of post-bronchoscopy fever in a patient with CF and lung transplantation. Clinical laboratory analysis, imaging, and culture results could not confirm the presence of an active infection but could not exclude it either. Uncertainty about the presence of active infection in the setting of colonization led the clinical team to treat this patient with a prolonged course of broad spectrum antimicrobials. This patient represents a population at high risk for the acquisition of antibiotic resistant infections, significantly increasing the risk of morbidity and mortality from both the infection and the antibiotics used to treat it [22]. However, delaying antibiotics or undertreating a bacterial infection in an immunocompromised patient could have dire consequences. Clinicians commonly face the need to limit antibiotic use to avoid antibiotic resistance, but they lack sufficiently sensitive diagnostics to exclude the presence of an active bacterial infection. Our results demonstrate that use of direct pathogen sequencing can augment routine testing for bacterial pathogens and offer timely solutions to direct treatment and guide more appropriate antimicrobial use.
Etiologies of infection in CF patients are broad and often complicated due to chronic colonization of multiple opportunistic microbes [22]. Identification of organisms, especially from non-sterile sites such as sputum, may simply represent commensal colonization rather than infection. As demonstrated here and by others, unbiased metagenomic sequencing can be used to characterize complex disease landscapes, especially for diagnostically challenging pulmonary diseases in which multiple organisms contribute to a pathogenic microbiome [23, 24]. In addition to being unbiased, direct sequencing from blood is less invasive than routine bronchoscopy, mitigating associated procedural risks. Importantly, detection of bacterial agents from a nonsterile source, such as sputum or bronchoalveolar lavage, does not necessarily indicate infection and could be representative of the microbiota. However, direct sequencing of pathogenic bacteria from blood, which is expected to be sterile, is more indicative of infection. Furthermore, metagenomic sequencing has demonstrated utility for diagnosing infections, yielding more sensitive and specific results than standard of care methods [25].
This case report demonstrates that multi-tool bioinformatics analysis followed by statistical mathematical normalization can bolster confidence for diagnostic determination and minimizing false positive results. Combining results from multiple tools has been demonstrated to increase the likelihood of detecting true positives in metagenomic datasets because the overlap in detection between sets of tools almost always increases precision when compared to using tools individually in benchmarking studies [26]. Two replicates were sequenced and processed by four separate taxonomic classifiers that vary in sensitivity and specificity. Results demonstrate that three of four detection methods agreed; MetaPhlAn did not detect S. maltophilia in Replicate 1. MetaPhlAn is one of the most specific community profiling tools, and detection in one sample only is likely a result of a difference in the depth of sequencing achieved on the independent runs. Given the output from all other tools, this result demonstrates the utility of replicates and a multi-tool approach for metagenomics-aided diagnostics.
A common problem faced by clinicians caring for CF patients is distinguishing colonization from infection. In this patient, rare growth of Pseudomonas aeruginosa, Staphylococcus aureus, and Aspergillus in bronchoalveolar lavage samples (Table 1) was observed. While both S. aureus and P. aeruginosa were detected at low levels in blood samples by BWA, neither bacterial species were detected in quantities deemed significant by SIQ analysis, further arguing against an active P. aeruginosa or S. aureus infection. Sequences corresponding to Propionibacterium acnes and Escherichia coli were also prevalent at significant levels in one replicate. While P. acnes is considered an opportunistic pathogen, Propionibacterium species found in blood cultures are often determined to be contaminants especially when found in only a single replicate ([27, 28],(J. R [29].). Furthermore, E. coli DNA is known to contaminate sequencing reagents and is frequently detected in sequence-based studies [5]. Thus, S. maltophilia was confidently identified as the singular dominant microbe in the patient’s blood samples. Moreover, detection as the dominate microbe through analysis of RNA sequencing (e.g. replicating bacterial transcripts) by multiple algorithms suggests an active infection in the patient’s blood.
S. maltophilia is a rare but serious cause of infection and is associated with a 21–69% fatality rate in immunocompromised individuals with bacteremia [30,31,32,33]. It is a gram-negative pathogen that evades immune clearance due to its intracellular localization. It typically forms biofilms on tubing and on the respiratory epithelium of dysfunctional airways. Interestingly, S. maltophilia did not grow in BAL cultures at the time of enrollment but was detected in two independent blood samples by direct-pathogen sequencing. This can be explained, in part, by the fact that S. maltophilia has a relatively slow growth rate and can be missed if growth plates are not carefully scrutinized. The presence of S. maltophilia in BAL cultures prior to and after enrollment indicated this bacteria was part of this patient’s complex lung microbiota. Invasive procedures, such as BAL with biopsies, as in this case, can increase the risk for bacterial translocation into the blood stream [34,35,36], and detection from two independent blood samples reveals that translocation of S. maltophilia into the bloodstream occurred in this case. Importantly, the presence of bacteremia was undetected with clinically available cultures. Clinical knowledge of these results could have allowed pathogen directed antimicrobial therapy instead of a prolonged course of broad spectrum antibacterial and antifungal agents. This knowledge is more critical in the age of antibiotic resistance, where clear detection of a pathogen could facilitate appropriate treatment early in the clinical course avoiding morbidity and mortality. For example, S. maltophilia has a β-metallocarbapenemase that makes it constitutively resistant to carbapenems, an antibiotic class commonly employed in this patient population. Fortunately, the broad spectrum antibiotics used in this case were effective against S. maltophilia, although they were not the treatment of choice.
This case highlights the importance of developing improved diagnostics for identification of the presence of infection and delineation of the pathogens involved. Patients are often exposed to over treatment with broad spectrum antimicrobials, even when the presence of infection is unclear. More importantly, the choice of treatment may be incorrect as infections are caused by increasingly diverse and resistant pathogens. Metagenomic sequencing-based diagnostics complement existing pathogen detection techniques, but may provide a new agnostic method that is not prone to the pitfalls of culture. Another advantage to metagenomics technologies is the ability to detect emerging pathogens due to its unbiased nature. The cost and turnaround time for metagenomic diagnostics are continuously improving, making extension to clinical use an increasingly viable option. Clinical laboratories have published case reports presenting sample-to-answer metagenomic sequencing protocols that can be performed routinely in a 30–48 h time frame [8, 21, 37]. Despite its advantages, interpreting raw shotgun sequencing data for infectious diseases remains challenging. Two primary risks of sequence-based diagnostics are false positives and false negatives resulting in inappropriate or inadequate treatment, respectively. One method to circumvent the aforementioned risk is to confirm agreement among several bioinformatics tools that screen sequences against comprehensive genomic databases with varying degrees of sensitivity and specificity. Mathematical approaches such as the SIQ score [21] and weighted z-score [38] have also been developed to simplify and prioritize microbes identified by bioinformatics algorithms.
In conclusion, rapid pathogen sequencing techniques are poised to fill the gap in pathogen identification, allowing early identification and customization of treatment once the costs, speed, and availability of sequencing further improves. This strategy is not unique to bacterial pathogen identification but applies to any pathogen present in a particular sample including viruses, fungi, or parasites.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ACESO:
-
Austere environments Consortium for Enhanced Sepsis Outcomes
- ACR stage:
-
Acute cellular rejection
- ANA:
-
Anti-nuclear antibody
- BAL:
-
Bronchoalveolar lavage
- BOLT:
-
Bilateral orthotopic lung transplant
- BUN:
-
Blood urea nitrogen
- CF:
-
Cystic fibrosis
- CMV:
-
Cytomegalovirus
- CRP:
-
c-reactive protein
- EBV:
-
Epstein Barr virus
- ED:
-
Emergency department
- ESR:
-
Erythrocyte sedimentation rate
- FOB:
-
Fiberoptic bronchoscopy
- L:
-
Liter
- mmol:
-
millimol
- MRSA:
-
Methicillin Resistant Staphylococcus aureus
- SIQ:
-
Sepsis indicating quantifier
- SIRS:
-
Systemic inflammatory response syndrome
- uL:
-
Microliter
- WBC:
-
White blood count
References
Spellberg, B., Guidos, R., Gilbert, D., Bradley, J., Boucher, H. W., Scheld, W. M.,Infectious diseases society of, A. (2008). The epidemic of antibiotic-resistant infections: a call to action for the medical community from the Infectious Diseases Society of America. Clin Infect Dis, 46(2), 155–164. doi:https://doi.org/10.1086/524891.
O'Neill JC. Tackling drug-resistant infections globally: final report and recommendations. The Review on Antimicrobial Resistance. 2016:1–84.
Kumar A. Early antimicrobial therapy in severe sepsis and septic shock. Curr Infect Dis Rep. 2010;12(5):336–44. https://doi.org/10.1007/s11908-010-0128-x.
Banerjee, R., Teng, C. B., Cunningham, S. A., Ihde, S. M., Steckelberg, J. M., Moriarty, J. P., . . . Patel, R. (2015). Randomized trial of rapid multiplex polymerase Chain reaction-based blood culture identification and susceptibility testing. Clin Infect Dis, 61(7), 1071–1080. doi:https://doi.org/10.1093/cid/civ447.
Salter, S. J., Cox, M. J., Turek, E. M., Calus, S. T., Cookson, W. O., Moffatt, M. F, Walker, A. W. (2014). Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol, 12, 87. doi:https://doi.org/10.1186/s12915-014-0087-z.
Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40(1):e3. https://doi.org/10.1093/nar/gkr771.
Li PE, Lo CC, Anderson JJ, Davenport KW, Bishop-Lilly KA, Xu Y, Chain PS. Enabling the democratization of the genomics revolution with a fully integrated web-based bioinformatics platform. Nucleic Acids Res. 2017;45(1):67–80. https://doi.org/10.1093/nar/gkw1027.
Wilson, M. R., Naccache, S. N., Samayoa, E., Biagtan, M., Bashir, H., Yu, G., . . . Chiu, C. Y. (2014). Actionable diagnosis of neuroleptospirosis by next-generation sequencing. N Engl J Med, 370(25), 2408–2417. doi:https://doi.org/10.1056/NEJMoa1401268.
Fonseca MT, Camargos PA, Abou Taam R, Le Bourgeois M, Scheinmann P, de Blic J. Incidence rate and factors related to post-bronchoalveolar lavage fever in children. Respiration. 2007;74(6):653–8. https://doi.org/10.1159/000107737.
Pereira W, Kovnat DM, Khan MA, Iacovino JR, Spivack ML, Snider GL. Fever and pneumonia after flexible fiberoptic bronchoscopy. Am Rev Respir Dis. 1975;112(1):59–64. https://doi.org/10.1164/arrd.1975.112.1.59.
Picard E, Schwartz S, Goldberg S, Glick T, Villa Y, Kerem E. A prospective study of fever and bacteremia after flexible fiberoptic bronchoscopy in children. Chest. 2000;117(2):573–7.
Sinha S, Guleria R, Pande JN, Pandey RM. Bronchoscopy in adults at a tertiary care Centre: indications and complications. J Indian Med Assoc. 2004;102(3):152–4 156.
Strumpf IJ, Bacher JD, Gadek JE, Morin ML, Crystal RG. Flexible fiberoptic bronchoscopy of the rhesus monkey (Macaca mulatta). Lab Anim Sci. 1979;29(6):785–8.
Um SW, Choi CM, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Prospective analysis of clinical characteristics and risk factors of postbronchoscopy fever. Chest. 2004;125(3):945–52.
Wainwright CE, Grimwood K, Carlin JB, Vidmar S, Cooper PJ, Francis PW, Robertson CF. Safety of bronchoalveolar lavage in young children with cystic fibrosis. Pediatr Pulmonol. 2008b;43(10):965–72. https://doi.org/10.1002/ppul.20885.
Wainwright CE, Grimwood K, Carlin JB, Vidmar S, Cooper PJ, Francis PW, Robertson CF. Safety of bronchoalveolar lavage in young children with cystic fibrosis. Pediatr Pulmonol. 2008a;43(10):965–72. https://doi.org/10.1002/ppul.20885.
Alexander WJ, Baker GL, Hunker FD. Bacteremia and meningitis following fiberoptic bronchoscopy. Arch Intern Med. 1979;139(5):580–3.
Standiford TJ, Kunkel SL, Strieter RM. Elevated serum levels of tumor necrosis factor-alpha after bronchoscopy and bronchoalveolar lavage. Chest. 1991;99(6):1529–30.
Krause A, Hohberg B, Heine F, John M, Burmester GR, Witt C. Cytokines derived from alveolar macrophages induce fever after bronchoscopy and bronchoalveolar lavage. Am J Respir Crit Care Med. 1997;155(5):1793–7. https://doi.org/10.1164/ajrccm.155.5.9154894.
Bone RC, Sibbald WJ, Sprung CL. The ACCP-SCCM consensus conference on sepsis and organ failure. Chest. 1992;101(6):1481–3.
Grumaz, S., Stevens, P., Grumaz, C., Decker, S. O., Weigand, M. A., Hofer, S., . . . Sohn, K. (2016). Next-generation sequencing diagnostics of bacteremia in septic patients. Genome Med, 8(1), 73. doi:https://doi.org/10.1186/s13073-016-0326-8.
Filkins LM, O'Toole GA. Cystic fibrosis lung infections: Polymicrobial, complex, and hard to treat. PLoS Pathog. 2015;11(12):e1005258. https://doi.org/10.1371/journal.ppat.1005258.
Zinter MS, Dvorak CC, Mayday MY, Iwanaga K, Ly NP, McGarry ME, DeRisi JL. Pulmonary metagenomic sequencing suggests missed infections in immunocompromised children. Clin Infect Dis. 2018. https://doi.org/10.1093/cid/ciy802.
Dunlap DG, Marshall CW, Fitch A, Rapport SF, Cooper VS, McVerry BJ, Kitsios GD. Improved detection of culprit pathogens by bacterial DNA sequencing affects antibiotic management decisions in severe pneumonia. Am J Case Rep. 2018;19:1405–9. https://doi.org/10.12659/AJCR.912055.
Brenner, T., Decker, S. O., Grumaz, S., Stevens, P., Bruckner, T., Schmoch, T., . . . Group, T. I. C. C. T. (2018). Next-generation sequencing diagnostics of bacteremia in sepsis (next GeneSiS-trial): study protocol of a prospective, observational, noninterventional, multicenter, clinical trial. Medicine (Baltimore), 97(6), e9868. doi:https://doi.org/10.1097/MD.0000000000009868.
McIntyre ABR, Ounit R, Afshinnekoo E, Prill RJ, Henaff E, Alexander N, Mason CE. Comprehensive benchmarking and ensemble approaches for metagenomic classifiers. Genome Biol. 2017;18(1):182. https://doi.org/10.1186/s13059-017-1299-7.
Muttaiyah S, Paviour S, Buckwell L, Roberts SA. Anaerobic bacteraemia in patients admitted to Auckland City Hospital: its clinical significance. N Z Med J. 2007;120(1265):U2809.
Weinstein MP, Towns ML, Quartey SM, Mirrett S, Reimer LG, Parmigiani G, Reller LB. The clinical significance of positive blood cultures in the 1990s: a prospective comprehensive evaluation of the microbiology, epidemiology, and outcome of bacteremia and fungemia in adults. Clin Infect Dis. 1997;24(4):584–602. https://doi.org/10.1093/clind/24.4.584.
Wilson JR, Limaye AP. Risk factors for mortality in patients with anaerobic bacteremia. Eur J Clin Microbiol Infect Dis. 2004;23(4):310–6. https://doi.org/10.1007/s10096-004-1111-y.
Paez JI, Costa SF. Risk factors associated with mortality of infections caused by Stenotrophomonas maltophilia: a systematic review. J Hosp Infect. 2008;70(2):101–8. https://doi.org/10.1016/j.jhin.2008.05.020.
Paez JI, Tengan FM, Barone AA, Levin AS, Costa SF. Factors associated with mortality in patients with bloodstream infection and pneumonia due to Stenotrophomonas maltophilia. Eur J Clin Microbiol Infect Dis. 2008;27(10):901–6. https://doi.org/10.1007/s10096-008-0518-2.
Senol E, DesJardin J, Stark PC, Barefoot L, Snydman DR. Attributable mortality of Stenotrophomonas maltophilia bacteremia. Clin Infect Dis. 2002;34(12):1653–6. https://doi.org/10.1086/340707.
Wang WS, Liu CP, Lee CM, Huang FY. Stenotrophomonas maltophilia bacteremia in adults: four years' experience in a medical center in northern Taiwan. J Microbiol Immunol Infect. 2004;37(6):359–65.
Krentz T, Allen S. Bacterial translocation in critical illness. J Small Anim Pract. 2017;58(4):191–8. https://doi.org/10.1111/jsap.12626.
Wiest R, Lawson M, Geuking M. Pathological bacterial translocation in liver cirrhosis. J Hepatol. 2014;60(1):197–209. https://doi.org/10.1016/j.jhep.2013.07.044.
Nagpal R, Yadav H. Bacterial translocation from the gut to the distant organs: an overview. Ann Nutr Metab. 2017;71(Suppl 1):11–6. https://doi.org/10.1159/000479918.
Horiba, K., Kawada, J. I., Okuno, Y., Tetsuka, N., Suzuki, T., Ando, S., . . . Ito, Y. (2018). Comprehensive detection of pathogens in immunocompromised children with bloodstream infections by next-generation sequencing. Sci Rep, 8(1), 3784. doi:https://doi.org/10.1038/s41598-018-22133-y.
Wilson MR, O'Donovan BD, Gelfand JM, Sample HA, Chow FC, Betjemann JP, DeRisi JL. Chronic meningitis investigated via metagenomic next-generation sequencing. JAMA Neurol. 2018;75(8):947–55. https://doi.org/10.1001/jamaneurol.2018.0463.
Acknowledgements
No acknowledgments.
About the authors
Drs. Philipson, Schully and Bishop-Lilly are employees of the U.S. Government. This work was prepared as part of their official duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties.
Funding
Funding was provided by the Defense Threat Reduction Agency Contract # HDTRA139501. The funder had no role in the design of the study or preparation of this manuscript.
Author information
Authors and Affiliations
Contributions
KS, DC, CW, ET, EK were involved in concept, supervision, writing. CP, KBL, RC, LV, and TB were involved in data analysis and writing. All authors have read and approved the submission of this manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Studies were approved by relevant Institutional Review Boards (IRB) and in accordance with the Declaration of Helsinki. Specifically, the ACESO study was approved by the Duke IRB who reviewed all study protocols, IRB authorizations, and all study procedures and ultimately gave approval for the work to be done. The study protocol was approved by the Naval Medical Research Center Institutional Review Board in compliance with all applicable Federal regulations governing the protection of human subjects.
Consent for publication
The subject in this case report provided written informed consent for publication of research results.
Competing interests
None of the authors had conflicts of interest or disclosures to report.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ko, E.R., Philipson, C.W., Burke, T.W. et al. Direct-from-blood RNA sequencing identifies the cause of post-bronchoscopy fever. BMC Infect Dis 19, 905 (2019). https://doi.org/10.1186/s12879-019-4462-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12879-019-4462-9