
Abstract
This review provides an in-depth discussion of the development, principles and utility of nanopore sequencing technology and its diverse applications in the identification of various pulmonary pathogens. We examined the emergence and advancements of nanopore sequencing as a significant player in this field. We illustrate the challenges faced in diagnosing mixed infections and further scrutinize the use of nanopore sequencing in the identification of single pathogens, including viruses (with a focus on its use in epidemiology, outbreak investigation, and viral resistance), bacteria (emphasizing 16S targeted sequencing, rare bacterial lung infections, and antimicrobial resistance studies), fungi (employing internal transcribed spacer sequencing), tuberculosis, and atypical pathogens. Furthermore, we discuss the role of nanopore sequencing in metagenomics and its potential for unbiased detection of all pathogens in a clinical setting, emphasizing its advantages in sequencing genome repeat areas and structural variant regions. We discuss the limitations in dealing with host DNA removal, the inherent high error rate of nanopore sequencing technology, along with the complexity of operation and processing, while acknowledging the possibilities provided by recent technological improvements. We compared nanopore sequencing with the BioFire system, a rapid molecular diagnostic system based on polymerase chain reaction. Although the BioFire system serves well for the rapid screening of known and common pathogens, it falls short in the identification of unknown or rare pathogens and in providing comprehensive genome analysis. As technological advancements continue, it is anticipated that the role of nanopore sequencing technology in diagnosing and treating lung infections will become increasingly significant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Nanopore sequencing technology plays a valuable role in the rapid and accurate detection of pathogens that are difficult to identify using conventional methods and the quick detection of antimicrobial resistance, thus guiding appropriate antimicrobial therapy. |
We have also illustrated the scope of the utility of nanopore sequencing in metagenomics and epidemiology. |
However, we also acknowledge the limitations of this technology, including its inherently high error rate and complexity of data processing. |
1 Third-Generation Sequencing and Principles of Nanopore Sequencing
Compared with conventional sequencing, third-generation sequencing is characterized by it not requiring polymerase chain reaction (PCR) amplification, which prevents the introduction of amplification bias, and third-generation sequencing can sequence each DNA molecule individually. Therefore, it is also known as single-molecule sequencing [1]. The major methods include single-molecule real-time (SMRT) sequencing, developed by Pacific Biosciences of California, Inc. (PacBio, Menlo Park, USA), and nanopore sequencing developed by Oxford Nanopore Technologies Limited (ONT, Oxford, UK) [2].
1.1 SMRT Sequencing
The PacBio SMRT chip contains nano-sized zero-mode waveguides, which anchor DNA polymerases (DNAPs) to the bottom of the pores. When single-molecule sequencing is performed, the fluorescence signals of the bases inserted during DNA elongation are excited by the laser emitted from the bottom of the zero-mode waveguides and are then detected [3]. Simultaneously, modified bases are detected based on the DNAP binding time being longer for paired deoxynucleoside triphosphates (dNTPs) than that for free dNTPs [4]. Therefore, the sequencing read length is associated with the activity of DNAPs, and the activity of DNAPs is influenced by the duration of laser irradiation. SMRT can achieve ultralong read lengths of up to 20 kb, which is approximately 100 times longer than that of second-generation sequencing, with a run time of several hours [5]. Therefore, it has a considerable advantage over second-generation sequencing for the identification of the loci of disease-causing genes in fields such as oncology and genetics [6, 7]. However, SMRT sequencing still relies on DNAPs. This process requires waiting for the polymerase to synthesize a new DNA strand and record all fluorescence signals before obtaining complete sequencing information; thus it cannot achieve real-time sequencing. The high cost and a relatively high single-read error rate of approximately 15% are also important limitations.
1.2 Nanopore Sequencing
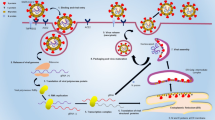
Compared with other third-generation sequencing technologies, a key feature of nanopore sequencing is that it does not rely on DNAPs during the sequencing process but directly reads DNA or RNA molecules through nanopores, achieving real-time sequencing in its true sense. Nanopore sequencing is performed by immersing a biological membrane containing nanopores in an ionic solution, applying a voltage to both sides of the phospholipid bilayer to generate an ionic current through the nanopore channels [8], and then driving the uncoiled single-stranded molecules to be tested through the nanopores (Fig. 1). For example, the most commonly used α-hemolysin nanopore channels with a pore size of 2.6 nm only allow the passage of single-stranded RNA or DNA. Thus, each polymer, which is an extended strand, blocks a channel when crossing the membrane, causing a change in the ionic current level [9]. Based on the measured changes in the current level, the information of the bases can be read, and epigenetic modifications, such as DNA methylation, can be detected directly [10]. As this process can be performed in real time, data can be generated and analyzed simultaneously.

Principle of nanopore sequencing. Nanopore sequencing technology is based on the utilization of nanoscale protein pores, referred to as nanopores, as biosensors embedded within an insulating polymer membrane. By applying a constant voltage across the polymer membrane, negatively charged single-stranded DNA or RNA molecules are driven from the negatively charged side (cis) to the positively charged side (trans) of the membrane. The motor protein possesses helicase activity that unwinds the double-stranded DNA or DNA–RNA duplex into single strands that pass through the nanopore. The migration rate of the nucleic acid strands within the nanopore is also controlled by the motor protein. As the nucleic acid strand translocates through the nanopore, different bases induce different changes in the electric current. These changes are read by the signal receptor within the nanopore, and subsequent base recognition is accomplished via computational algorithms
However, the identification of DNA passing through nanopores at ultra-fast speeds is a challenge for the current detection capabilities of optoelectronic technology, resulting in low accuracy of nanopore sequencing [11]. To this end, many studies have used active and passive methods to slow the passage of DNA. Among them, some researchers have used nanopores made of Mycobacterium smegmatis porin A (MspA) in combination with phi29 DNAP to slow down the passage of DNA through nanopores [12], achieving good changes in ionic current level to read the DNA sequences.
The sequencing platform MinION [13], developed by ONT, could generate 50 GB of data in a 72 h run in 2014, and the PromethION platform updated in 2015 can generate up to 7.6 TB of data. Compared with the mainstream Illumina sequencing platform of second-generation sequencing, which has a run time of more than 16 h and takes 48–72 h from sampling to obtain results, the MinION sequencer can rapidly detect microorganisms within a few minutes of starting sequencing, and the entire sequencing process takes less than 6 h [14]. In addition, MinION is the first handheld sequencer, with a size similar to that of a cell phone [15], and it can transfer data to a computer for result analysis with a USB cable [16]. Its compact size and portability enable real-time sequencing in settings and environments with restricted experimental conditions, such as the Arctic [17], military training grounds [18], and space stations [19]. In 2016, a major Ebola virus epidemic in West Africa caused the death of tens of thousands of people, and performing pathogen sequencing was difficult because of the lack of local equipment and limited research capacity. Quick et al. [20] solved these challenges by bringing the compact MinION sequencer in luggage to the frontline of the outbreak and successfully sequenced samples from patients with Ebola in an environment with limited resources. The entire sequencing process took only 15–60 min, with a turnaround time of less than 24 h. Accurate genotypes were identified at a sequencing depth of 25 times, and a new transmission chain was identified, providing valuable information on the epidemiology of the Ebola outbreak.
Its advantages of ultralong read length, lightness, portability, and real-time access to sequencing results have revolutionized the genetic testing field, and some industry insiders refer to it as the fourth-generation sequencing technology [15]. Although nanopore sequencing technology could not replace traditional sequencing methods when it was first introduced, due to its high error rates of 5–15%, an accuracy of up to 99% can now be achieved owing to increased sequencing depth, optimized algorithms, and improved nanopore structure [21, 22].
2 Recent Advances in Nanopore Sequencing Technology
A significant challenge for nanopore sequencing is its relatively high error rate, which, in the early days, was as high as 15%, predominantly constituted by insertions and deletions (indels). To improve the accuracy of the results, scientists often use a method known as hybrid error correction, integrating high-accuracy short reads (e.g., Illumina short reads) with long reads from nanopore sequencing. This method employs the precision of short reads to rectify errors in long reads, thereby obtaining more accurate long reads [23]. Hence, by using short reads to correct errors in long reads, the benefits of long reads of nanopore sequencing are combined with the high accuracy of short-read sequencing.
Nanopore sequencing technology has been continually evolving and improving with enhanced data quality and accuracy. Recently, ONT introduced a novel R10.4 chip, which has a higher sequencing accuracy than its predecessor, R9.4.1, primarily owing to its enhanced ability to identify homopolymer repeats. Homopolymer repeats, which are consecutive repeat sequences of the same base, are ubiquitous in microbial genomes and frequently cause errors in nanopore sequencing runs using earlier versions of the technology. The R10.4 chip optimizes identification algorithms and hardware design to generate accurate microbial genomes without the need for short-read or reference genome correction [24].
The advent of the R10.4 chip marks a significant breakthrough in the accuracy and complex sequence processing of nanopore sequencing technology. This development will greatly improve the reliability of microbial genome sequencing, thereby more accurately revealing the characteristics and functions of microbial genomes. These technological advances carry significant implications for scientific research and open new possibilities for the diagnosis and treatment of microbial diseases.
Q20+ is a sequencing technology that combines the latest Q20+ chemical reagent, a new reagent kit (Kit12) supporting duplex sequencing, and the most recent R10.4 chip. It utilizes a new reaction buffer and a new data processing algorithm to achieve longer read lengths and higher coverage, thereby exhibiting an improved ability to rectify errors that occur during the sequencing process. Under the Q20+ mode, multiple copies of a particular DNA/RNA region are stacked, implying that random errors are averaged out, yielding a more accurate consensus sequence. For example, in bacterial genome sequencing, achieving a quality score of Q50 is challenging, even at high depth in the standard mode. However, Q20+ can achieve a Q50 quality (99.999% accuracy) at a sequencing depth of 20×. The advent of Q20+ has enabled nanopore sequencing technology to reduce its average error rate to less than 1%, achieving > 99% original read (single-strand) accuracy, or approximately Q30 duplex accuracy, and enhancing the precision of shared sequence sequencing and variant identification. As such, nanopore sequencing has reached an important milestone in accuracy [25].
Q20+ allows high-resolution bacterial typing and genetic variation analysis through highly accurate long reads, an aspect that traditional nanopore sequencing technology struggles to achieve. This improvement not only enhances the accuracy of sequencing results but also broadens the application scope of nanopore sequencing, enabling it to compete with short-read sequencing in tasks that require high resolution and accuracy. Real-time nanopore Q20+ sequencing can be used for rapid and precise bacterial pathogen monitoring, including core genome multi-locus sequence typing (cgMLST), virulence-factor screening, and antibiotic-resistance gene screening. Analyzing the genetic relationships and evolutionary history among different strains can help clinicians better understand the epidemiological characteristics, transmission routes, and drug resistance of pathogenic microbes, thereby guiding the formulation of clinical treatments and preventive measures. For example, cgMLST can identify genetic differences and relationships among different strains, subsequently determining whether a cluster infection or epidemiological link exists, and promptly implementing appropriate isolation, disinfection, and treatment measures. Additionally, by sequencing and comparing the genomes of different strains, important genes, such as antibiotic-resistance genes and virulence factors, can be identified, and their possible expression levels and functions can be predicted, providing a basis for personalized treatment.
Although the emerging Q20+ nanopore sequencing technology has led to improvements in read length, coverage, and accuracy, it cannot yet fully replace short-read technologies. Q20+ offers enhanced read length and accuracy, undeniably beneficial for whole-genome sequencing, chromosomal structural variation detection, and other applications. In these areas, Q20+ has the potential to become the preferred sequencing method. Although the Q20+ error rate has been reduced to less than 1%, this rate is still higher than that of some second-generation sequencing platforms (e.g., Illumina) [26]. Moreover, the approach to select the most appropriate data analysis strategy and resolve potential issues of host DNA interference requires further investigation. Therefore, in applications that require very high accuracy or large-scale sample sequencing, such as single-nucleotide variation detection or extensive population genetic studies, short-read sequencing may still be the preferred option.
Table 1 expounds on the operational principles of the first, second, and third-generation sequencing technologies, showcasing the field's technical progression. Subsequently, Table 2 presents a thorough comparison of these generations, focusing on Sanger's 3730xl, Illumina's MiSeq, and ONT's MinION. The comparison is structured around critical metrics, such as read length, maximum throughput, data volume, cost, time to data, run duration, device size, and consensus sequence accuracy. Lastly, Table 3 details the features and specifications of four Oxford nanopore sequencing platforms: Flongle, MinION, GridION, and PromethION. The compared parameters include read length, number of flow cells, independence of flow cells, maximum theoretical output, real-time data availability, run time for maximum output, machine size, weight, test cost, and hardware requirements.
3 Introduction to Nanopore Materials
Nanopore sequencing is a single-molecule sequencing technology that determines the sequence of DNA or RNA molecules by monitoring the changes in electric current as the molecule passes through a nanopore. Nanopores can be divided into two categories: biological nanopores and solid-state nanopores, and they vary in their structure, mechanism, advantages, and limitations.
3.1 Biological Nanopores
Biological nanopores are channels formed by protein molecules through which molecules are driven by voltage to achieve detection and sequencing. They typically have a small pore size, usually 1–2 nm, and can be used to detect biological molecules, such as DNA, RNA, and proteins. Examples include α-hemolysin, MspA, bacteriophage phi29, and aerolysin nanopores. Among them, α-hemolysin has become the preferred material for commercial nanopore sequencing equipment owing to its strong acidic nature and heat resistance [31].
3.2 Solid-State Nanopores
Solid-state nanopores are prepared using solid materials, such as silicon, silicon nitride, and aluminum oxide, and use voltage to drive molecules through the pores for detection and sequencing. These nanopores allow for precise control of pore diameter and thickness and have high stability and adjustability. Solid-state nanopores typically have a larger pore size, generally 3–1000 nm, and can be used to detect biological molecules, such as DNA, RNA, and proteins, as well as non-biological molecules, such as nanoparticles and ions, and have the advantage of multifunctionality [32].
4 Bioinformatics Tools for Processing Nanopore Sequencing Data
The processing of data generated by nanopore sequencing, especially for metagenomic analysis, involves the use of an array of bioinformatics tools and methods. The raw sequencing signal data requires initial quality control and filtering, a task that can be accomplished by tools such as Cramino or Kyber [33]. Furthermore, the interpretation of raw current signals from nanopore sequencing, thereby enhancing sequence accuracy, can be facilitated by Nanopolish [34].
After initial quality control and filtering, the alignment and assembly of reads using reference databases is a critical step. This process can be efficiently performed using minimap2 [35] or NGMLR [36], both capable of handling the unique attributes of long-read data from nanopore sequencing.
Unicycler [37] emerges as a potent assembly tool specifically designed for bacterial genomics. It employs a unique algorithm combining short, accurate reads from Illumina sequencing with long reads from nanopore sequencing to produce high-quality hybrid assemblies. The algorithm conducts multiple rounds of read alignments and assembly graph cleaning, thereby accurately resolving repeats in bacterial genomes, making Unicycler a valuable asset for nanopore sequencing data processing.
For read assembly, other tools, such as GoldRush [38] and Shasta [39], can offer effective solutions, while gene prediction and annotation, crucial parts of nanopore sequencing data processing, can be conducted rapidly and accurately by tools like Prokka [40].
In addition to the above, BacWGSTdb 2.0 [41] serves as a comprehensive platform for bacterial whole-genome sequence analysis and source tracking. This user-friendly tool integrates an extensive range of bacterial genome sequencing data and associated metadata. It incorporates specialized features for multiple genome analysis, bacterial isolate characterization, and user-uploaded sequence comparison, making it an invaluable resource for downstream analysis following nanopore sequencing.
For metagenomic analysis, specialized tools, such as MetaBAT [42], prove to be essential. These tools can separate the genomes of individual species from metagenomic sequence data, thus elucidating the composition of microbial communities.
In terms of de novo assembly of nanopore sequencing data, Flye and Canu [43] are remarkable tools. Flye specializes in long-read assembly and excels in constructing high-quality, particularly circular, bacterial genome sequences. Canu, on the other hand, adeptly handles both long and short reads and is particularly effective in hybrid data assembly. Despite differences in their strengths and operational requirements, both tools manage the high error rates and variability in read lengths, delivering robust genome assembly tailored to the specific research objectives and data types.
Lastly, for the taxonomic classification of metagenomic data, Kraken 2 [44] has emerged as an exceptionally useful tool. With its high-speed operation, accuracy, and parameter optimization flexibility, Kraken 2 is positioned as a vital asset in the classification and identification of sequences within nanopore sequencing data analysis.
5 Application of Nanopore Sequencing in the Identification of Respiratory Pathogens
This section discusses the application of nanopore sequencing for the identification of specific respiratory pathogens.
5.1 Challenges with the Application of Nanopore Sequencing in Mixed Infections
In clinical diagnostics, particularly in respiratory infections, mixed infections with multiple pathogens are a common occurrence. Mixed infection can provide a diagnostic challenge due to overlapping symptoms and the complexity of distinguishing pathogens from commensals. This section explores how nanopore sequencing with its high-resolution perspective is used to identify multiple infections in hosts with mixed infections and how it addresses related challenges.
One of the primary challenges in the context of mixed infections is differentiating between colonization and infection. Many respiratory infections, especially in individuals with compromised immune systems or chronic lung disease, are characterized by the colonization of multiple microorganisms, which could potentially include pathogens [45]. Nanopore sequencing, through metagenomic sequencing, can generate comprehensive genomic data from clinical samples, thus enabling the identification of a myriad of organisms. However, distinguishing pathogens from commensals solely based on sequencing data can be challenging. Approaches such as determining relative abundance at the genus level, normalizing to the number of sequences per million, comparing the number of sequences aligned to the target microbe, and non-overlapping genomic coverage area can aid in distinguishing infection from colonization [46]. Furthermore, z-scores can be used in the bioinformatics workflow to differentiate pathogens and background microbes [47]. Additionally, researchers have developed methods to distinguish potential pathogens and respiratory commensals using rule-based models and logistic regression models, both achieving 95.5% accuracy in the validation cohorts [45]. Another challenge faced by nanopore sequencing in the context of mixed infections is the presence of host DNA, which is discussed in Sect. 4.
5.2 Application of Nanopore Sequencing in Viral Respiratory Infections
Respiratory viruses can be transmitted via the airborne route in the form of small droplet nuclei (< 5 μm). These droplet nuclei can remain suspended for a prolonged period and can be inhaled into the lower respiratory tract, causing pulmonary infections [48]. Previously, the lack of appropriate diagnostic methods led to an underestimation of the role of viruses in pneumonia. Influenza A virus and respiratory syncytial virus are the most common causes of viral pneumonia, followed by adenoviruses, parainfluenza viruses types 1–3, and influenza B virus [49]. These common lower respiratory viruses are RNA viruses [50]. Traditional sequencing methods require reverse transcription before RNA viruses can be sequenced, whereas nanopore sequencing allows direct detection of RNA viruses, simplifying the sequencing steps. One study performed direct RNA sequencing of the influenza A virus using the MinION platform and obtained its full-length genome sequence [51].
However, RNA viruses have a higher replication error rate and are more prone to mutations than DNA viruses [52]. The ultralong read length of nanopore sequencing is advantageous for monitoring mutations. During the coronavirus disease 2019 (COVID-19) pandemic, Li et al. [53] successfully identified and analyzed 33 mutations in 29 copies of the complete genome of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) using nanopore sequencing technology. Another study compared whole-genome sequencing using ONT and Illumina platforms on samples from patients with SARS-CoV-2 infection. ONT achieved > 99% sensitivity and accuracy with coverage depths above 60 times and identified a diversity of structural variations in patients with SARS-CoV-2 infection [54].
Early treatment with antiviral drugs can shorten the duration of symptoms and reduce the mortality rate of high-risk individuals [55]. The current tests for lower respiratory viral infections rely on viral antigen tests and reverse transcription quantitative PCR (RT-qPCR) [56], but antigen testing has low sensitivity and RT-qPCR is time-consuming [57], as illustrated by the SARS-CoV-2 RT-qPCR kit currently used in clinical practice, which has a high false-negative rate [58]. This limits the accuracy of early clinical triage of patients and increases the potential for disease transmission during the diagnostic process. Wang et al. [58] performed nanopore sequencing on 61 nucleic acid samples from patients with suspected COVID-19, and found that 22 (36%) of the samples that had tested negative or uncertain for SARS-CoV-2 using RT-qPCR were identified as positive using nanopore sequencing. In addition, the investigators successfully detected four respiratory viruses, influenza A virus, influenza B virus, respiratory syncytial virus, and rhinovirus, within 10 min using nanopore sequencing. Real-time sequencing and data processing substantially reduce the analysis time, serving as an effective supplement for patients who cannot be accurately assessed using antigen testing and RT-qPCR. This quick turnaround aids clinical practitioners in the rapid identification of pathogens, thus supporting the development of more effective treatment plans.
5.2.1 Sequencing Viruses in Epidemiological Research and Outbreak Surveillance
Antigen testing and RT-qPCR, although effective in their roles, do not offer insights into viral evolution. Nanopore sequencing can address this gap and is a useful tool for determining the genomic characteristics and origins of viruses, and thus can be used to trace pathogen transmission and identify the sources of outbreaks. A case in point is the widespread utilization of nanopore sequencing during the COVID-19 pandemic where its real-time and high-throughput attributes proved invaluable for monitoring the mutations and transmission dynamics of the virus [59]. The provision for long-read capabilities uniquely equips this technology for whole-genome sequencing, thereby facilitating the detection of structural variations within viral genomes and offering a potential avenue for the precise tracing of viral strains [60].
In an investigation of an outbreak of human adenovirus type 55 infection in Hubei Province, China, in 2019, Li et al. [61] sequenced nasopharyngeal swab samples from nine patients using MinION and detected human adenovirus within 13–20 min. The results were confirmed by Reverse transcription polymerase chain reaction (RT-PCR). After further metagenomic testing, the investigators found that the strain responsible for the Hubei Province outbreak had a close genetic relationship to a strain from Sichuan Province.
Nanopore sequencing technology confers a distinct advantage with its capacity to produce long reads, thereby exposing the intricate structures embedded within genomes, particularly those components, such as repeat and insert sequences, that imbue microbial genomes with their complexity. Its ability to operate in real time and its inherent portability make nanopore sequencing highly suitable for field research, including surveillance of disease outbreaks in isolated regions. The relatively high error rate within raw reads exhibited by this technology can limit its ability to detect genomic variants. However, technology's relentless advance has seen the introduction of hardware innovations, such as the R10.4 chip, and software enhancements that have considerably increased sequencing accuracy. This, in turn, has widened the application spectrum of nanopore sequencing in epidemiological investigations.
5.2.2 Nanopore Sequencing in Antiviral Resistance Research
Viruses may be resistant to drugs. This is particularly true for RNA viruses, which have the innate ability to evade antiviral treatment through resistance mutations. Nanopore sequencing can detect viral resistance, whereas current antigen tests and RT-qPCR have limited ability to detect resistance. Influenza A virus is an encapsulated virus of the family Orthomyxoviridae that causes seasonal epidemics [62]. During the influenza virus pandemic season in 2019, researchers performed nanopore metagenomic sequencing of 180 copies of respiratory samples from a hospital in the United Kingdom. The results showed a sensitivity of 83% and specificity of 93% for detecting influenza A virus and identified mutant genomes that were resistant to antiviral drugs, such as amantadine and oseltamivir [63].
These studies illustrate the utility of applying nanopore sequencing technology to viral respiratory infections.
5.3 Nanopore Sequencing in Bacterial Respiratory Infections
Traditional diagnostic methods for bacterial pulmonary infections, which include direct smear microscopy, routine culture, and serological tests, have low sensitivity and are time-consuming. Additionally, the misuse of broad-spectrum antibiotics has made culturing pathogens increasingly difficult. Nanopore sequencing with real-time data generation and an extremely short turnaround time can greatly reduce the time required to adjust the initial empirical antibiotic regimen and is a rapid and comprehensive diagnostic tool for severe pneumonia, thereby providing an advantage over traditional methods of diagnosis.
5.3.1 Nanopore-Based 16S Targeted Sequencing
In the last 2 decades, amplicon sequencing, which targets the bacterial 16S ribosomal RNA (rRNA) gene, has been used to identify pathogens causing rare bacterial infections that are not detected by standard diagnostic tests in clinical settings to help clinicians choose appropriate antibiotics [64]. The 16S rRNA gene is present in the genomes of all bacteria and is divided into conserved regions that are common to all bacteria, and variable regions that vary among bacteria [65]. Amplicon sequencing of 16S identifies the bacterial strains by amplifying the variable regions and is considered the most promising and sensitive technique for the identification of bacterial communities present in environmental samples. However, owing to the short read length of second-generation sequencing, the sequence of the entire 16S rRNA gene (1500 bp) cannot be obtained at once [66], and thus, it is prone to mismatches during the splicing of different variable regions due to intra-genus similarities, which in turn leads to incorrect classification and misidentification of microorganisms [64]. In contrast, nanopore sequencing technology offers an ultralong read length of 2 Mbp [67], enabling the coverage of the entire 16S rRNA gene in a single sequence during the 16S amplicon sequencing of a particular strain. Recent advancements, such as the use of Emu, an expectation-maximization algorithm-based approach, have further improved the accuracy and reliability of nanopore sequencing, effectively leveraging the ultralong read length to generate accurate microbial community profiles even with high error rates in input sequences [68].
In pulmonary infections, sputum is the most easily obtained specimen, but it is susceptible to contamination, and it can be difficult to obtain acceptable quality sputum specimens to perform sputum culture [69]. In a study conducted by Moon et al. [70], MinION-based 16S rRNA gene amplicon sequencing was performed on sputum samples from patients with severe pneumonia who had been treated with cefuroxime as empirical antibiotic therapy. A total of 122,722 reads were obtained, with 98.1% aligned with Haemophilus spp., which were 100-fold more abundant than other aligned commensal bacteria. Sufficient reads for the identification of the pathogen were obtained within 10 min of starting the sequencing, and the number of reads was similar to that obtained at 1 h. The diagnosis of pneumonia caused by Haemophilus influenzae was made based on the sequencing results and confirmed by qPCR. This study showed that nanopore-based 16S amplicon sequencing could distinguish between pathogens and commensal bacteria in sputum wells, and the results were not affected by the patient’s use of antibiotics.
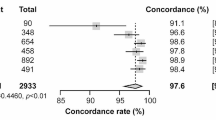
Baldan et al. [71] also performed nanopore-based 16S rRNA gene sequencing (Np16S) of respiratory specimens from patients with severe bacterial pneumonia in the intensive care unit. A total of 140 of 167 (84%) strains were correctly identified after 1 h. The results were consistent with those of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) (discussed in Sect. 5.3.2), and the sequencing results obtained at 1 h were not significantly different from those obtained at 16 h. Sanger-based 16S sequencing in this study required a turnaround time of 2–3 days and could not guide the initial antibiotic therapy. In contrast, the rapid 1-h sequencing using Np16S can guide the choice of the initial antibiotics in patients with acute pulmonary infections.
5.3.2 Comparison of Nanopore Sequencing with MALDI-TOF MS
Anhalt and Fenselau [72] first proposed mass spectrometry in 1975 for bacterial identification by recognizing characteristic spectra of various bacteria. The advent of MALDI-TOF MS enabled researchers to swiftly identify most bacteria at the genus and species level through proteomics. Its strength lies in the ability to achieve highly accurate bacterial species identification within minutes by comparison with known databases [73]. However, the MALDI-TOF MS system has some limitations: it can only identify microorganisms contained within the spectral database fingerprints, and it may not distinguish subspecies or strain variations. In discovering new species and undescribed variants, nanopore 16S rRNA gene sequencing, which can match and compare similar sequences, clearly has an advantage [65]. Moreover, the accuracy of MALDI-TOF MS can be affected by the presence of mixed infections with fewer or overlapping species. For certain bacterial species, high similarities may exist in spectral patterns, resulting in uncertainties in the identification results. For example, MALDI-TOF MS may misidentify encapsulated microorganisms, such as Streptococcus pneumoniae, Streptococcus viridans, Klebsiella pneumoniae, and H. influenzae [74]. In contrast, nanopore sequencing not only identifies the bacterial species but also applies whole-genome and metagenomic sequencing, covering more biological information.
A study compared two 16S rRNA gene sequencing tests based on Illumina and nanopore sequencing technologies. They identified 172 clinically isolated strains that MALDI-TOF MS failed to recognize. The time taken by Illumina sequencing was 78 h, whereas nanopore sequencing required 8.25 h. Furthermore, the study found that although Illumina sequencing had high accuracy in base recognition, nanopore sequencing provided a higher taxonomic resolution at the species level. Therefore, compared with MALDI-TOF MS and Illumina sequencing, nanopore sequencing technology has a higher species identification ability, despite its higher sequencing error rate [75].
Therefore, the MALDI-TOF MS system provides a quick, accurate, and widely commercialized advantage in bacterial identification, whereas nanopore sequencing provides high resolution and has more extensive application potential. Researchers need to choose the technology according to the specific requirements and purpose of the research, and in some situations, combining both technologies may be the best option.
5.3.3 Nanopore Sequencing in the Diagnosis of Rare Bacterial Pulmonary Infections
Studies have confirmed the value of nanopore sequencing technology in the diagnosis of rare bacterial pulmonary infections. Guo et al. [76] performed nanopore sequencing of bronchoalveolar lavage fluid (BALF) from patients with interstitial lung disease of unknown origin. Tropheryma whipplei, which rarely affects the lungs, was detected within 6–8 h, and the results were validated by PCR and Sanger sequencing. Watanabe et al. [77] reported a pulmonary infection caused by Nocardia spp., which is a rare pathogen, that was rapidly identified using nanopore sequencing. Although targeted PCR has a rapid turnaround time, it can only identify established pathogens and has limitations in the testing range, which can be overcome using nanopore sequencing.
5.3.4 Nanopore Sequencing in Antimicrobial Resistance Research
Antimicrobial resistance has become a public health challenge worldwide, and the shift in drug-resistance genes in pathogens has made the treatment of severe infections increasingly difficult [78]. The ultralong read length of nanopore sequencing technology has promising application prospects for exploring the mechanisms of antimicrobial resistance.
Recently, resistance to tigecycline, which is a last-line antibiotic for multidrug-resistant Enterobacteriaceae, has been widely observed in patients with pneumonia caused by K. pneumoniae in China. Liu et al. [79] used nanopore sequencing to identify three new drug-resistant strains and identified the drug-resistance gene corresponding to the tigecycline-resistant phenotype. The investigators found that tmexd1-toprj1-positive pneumonia caused by K. pneumoniae in humans was closely associated with K. pneumoniae in animal-source foods and related environments. This study showed that K. pneumoniae containing the tmexd1-toprj1 gene cluster was widely disseminated and highlighted the urgent need for active surveillance of tigecycline-resistant K. pneumoniae in the food animal production industry and healthcare settings. Wang et al. [80] demonstrated a rapid detection of drug-resistance genes implicated in pneumonia caused by K. pneumoniae in a turnaround time of only 1.5 h. This is a substantial advance on traditional culture and resistance testing, which are relatively slow. By expeditious identification of these drug-resistance genes, this approach holds the potential to facilitate more timely and appropriate choices regarding antibiotic selection, subsequently mitigating the risk of antibiotic misuse and optimizing patient outcomes.
Mycobacterium kubicae, which is part of the environmental nontuberculous mycobacteria group, can cause severe pulmonary infections in humans under certain conditions. Hendrix et al. [81] conducted a comprehensive genomic analysis of M. kubicae strains using the MinION platform in order to delineate the chromosomal and plasmid sequences pertinent to its drug resistance, virulence, and persistence, and to deepen the understanding of its opportunistic pathogenic mechanisms. They harnessed the respective strengths of Illumina, characterized by a low error rate but shorter read length, and nanopore sequencing, known for longer read length but a heightened error rate, to yield a genome assembly that exhibited superior continuity and precision. Notably, their findings indicated the existence of single-nucleotide polymorphisms (SNPs) and small indels between the hybrid assemblies and the corresponding Illumina read sets. This could be indicative of certain inaccuracies inherent in the ONT reads, which were rectified during the correction process using Illumina reads. This study underscores the crucial role that nanopore sequencing could potentially play in antimicrobial-resistance research. Although nanopore sequencing offers the advantage of long reads and high genome assembly continuity, it is also characterized by a higher error rate, which can introduce inaccuracies into the assembled genome. To ensure an accurate genome sequence, it is imperative to correct these potential inaccuracies using more precise sequencing techniques, such as Illumina.
5.4 Nanopore Sequencing in Fungal Respiratory Infections
Due to the long duration and low sensitivity and specificity of traditional fungal cultivation, the diagnosis of pulmonary fungal infections remains a significant challenge [82].
5.4.1 ITS Sequencing Based on Nanopore Technology
The internal transcribed spacer (ITS) is a non-coding region in ribosomal DNA (rDNA), comprising ITS1, 5.8S rDNA, and ITS2. These display remarkable sequence differences among different types of fungi, rendering ITS to be extensively used as a “universal DNA barcode for fungi” in mycological taxonomy and identification studies [83].
ITS sequencing is a widely used method in fungal diagnosis. However, PCR amplification and short read length sequencing methods such as Illumina have their limitations, such as the potential loss of variability in long ITS regions and the PCR bias can affect the assessment of the true mycobiome. In contrast, due to its long read length advantage, nanopore sequencing can sequence the entire ITS region in a single pass, thereby obtaining more comprehensive information. Additionally, because nanopore sequencing technology performs direct sequencing without the need for PCR amplification, it avoids the problem of PCR bias.
The real-time nature of nanopore sequencing technology also offers the potential for rapid fungal diagnosis. Traditional ITS-region sequencing requires waiting for the complete sequencing run to end before results can be obtained, whereas nanopore sequencing technology can generate data in real time during the sequencing process. This allows for the identification of fungi to be completed in a short time, which is useful in clinical situations requiring rapid diagnosis.
Although the precision of identification is limited for some fungal species with high homology, the recent development of Q20+ nanopore sequencing technology can effectively address this issue.
5.4.2 Nanopore Sequencing in the Diagnosis of Fungal Infections
Talaromyces marneffei infections occur primarily in individuals with immunodeficiencies, particularly HIV infection. However, recent studies have shown an increasing incidence among individuals without HIV infection. T. marneffei can invade the lungs, causing pneumonia [84].
In a reported case of T. marneffei infection, researchers conducted nanopore genomic sequencing on the patient's blood sample, obtaining 84,000 reads within 3.5 h. The average sequence length was 7088.7 bases, with the longest sequence reaching 87,471 bases. By comparison with a database, 13 homologous sequences of T. marneffei were identified. This outcome was further confirmed through T. marneffei-specific real-time qPCR, fungal ITS sequencing, and biphasic fungal culture [85]. This study revealed the potential value of nanopore sequencing technology in diagnosing fungal infections, particularly T. marneffei infections. Despite the common occurrence of this infection in certain regions, T. marneffei infections are often misdiagnosed, and nanopore sequencing technology offers a more accurate and efficient method to identify this pathogen. By obtaining a vast amount of genomic sequence data in a short time, the presence of T. marneffei can rapidly be detected in blood samples from patients with infection. Nanopore sequencing technology provides new possibilities for the identification and diagnosis of fungal infections.
Additionally, the emergence of nanopore sequencing technology enables direct identification and characterization of fungi in metagenomic detection through its capability of generating ultralong reads. Pneumocystis pneumonia is caused by Pneumocystis jirovecii, an opportunistic pathogen that infects immunocompromised patients, such as individuals with HIV infection and organ transplant recipients. Pneumocystis pneumonia has a high mortality rate. Rapid and accurate testing for fungal pathogens and early administration of targeted antifungal therapy can help to improve prognosis [86]. Irinyi et al. [87] used MinION to perform metagenomic sequencing on BALF and induced sputum specimens from three patients with clinically confirmed Pneumocystis pneumonia, and P. jirovecii was identified in all samples.
By facilitating the production of extended read lengths, nanopore sequencing enables in-depth investigation of complex metagenomic samples. The coupling of nanopore sequencing with two distinct analytic techniques, What’s In My Pot (WIMP) and Basic Local Alignment Search Tool (BLAST), significantly enhances the precision of fungal identification. However, a relatively high error rate, inherent to nanopore sequencing, can trigger the misclassification of reads. Furthermore, the inclination of classification methodologies to misidentify plant-associated fungi, in tandem with the additional computational exertion necessitated by the cross-validation of WIMP and BLAST results, amplifies the complexity and time required for the analysis. Although nanopore sequencing is a promising tool for diagnosing fungal infections, certain technical challenges still need to be overcome for successful practical implementation.
Zhang et al. [88] performed simultaneous Illumina- and nanopore-based metagenomic sequencing of BALF from 66 patients with suspected community-acquired pneumonia. The results showed that both had similar rates of pathogen detection (56% and 58%, respectively) in clinical diagnosis and outperformed conventional culture (24% positivity), but nanopore sequencing detected more species than Illumina, especially in patients with fungal, viral, and mycobacterial infections. In addition, the turnaround time of nanopore sequencing was significantly shorter than that of Illumina sequencing, and the results detected at 1 and 4 h were similar.
5.5 Nanopore Sequencing in Tuberculosis
Tuberculosis (TB) is one of the most common infectious causes of death worldwide, causing 1.7 million deaths per year [89], and drug-resistant TB is a major public health problem. Previously, the identification of clinically relevant drug resistance relied mainly on laboratory culture techniques. However, traditional culture methods are time-consuming (11.5 days on average) [90], and rapid prediction of drug resistance is crucial for selecting anti-TB drugs, which can significantly increase the cure rate. Several studies have applied nanopore sequencing to TB drug-resistance testing.
Zhao et al. [91] performed nanopore sequencing and Sanger sequencing of sputum specimens from patients with TB to identify drug resistance. The results showed 100% identity between the two methods of identification of drug-resistance genes, with a sequencing time of 3 h, data analysis of 1 h, and turnaround of less than 12 h using nanopore sequencing, and several days using Sanger sequencing, demonstrating the potential of nanopore sequencing for rapid assessment of drug susceptibility prior to starting treatment, enabling appropriate antibiotic selection.
Smith et al. [92] also compared the ability of nanopore sequencing using MinION and second-generation sequencing using MiSeq for taxonomic identification of Mycobacterium tuberculosis. The results showed that MinION sequencing correctly identified species levels in 98.5% of the samples and specific lineages of M. tuberculosis in 99.5% of the samples. Moreover, the cost of sequencing per sample was lower with the MinION platform than with the MiSeq platform. Furthermore, the utilization of the MinION platform enabled the generation of whole-genome sequencing results within 2–4 days, which is shorter than the minimum time of 3 days required using the MiSeq platform.
5.6 Nanopore Sequencing for the Identification of Atypical Pathogens
A recent study by Sharda et al. [93] found that nanopore sequencing was useful in the diagnosis of scrub typhus. The investigators used MinION to perform 16S rRNA gene amplicon sequencing of blood samples from a patient with sepsis and multiple organ failure. A total of 44,747 reads were obtained, with an average read length of 1368 bp, and 98.7% of the reads aligned with Orientia tsutsugamushi, the scrub typhus pathogen. The results were validated using enzyme-linked immunosorbent assay and PCR. Additionally, the cost of sequencing per sample in this study was only $13 (USD).
In a study conducted by Baldan et al. [71], Chlamydia psittaci (C. psittaci) was the only important pathogen that was left out in the sequencing results obtained within 1 h of Np16S targeted sequencing. This was probably due to a mismatch between primers and templates. Currently, there is still limited evidence available on the application of nanopore sequencing technology in detecting C. psittaci, and further research is required on the application of nanopore sequencing technology to detecting atypical pathogens, such as C. psittaci.
6 Application of Nanopore Sequencing in Metagenomics
Second-generation sequencing, with its high-throughput, is widely used in metagenomics, also known as metagenomic NGS (mNGS). Metagenomic sequencing involves sequencing all microbial nucleic acids in environmental samples. When applied clinically, its greatest advantage lies in its unbiased detection of all pathogens, including bacteria, fungi, viruses, and atypical pathogens, and its potential to discover unknown pathogens. Due to the large total genome volume, the shotgun method is used to break up the target DNA fragments, which are then sequenced by a computer assembly [94]. Eukaryotic organisms often contain a large number of repeat sequences, and second-generation sequencing, with its short read length, introduces more misalignments when dealing with complex repeat sequences, and the assembly process is more time-consuming. Nanopore sequencing has shown great potential in metagenomic studies. Its long read length can provide a more complete and continuous genome assembly [95]; it also has advantages in sequencing genome repeat areas and structural variant regions.
However, for samples containing host organisms, the removal of host DNA poses a major challenge. For example, when pathogenic microorganisms infect the lungs, 99% of the nucleic acids extracted from samples taken from the infected area come from the human host, vastly outnumbering the DNA of the pathogenic microorganisms and limiting the sensitivity of detection [94, 96]. Although this inherent drawback can be improved by methods such as host gene depletion by DNA exhaustion and differential lysis [97, 98], the process of removing or reducing host DNA to enrich pathogenic microbial DNA may lead to the loss or bias of pathogenic microbial DNA in the sample, thereby affecting subsequent genomic analysis results [99].
Compared with second-generation sequencing technology, traditional nanopore sequencing has a relatively high error rate, which may lead to the misidentification or omission of certain microbial groups when dealing with regions of host and microbial DNA that are extremely similar [100]. However, the recent R10.4 chip developed by ONT enhances the recognition of homopolymeric compounds and can generate accurate microbial genomes without the need for short-read or reference genome correction.
Although nanopore sequencing faces some technical and data analysis challenges in dealing with host DNA removal in metagenomic samples, the latest technological advancements, especially the emergence of the R10.4 chip, provide new possibilities for solving these problems [24].
Mu et al. [101] collected BALF and sputum specimens from hospitalized patients with suspected lower respiratory tract infections and performed metagenomic tests using nanopore sequencing and confirmed the results using qPCR and Sanger sequencing. The turnaround time from sampling to obtaining results was approximately 6 h. In contrast, the turnaround time of conventional culture was approximately 94 h. Compared with conventional culture and real-time PCR diagnostic tests, rapid metagenomics achieved 96.6% sensitivity and 88.0% specificity. Among the five diseases caused by lower respiratory tract infections, the diagnostic accuracy was the highest in patients with community-acquired pneumonia, with 97.6% sensitivity and 90.2% specificity. The investigators successfully identified 63 pathogens in 161 culture-negative samples. Wang et al. [80] performed nanopore sequencing on culture-negative pulmonary tissue biopsy specimens from patients with severe pneumonia who had been treated with empirical antibiotics. K. pneumoniae was rapidly identified within 1 min through a specific sequence of 823 bp. In this study, the use of antibiotics prior to sample acquisition reduced the sensitivity of culture, whereas nanopore sequencing could still rapidly detect the pathogen with a small number of sequences. These studies demonstrate that nanopore sequencing-based metagenomic testing has advantages over conventional culture in terms of rapid turnaround time and sensitivity.
7 Comparison of Nanopore Sequencing with the BioFire System
The BioFire system is a product series from BioFire Diagnostics (Salt Lake City, USA), which includes the FilmArray Pneumonia Panel, FilmArray Respiratory Panel, and several other testing chips. This system is a rapid molecular diagnostic system based on PCR, equipped with pre-designed nested multiplex PCR kits that can perform simultaneous detection of up to 20 respiratory infection pathogens within 1 h [102].
The BioFire system also has its limitations. First, the kits of the BioFire system are pre-designed, mainly for the detection of specific pathogens or combinations of pathogens. Therefore, its target detection range is restricted and cannot cover all possible pathogens. Second, although the BioFire system can simultaneously detect multiple pathogens, its throughput is relatively low. Nanopore sequencing, on the contrary, has higher throughput and can handle a larger number of samples. Finally, the BioFire system focuses on the detection of specific gene fragments and cannot provide complete genomic information, whereas nanopore sequencing provides more comprehensive genomic sequencing and analysis, including the detection of unknown sequences and structural variants [63].
However, the higher sequencing error rate of nanopore sequencing technology might affect certain analyses that require high-precision sequences. In addition, nanopore sequencing data processing and analysis require certain bioinformatics knowledge. Compared with the BioFire system, the operation and data processing procedures of nanopore sequencing are more complex.
In summary, the BioFire system is more suited for rapid screening of known and common pathogens in clinical environments; whereas, nanopore sequencing is more appropriate for the identification of unknown or rare pathogens and in-depth and comprehensive genome analyses, such as drug-resistance detection and gene-expression analysis.
8 Summary and Vision
The exceptional read length of nanopore sequencing technology offers new possibilities for optimizing metagenomic sequencing and 16S rRNA targeted sequencing protocols. Particularly in dealing with the diagnosis and treatment of rare and emerging pulmonary infections, it provides clinicians with richer and more in-depth information. The relatively low startup cost and rapid turnaround time render it particularly suitable for the detection of pathogens in acute and severe pulmonary infections in clinical settings. In addition, its portability enables bedside testing and rapid detection in resource-limited environments, including in field investigations of infectious disease outbreaks.
However, nanopore sequencing technology also faces certain challenges. Although recent technological advancements, such as the optimization of algorithms and nanopore structures, and improvements in reagents, have significantly ameliorated the issue of high error rates that were present in its early iterations, it currently cannot entirely replace shorter-read technologies with higher accuracy. Appropriate diagnostic tools should be selected according to research needs.
Looking forward, nanopore sequencing technology has the potential to overcome current bottlenecks in molecular diagnostics. Combined with metagenomics, amplicon sequencing, PCR, and mass spectrometry, among other technologies, nanopore sequencing could jointly promote the development of diagnostic and therapeutic technologies for pulmonary infections.
References
Heather JM, Chain B. The sequence of sequencers: the history of sequencing DNA. Genomics. 2016;107:1–8. https://doi.org/10.1016/j.ygeno.2015.11.003.
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-time DNA sequencing from single polymerase molecules. Science. 2009;323:133–8. https://doi.org/10.1126/science.1162986.
Travers KJ, Chin CS, Rank DR, Eid JS, Turner SW. A flexible and efficient template format for circular consensus sequencing and SNP detection. Nucleic Acids Res. 2010;38: e159. https://doi.org/10.1093/nar/gkq543.
Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7:461–5. https://doi.org/10.1038/nmeth.1459.
van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30:418–26. https://doi.org/10.1016/j.tig.2014.07.001.
Mizuguchi T, Okamoto N, Yanagihara K, Miyatake S, Uchiyama Y, Tsuchida N, et al. Pathogenic 12-kb copy-neutral inversion in syndromic intellectual disability identified by high-fidelity long-read sequencing. Genomics. 2021;113(1 Pt 2):1044–53. https://doi.org/10.1016/j.ygeno.2020.10.038.
Vasan N, Razavi P, Johnson JL, Shao H, Shah H, Antoine A, et al. Double PIK3CA mutations in cis increase oncogenicity and sensitivity to PI3Kα inhibitors. Science. 2019;366:714–23. https://doi.org/10.1126/science.aaw9032.
Branton D, Deamer DW, Marziali A, Bayley H, Benner SA, Butler T, et al. The potential and challenges of nanopore sequencing. Nat Biotechnol. 2008;26:1146–53. https://doi.org/10.1038/nbt.1495.
Kasianowicz JJ, Brandin E, Branton D, Deamer DW. Characterization of individual polynucleotide molecules using a membrane channel. Proc Natl Acad Sci U S A. 1996;93:13770–3. https://doi.org/10.1073/pnas.93.24.13770.
Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol. 2009;4:265–70. https://doi.org/10.1038/nnano.2009.12.
Venkatesan BM, Bashir R. Nanopore sensors for nucleic acid analysis. Nat Nanotechnol. 2011;6:615–24. https://doi.org/10.1038/nnano.2011.129.
Manrao EA, Derrington IM, Laszlo AH, Langford KW, Hopper MK, Gillgren N, et al. Reading DNA at single-nucleotide resolution with a mutant MspA nanopore and phi29 DNA polymerase. Nat Biotechnol. 2012;30:349–53. https://doi.org/10.1038/nbt.2171.
Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Methods. 2015;12:351–6. https://doi.org/10.1038/nmeth.3290.
Gu W, Deng X, Lee M, Sucu YD, Arevalo S, Stryke D, et al. Rapid pathogen detection by metagenomic next-generation sequencing of infected body fluids. Nat Med. 2021;27:115–24. https://doi.org/10.1038/s41591-020-1105-z.
Feng Y, Zhang Y, Ying C, Wang D, Du C. Nanopore-based fourth-generation DNA sequencing technology. Genom Proteom Bioinform. 2015;13:4–16. https://doi.org/10.1016/j.gpb.2015.01.009.
Runtuwene LR, Tuda JSB, Mongan AE, Suzuki Y. On-site MinION sequencing. Adv Exp Med Biol. 2019;1129:143–50. https://doi.org/10.1007/978-981-13-6037-4_10.
Goordial J, Altshuler I, Hindson K, Chan-Yam K, Marcolefas E, Whyte LG. In situ field sequencing and life detection in remote (79°26’N) Canadian high arctic permafrost ice wedge microbial communities. Front Microbiol. 2017;8:2594. https://doi.org/10.3389/fmicb.2017.02594.
Walter MC, Zwirglmaier K, Vette P, Holowachuk SA, Stoecker K, Genzel GH, et al. MinION as part of a biomedical rapidly deployable laboratory. J Biotechnol. 2017;250:16–22. https://doi.org/10.1016/j.jbiotec.2016.12.006.
Castro-Wallace SL, Chiu CY, John KK, Stahl SE, Rubins KH, McIntyre ABR, et al. Nanopore DNA sequencing and genome assembly on the International Space Station. Sci Rep. 2017;7:18022. https://doi.org/10.1038/s41598-017-18364-0.
Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530:228–32. https://doi.org/10.1038/nature16996.
Sahlin K, Medvedev P. Error correction enables use of Oxford Nanopore technology for reference-free transcriptome analysis. Nat Commun. 2021;12:2. https://doi.org/10.1038/s41467-020-20340-8.
Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol. 2018;36:338–45. https://doi.org/10.1038/nbt.4060.
Fu S, Wang A, Au KF. A comparative evaluation of hybrid error correction methods for error-prone long reads. Genome Biol. 2019;20(1):26. https://doi.org/10.1186/s13059-018-1605-z.
Sereika M, Kirkegaard RH, Karst SM, Michaelsen TY, Sørensen EA, Wollenberg RD, et al. Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat Methods. 2022;19:823–6. https://doi.org/10.1038/s41592-022-01539-7.
Wagner GE, Dabernig-Heinz J, Lipp M, Cabal A, Simantzik J, Kohl M, et al. Real-time nanopore Q20+ sequencing enables extremely fast and accurate core genome MLST typing and democratizes access to high-resolution bacterial pathogen surveillance. J Clin Microbiol. 2023;61: e0163122. https://doi.org/10.1128/jcm.01631-22.
Pfeiffer F, Gröber C, Blank M, Händler K, Beyer M, Schultze JL, et al. Systematic evaluation of error rates and causes in short samples in next-generation sequencing. Sci Rep. 2018;8:10950. https://doi.org/10.1038/s41598-018-29325-6.
Chidgeavadze ZG, Beabealashvilli RS, Atrazhev AM, Kukhanova MK, Azhayev AV, Krayevsky AA. 2’,3’-Dideoxy-3’ aminonucleoside 5’-triphosphates are the terminators of DNA synthesis catalyzed by DNA polymerases. Nucleic Acids Res. 1984;12:1671–86. https://doi.org/10.1093/nar/12.3.1671.
Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. https://doi.org/10.1038/nbt1486.
Metzker ML. Sequencing technologies—the next generation. Nat Rev Genet. 2010;11:31–46. https://doi.org/10.1038/nrg2626.
Tucker T, Marra M, Friedman JM. Massively parallel sequencing: the next big thing in genetic medicine. Am J Hum Genet. 2009;85:142–54. https://doi.org/10.1016/j.ajhg.2009.06.022.
Cherf GM, Lieberman KR, Rashid H, Lam CE, Karplus K, Akeson M. Automated forward and reverse ratcheting of DNA in a nanopore at 5-Å precision. Nat Biotechnol. 2012;30:344–8. https://doi.org/10.1038/nbt.2147.
Chen P, Sun Z, Wang J, Liu X, Bai Y, Chen J, et al. Portable nanopore-sequencing technology: trends in development and applications. Front Microbiol. 2023;14:1043967. https://doi.org/10.3389/fmicb.2023.1043967.
De Coster W, Rademakers R. NanoPack2: Population scale evaluation of long-read sequencing data. Bioinformatics. 2023;39(5):btad311. https://doi.org/10.1093/bioinformatics/btad311.
Wu YW, Simmons BA, Singer SW. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics. 2016;32:605–7. https://doi.org/10.1093/bioinformatics/btv638.
Li H. New strategies to improve minimap2 alignment accuracy. Bioinformatics. 2021;37:4572–4. https://doi.org/10.1093/bioinformatics/btab705.
Sedlazeck FJ, Rescheneder P, Smolka M, Fang H, Nattestad M, von Haeseler A, et al. Accurate detection of complex structural variations using single-molecule sequencing. Nat Methods. 2018;15:461–8. https://doi.org/10.1038/s41592-018-0001-7.
Wick RR, Judd LM, Gorrie CL, Holt KE. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 2017;13(6): e1005595. https://doi.org/10.1371/journal.pcbi.1005595.
Wong J, Coombe L, Nikolić V, Zhang E, Nip KM, Sidhu P, Warren RL, Birol I. Linear time complexity de novo long read genome assembly with GoldRush. Nat Commun. 2023;14:2906. https://doi.org/10.1038/s41467-023-38716-x.
Shafin K, Pesout T, Lorig-Roach R, Haukness M, Olsen HE, Bosworth C, et al. Nanopore sequencing and the Shasta toolkit enable efficient de novo assembly of eleven human genomes. Nat Biotechnol. 2020;38:1044–53. https://doi.org/10.1038/s41587-020-0503-6.
Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–9. https://doi.org/10.1093/bioinformatics/btu153.
Feng Y, Zou S, Chen H, Yu Y, Ruan Z. BacWGSTdb 2.0: a one-stop repository for bacterial whole-genome sequence typing and source tracking. Nucleic Acids Res. 2021;49(D1):D644–50. https://doi.org/10.1093/nar/gkaa821.
Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, et al. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ. 2019;7: e7359. https://doi.org/10.7717/peerj.7359.
Huang F, Xiao L, Gao M, Vallely EJ, Dybvig K, Atkinson TP, Waites KB, Chong Z. B-Assembler: a circular bacterial genome assembler. BMC Genom. 2022;23(Suppl 4):361. https://doi.org/10.1186/s12864-022-08577-7.
Liu Y-X, Qin Y, Chen T, Lu M, Qian X, Guo X, Bai Y. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell. 2021;12(5):315–30. https://doi.org/10.1007/s13238-020-00724-8.
Langelier C, Kalantar KL, Moazed F, Wilson MR, Crawford ED, Deiss T, et al. Integrating host response and unbiased microbe detection for lower respiratory tract infection diagnosis in critically ill adults. Proc Natl Acad Sci U S A. 2018;115:E12353–62. https://doi.org/10.1073/pnas.1809700115.
Diao Z, Han D, Zhang R, Li J. Metagenomics next-generation sequencing tests take the stage in the diagnosis of lower respiratory tract infections. J Adv Res. 2021;38:201–12. https://doi.org/10.1016/j.jare.2021.09.012.
Langelier C, Zinter MS, Kalantar K, Yanik GA, Christenson S, O’Donovan B, et al. Metagenomic sequencing detects respiratory pathogens in hematopoietic cellular transplant patients. Am J Respir Crit Care Med. 2018;197:524–8. https://doi.org/10.1164/rccm.201706-1097LE.
Subbarao K, Mahanty S. Respiratory virus infections: understanding COVID-19. Immunity. 2020;52:905–9. https://doi.org/10.1016/j.immuni.2020.05.004.
Marcos MA, Esperatti M, Torres A. Viral pneumonia. Curr Opin Infect Dis. 2009;22:143–7. https://doi.org/10.1097/QCO.0b013e328328cf65.
Sanjuán R, Domingo-Calap P. Mechanisms of viral mutation. Cell Mol Life Sci. 2016;73:4433–8. https://doi.org/10.1007/s00018-016-2299-6.
Keller MW, Rambo-Martin BL, Wilson MM, Ridenour CA, Shepard SS, Stark TJ, et al. Direct RNA sequencing of the coding complete influenza A virus genome. Sci Rep. 2018;8:14408. https://doi.org/10.1038/s41598-018-32615-8.
Mason S, Devincenzo JP, Toovey S, Wu JZ, Whitley RJ. Comparison of antiviral resistance across acute and chronic viral infections. Antivir Res. 2018;158:103–12. https://doi.org/10.1016/j.antiviral.2018.07.020.
Li J, Wang H, Mao L, Yu H, Yu X, Sun Z, et al. Rapid genomic characterization of SARS-CoV-2 viruses from clinical specimens using nanopore sequencing. Sci Rep. 2020;10:17492. https://doi.org/10.1038/s41598-020-74656-y.
Bull RA, Adikari TN, Ferguson JM, Hammond JM, Stevanovski I, Beukers AG, et al. Analytical validity of nanopore sequencing for rapid SARS-CoV-2 genome analysis. Nat Commun. 2020;11:6272. https://doi.org/10.1038/s41467-020-20075-6.
Muthuri SG, Venkatesan S, Myles PR, Leonardi-Bee J, Al Khuwaitir TS, Al Mamun A, et al. Effectiveness of neuraminidase inhibitors in reducing mortality in patients admitted to hospital with influenza A H1N1pdm09 virus infection: a meta-analysis of individual participant data. Lancet Respir Med. 2014;2:395–404. https://doi.org/10.1016/S2213-2600(14)70041-4.
Green DA, StGeorge K. Rapid antigen tests for influenza: rationale and significance of the FDA reclassification. J Clin Microbiol. 2018;56:e00711-e718. https://doi.org/10.1128/JCM.00711-18.
Uyeki TM, Bernstein HH, Bradley JS, Englund JA, File TM, Fry AM, et al. Clinical practice guidelines by the Infectious Diseases Society of America: 2018 update on diagnosis, treatment, chemoprophylaxis, and institutional outbreak management of seasonal influenza. Clin Infect Dis. 2019;68:895–902. https://doi.org/10.1093/cid/ciy874.
Wang Y, Kang H, Liu X, Tong Z. Combination of RT-qPCR testing and clinical features for diagnosis of COVID-19 facilitates management of SARS-CoV-2 outbreak. J Med Virol. 2020;92:538–9. https://doi.org/10.1002/jmv.25721.
Pater AA, Bosmeny MS, White AA, Sylvain RJ, Eddington SB, Parasrampuria M, et al. High throughput nanopore sequencing of SARS-CoV-2 viral genomes from patient samples. J Biol Methods. 2021;8(COVID 19 Spec Iss): e155. https://doi.org/10.14440/jbm.2021.360.
Wang M, Fu A, Hu B, Tong Y, Liu R, Liu Z, et al. Nanopore targeted sequencing for the accurate and comprehensive detection of SARS-CoV-2 and other respiratory viruses. Small. 2020;16: e2002169. https://doi.org/10.1002/smll.202002169.
Li P, Wang K, Qiu S, Lin Y, Xie J, Li J, et al. Rapid identification and metagenomics analysis of the adenovirus type 55 outbreak in Hubei using real-time and high-throughput sequencing platforms. Infect Genet Evol. 2021;93:104939. https://doi.org/10.1016/j.meegid.2021.104939.
Bouvier NM, Palese P. The biology of influenza viruses. Vaccine. 2008;26(Suppl 4):D49-53. https://doi.org/10.1016/j.vaccine.2008.07.039.
Xu Y, Lewandowski K, Downs LO, Kavanagh J, Hender T, Lumley S, et al. Nanopore metagenomic sequencing of influenza virus directly from respiratory samples: diagnosis, drug resistance and nosocomial transmission, United Kingdom, 2018/19 influenza season. Euro Surveill. 2021;26:2000004. https://doi.org/10.2807/1560-7917.ES.2021.26.27.2000004.
Woo PC, Lau SK, Teng JL, Tse H, Yuen KY. Then and now: use of 16S rDNA gene sequencing for bacterial identification and discovery of novel bacteria in clinical microbiology laboratories. Clin Microbiol Infect. 2008;14:908–34. https://doi.org/10.1111/j.1469-0691.2008.02070.x.
Church DL, Cerutti L, Gürtler A, Griener T, Zelazny A, Emler S. Performance and application of 16S rRNA gene cycle sequencing for routine identification of bacteria in the clinical microbiology laboratory. Clin Microbiol Rev. 2020;33:e00053-e119. https://doi.org/10.1128/CMR.00053-19.
Jeong J, Yun K, Mun S, Chung WH, Choi SY, Nam YD, et al. The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci Rep. 2021;11:1727. https://doi.org/10.1038/s41598-020-80826-9.
Miga KH, Koren S, Rhie A, Vollger MR, Gershman A, Bzikadze A, et al. Telomere-to-telomere assembly of a complete human X chromosome. Nature. 2020;585:79–84. https://doi.org/10.1038/s41586-020-2547-7.
Curry KD, Wang Q, Nute MG, Tyshaieva A, Reeves E, Soriano S, Wu Q, Graeber E, Finzer P, Mendling W, Savidge T, Villapol S, Dilthey A, Treangen TJ. Emu: Species-level microbial community profiling for full-length nanopore 16S reads. Nat Methods. 2022;19(7):845–53. https://doi.org/10.1038/s41592-022-01520-4.
Bartlett JG. Diagnostic tests for agents of community-acquired pneumonia. Clin Infect Dis. 2011;52 Suppl 4:S296-304. https://doi.org/10.1093/cid/cir045.
Moon J, Jang Y, Kim N, Park WB, Park KI, Lee ST, et al. Diagnosis of Haemophilus influenzae pneumonia by nanopore 16S amplicon sequencing of sputum. Emerg Infect Dis. 2018;24:1944–6. https://doi.org/10.3201/eid2410.180234.
Baldan R, Cliff PR, Burns S, Medina A, Smith GC, Batra R, et al. Development and evaluation of a nanopore 16S rRNA gene sequencing service for same day targeted treatment of bacterial respiratory infection in the intensive care unit. J Infect. 2021;83:167–74. https://doi.org/10.1016/j.jinf.2021.06.014.
Croxatto A, Prod’hom G, Greub G. Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. FEMS Microbiol Rev. 2012;36:380–407. https://doi.org/10.1111/j.1574-6976.2011.00298.x.
Angeletti S. Matrix assisted laser desorption time of flight mass spectrometry (MALDI-TOF MS) in clinical microbiology. J Microbiol Methods. 2017;138:20–9. https://doi.org/10.1016/j.mimet.2016.09.003.
Hou TY, Chiang-Ni C, Teng SH. Current status of MALDI-TOF mass spectrometry in clinical microbiology. J Food Drug Anal. 2019;27:404–14. https://doi.org/10.1016/j.jfda.2019.01.001.
Lao HY, Ng TT, Wong RY, Wong CS, Lee LK, Wong DS, et al. The clinical utility of two high-throughput 16S rRNA gene sequencing workflows for taxonomic assignment of unidentifiable bacterial pathogens in matrix-assisted laser desorption ionization-time of flight mass spectrometry. J Clin Microbiol. 2022;60: e0176921. https://doi.org/10.1128/JCM.01769-21.
Guo Y, Li L, Li Z, Sun L, Wang H. Tropheryma whipplei detection by nanopore sequencing in patients with interstitial lung disease. Front Microbiol. 2021;12:760696. https://doi.org/10.3389/fmicb.2021.760696.
Watanabe C, Kimizuka Y, Fujikura Y, Hamamoto T, Watanabe A, Yaguchi T, et al. Mixed Infection of cytomegalovirus and pulmonary nocardiosis caused by Nocardia elegans diagnosed using nanopore sequencing technology. Intern Med. 2022;61:1613–7. https://doi.org/10.2169/internalmedicine.7639-21.
Blair JM, Webber MA, Baylay AJ, Ogbolu DO, Piddock LJ. Molecular mechanisms of antibiotic resistance. Nat Rev Microbiol. 2015;13:42–51. https://doi.org/10.1038/nrmicro3380.
Liu C, Guo J, Lu M, Shen N, Du P. Dissemination of the mobilised RND efflux pump gene cluster tmexCD-toprJ among Klebsiella pneumoniae. Lancet Microbe. 2023;4: e135. https://doi.org/10.1016/S2666-5247(22)00325-1.
Wang K, Li P, Lin Y, Chen H, Yang L, Li J, et al. Metagenomic diagnosis for a culture-negative sample from a patient with severe pneumonia by nanopore and next-generation sequencing. Front Cell Infect Microbiol. 2020;10:182. https://doi.org/10.3389/fcimb.2020.00182.
Hendrix J, Epperson LE, Durbin D, Honda JR, Strong M. Intraspecies plasmid and genomic variation of Mycobacterium kubicae revealed by the complete genome sequences of two clinical isolates. Microb Genom. 2021;7:mgen000497. https://doi.org/10.1099/mgen.0.000497.
Kelly BT, Pennington KM, Limper AH. Advances in the diagnosis of fungal pneumonias. Expert Rev Respir Med. 2020;14:703–14. https://doi.org/10.1080/17476348.2020.1753506.
Schoch CL, Seifert KA, Huhndorf S, Robert V, Spouge JL, Levesque CA, Fungal Barcoding Consortium, et al. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc Natl Acad Sci U S A. 2012;109:6241–6. https://doi.org/10.1073/pnas.1117018109.
Cai DH, Wang J, Fang XL. Successful treatment of Talaromyces marneffei pneumonia in a HIV-negative renal transplantation recipient: A case report. Medicine (Baltimore). 2022;101: e30958. https://doi.org/10.1097/MD.0000000000030958.
Liu D, Ma L, Mao L, Wang Y, Ding L, Wang A. Identification of the first case of Penicillium marneffei infection in Shaanxi Province by nanopore sequencing. J Infect Dev Ctries. 2022;16:575–9. https://doi.org/10.3855/jidc.15348.
Catherinot E, Lanternier F, Bougnoux ME, Lecuit M, Couderc LJ, Lortholary O. Pneumocystis jirovecii pneumonia. Infect Dis Clin N Am. 2010;24:107–38. https://doi.org/10.1016/j.idc.2009.10.010.
Irinyi L, Hu Y, Hoang MTV, Pasic L, Halliday C, Jayawardena M, et al. Long-read sequencing based clinical metagenomics for the detection and confirmation of Pneumocystis jirovecii directly from clinical specimens: a paradigm shift in mycological diagnostics. Med Mycol. 2020;58:650–60. https://doi.org/10.1093/mmy/myz109.
Zhang J, Gao L, Zhu C, Jin J, Song C, Dong H, et al. Clinical value of metagenomic next-generation sequencing by Illumina and Nanopore for the detection of pathogens in bronchoalveolar lavage fluid in suspected community-acquired pneumonia patients. Front Cell Infect Microbiol. 2022;12:1021320. https://doi.org/10.3389/fcimb.2022.1021320.
Daley CL. The global fight against tuberculosis. Thorac Surg Clin. 2019;29:19–25. https://doi.org/10.1016/j.thorsurg.2018.09.010.
Libonati JP, Stager CE, Davis JR, Siddiqi SH. Direct antimicrobial drug susceptibility testing of Mycobacterium tuberculosis by the radiometric method. Diagn Microbiol Infect Dis. 1988;10:41–8. https://doi.org/10.1016/0732-8893(88)90125-3.
Zhao K, Tu C, Chen W, Liang H, Zhang W, Wang Y, et al. Rapid identification of drug-resistant tuberculosis genes using direct PCR amplification and Oxford Nanopore Technology sequencing. Can J Infect Dis Med Microbiol. 2022;2022:7588033. https://doi.org/10.1155/2022/7588033.
Smith C, Halse TA, Shea J, Modestil H, Fowler RC, Musser KA, et al. Assessing nanopore sequencing for clinical diagnostics: A comparison of next-generation sequencing (NGS) methods for Mycobacterium tuberculosis. J Clin Microbiol. 2020;59:e00583-e620. https://doi.org/10.1128/JCM.00583-20.
Sharda SC, Bisht K, Sharma V, Lakku PR, Bhatia MS, Sharma N, et al. Direct diagnosis of scrub typhus by full-length 16S ribosomal RNA gene analysis using Oxford Nanopore sequencing. Int J Infect Dis. 2022;125:132–4. https://doi.org/10.1016/j.ijid.2022.10.040.
Gu W, Miller S, Chiu CY. Clinical metagenomic next-generation sequencing for pathogen detection. Annu Rev Pathol. 2019;14:319–38. https://doi.org/10.1146/annurev-pathmechdis-012418-012751.
Tyson JR, O’Neil NJ, Jain M, Olsen HE, Hieter P, Snutch TP. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018;28:266–74. https://doi.org/10.1101/gr.221184.117.
Pogka V, Papadopoulou G, Valiakou V, Sgouras DN, Mentis AF, Karamitros T. Targeted virome sequencing enhances unbiased detection and genome assembly of known and emerging viruses—the example of SARS-CoV-2. Viruses. 2022;14:1272. https://doi.org/10.3390/v14061272.
Feehery GR, Yigit E, Oyola SO, Langhorst BW, Schmidt VT, Stewart FJ, et al. A method for selectively enriching microbial DNA from contaminating vertebrate host DNA. PLoS ONE. 2013;8: e76096. https://doi.org/10.1371/journal.pone.0076096.
Hasan MR, Rawat A, Tang P, Jithesh PV, Thomas E, Tan R, et al. Depletion of human DNA in spiked clinical specimens for improvement of sensitivity of pathogen detection by next-generation sequencing. J Clin Microbiol. 2016;54:919–27. https://doi.org/10.1128/JCM.03050-15.
Charalampous T, Kay GL, Richardson H, Aydin A, Baldan R, Jeanes C, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection. Nat Biotechnol. 2019;37:783–92. https://doi.org/10.1038/s41587-019-0156-5.
Senol Cali D, Kim JS, Ghose S, Alkan C, Mutlu O. Nanopore sequencing technology and tools for genome assembly: computational analysis of the current state, bottlenecks and future directions. Brief Bioinform. 2019;20:1542–59. https://doi.org/10.1093/bib/bby017.
Mu S, Hu L, Zhang Y, Liu Y, Cui X, Zou X, et al. Prospective evaluation of a rapid clinical metagenomics test for bacterial pneumonia. Front Cell Infect Microbiol. 2021;11:684965. https://doi.org/10.3389/fcimb.2021.684965.
Rhoads DD, Pournaras S, Leber A, Balada-Llasat JM, Harrington A, Sambri V, et al. Multicenter evaluation of the BIOFIRE Blood Culture Identification 2 panel for detection of bacteria, yeasts, and antimicrobial resistance genes in positive blood culture samples. J Clin Microbiol. 2023. https://doi.org/10.1128/jcm.01891-22.
Acknowledgements
We would like to thank Editage (www.editage.cn) for English language editing. This review did not receive any external funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were received for the preparation of this article.
Conflicts of Interest/Competing Interests
The authors FX and JC have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Code Availability
Not applicable.
Author Contributions
FX created the concept. JC performed the literature search and drafted the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Chen, J., Xu, F. Application of Nanopore Sequencing in the Diagnosis and Treatment of Pulmonary Infections. Mol Diagn Ther 27, 685–701 (2023). https://doi.org/10.1007/s40291-023-00669-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-023-00669-8