
Abstract
Background
Before studying gene expression of different organisms, it is important to determine the best reference gene. At present, the most accurate method of detecting gene expression is quantitative real-time PCR (RT-qPCR). With this method, reference genes that are stable in different biological systems and under different conditions can be obtained. Toona ciliata Roem (T. ciliata). is a valuable and fast-growing timber specie. In this study, 20 reference genes were identified using RT-qPCR, as a primary prerequisite for future gene expression analysis. Four different methods, geNorm, NormFinder, BestKeeper, and RankAggreg were used to evaluate the expression stability of the 20 candidate reference genes in various tissues under different conditions.
Results
The experimental results showed that TUB-α was the most stably expressed reference gene across all samples and UBC17 was the most stable in leaves and young stems under Hypsipyla robusta (H. robusta) and methyl jasmonate (MeJA) treatments. In addition, PP2C59 and UBC5B were the best-performing genes in leaves under H. robusta treatment, while HIS1 and ACT7 were the best reference genes in young stems. The two best reference genes were 60S-18 and TUB-α after treatment at 4 °C. The expression of HIS6 and MUB1 was the most stable under PEG6000 treatment. The accuracy of the selected reference genes was verified using the transcription factor MYB3 (TcMYB3) gene.
Conclusions
This is the first report to verify the best reference genes for normalizing gene expression in T. ciliata under different conditions, which will facilitate future elucidation of gene regulations in this species.
Similar content being viewed by others

Background
Toona ciliata Roem. belongs to the Meliaceae family, which is widely distributed in China, Australia, and India. Because of its straight trunks and russet wood, T. ciliata has the title of “Chinese Mahogany” [1]. But its population has declines sharply in the past century due to environmental degradation and destruction by humans, and it has been listed as one of the “National Class II Key Protected Endangered Plants” in China. T. ciliata has great economic value, for example, its wood is often used to produce high-end furniture, instruments and crafts [2]. More importantly, it is also a medicinal plant as a result of the rich chemical substances in its roots, stems and leaves [3]. Compounds that have been isolated from T. ciliata include ketones, steroids, and coumarins, many of which have antifungal, anti-glycation, or anti-tumor activities [4,5,6,7], and its flower extract has a therapeutic effect on gastric ulcers [8]. However, the yield of compounds isolated from T. ciliata is low. In addition, in previous research, it has been found that T. ciliata is very susceptible to the moth pest Hypsipyla robusta Moore [9] that eats mainly the young stems and causes the hollow branches to fail to grow and die in some cases. This pest is not only a regional issue in China, but also a worldwide problem. In some of the main areas where H. robusta is distributed, such as Australia and Brazil, T. ciliata also faces serious damage from H. robusta [10,11,12]. At present, there are no chemical or physical methods to prevent or control H. robusta effectively, and current pest control methods are time- and labor- consuming, thus not applicable to large-scale forest plantations [13]. It may help to pest control by obtaining insect-resistant plants through molecular breeding. In order to synthesize a desired compound related to the resistance mechanism, it is necessary to first explore the pathway and its related regulatory genes [14, 15]. Gene expression analysis is one of the most powerful tools to explore biosynthetic and insect-resistance mechanisms in T. ciliata. So far, the knowledge base ICG (http://icg.big.ac.cn) has collected reference genes from more than 120 plant species including Arabidopsis [16], peanut [17], cucumber [18], and soybean [19], except T. ciliata. Nor are there any literature references about the housekeeping genes in T. ciliata, which can be used for the standardization of gene expression.
RT-qPCR has good repeatability, high sensitivity, accurate quantification, and fast reaction, making it a powerful tool to carry out the entire PCR process and the most commonly used method of determining gene expression levels [20]. However, RT-qPCR can be affected by multiple sources of error, such as the amount of starting materials, the integrity of the RNA, and the efficiency of the enzymatic reactions. It is therefore necessary to introduce a stably expressed housekeeping gene as a reference for correction and standardization, so as to control the unnecessary errors generated within and between samples [21].
The commonly used housekeeping genes are those consistently express under all conditions, such as genes encoding actin (ACT), glyceraldehyde-3 phosphate dehydrogenase (GAPDH), and tubulin (TUB) [22]. However, more and more studies are now questioning the existence of genes that are stably expressed across different tissues, different experimental conditions, and different species. In order to ensure the accuracy of an experiment, it is important to select those suitable reference genes for specific experimental conditions [23]. Software packages, including geNorm [24], NormFinder [25], and BestKeeper [26], are widely used to assess the expression stability of candidate reference genes and determine the best choices. Many researchers have used these algorithms to successfully identify reference genes in different species [27, 28]. The use of reference genes in expression analysis has greatly facilitated research in plant development and evolutionary mechanisms in species where a reference genome sequence is available [29].
In this study, 20 candidate genes from T. ciliata transcriptome database generated by our group were investigated to determine the most suitable T. ciliata candidate gene(s) as the reference(s) for gene expression analysis using RT-qPCR technique under specific conditions including different tissues (mature leaves, young leaves, flowers, shoots and young stems) and treatments (4 °C, MeJA, PEG6000 and H. robusta), including actin 7 (ACT7), phosphoglycerate kinase (PGK), 60S ribosomal protein L13 (60S-13) and L18 (60S-18), histone deacetylase 1 (HIS1) and 6 (HIS6), protein phosphatase 2 C57 (PP2C57) and C59 (PP2C59), ubiquitin-conjugating enzyme E2 5B (UBC5B) and 17 (UBC17), S-adenosylmethionine decarboxylase proenzyme (SAMDC), elongation factor 1 (EF1) and 2 (EF2), peptidyl-prolyl cis-trans isomerase CYP95 (PPIA95) and CYP26–2 (PPIA26), 18S rRNA (18S), tubulin alpha-3 chain (TUB-α), tubulin beta-5 chain (TUB-β), membrane-anchored ubiquitin-fold protein 1 (MUB1), and TIP41-like protein (TIP41). In addition, the TcMYB3 gene was used to confirm the reliability and validity of the reference genes screened. MYB proteins, which constitute one of the largest family of transcription factors in plants, play important roles in plant growth and development, biotic and abiotic stress responses, and circadian rhythm regulation [30, 31]. For example, the R2R3 MYB transcription factor MdMYB30 modulates plant resistance against pathogens, and Arabidopsis transcription factor MYB102 increases plant susceptibility to aphids [32, 33]. Our research provided the best reference genes for RT-qPCR analysis of T. ciliata under different conditions, laying a foundation for studying molecular mechanisms in T. ciliata through gene expression analysis.
Results
Primer specificity, amplification efficiency, and expression profiles of candidate reference genes
Reverse-transcribed cDNA from each sample was used as a template with primers for standard PCR amplification. Electrophoresis verified all PCR products were specific with single bands in the gel (Fig. S1). The melting profiles of all amplified candidate reference genes using RT-qPCR showed single peaks (Fig. S2). A standard curve for each candidate was obtained by serial dilution, and their linear correlation coefficients were all greater than 0.99 (R2 > 0.99). The amplification efficiency for the 20 candidate reference genes ranged from 90.41% for PPIA95 to 102.44% for PGK. Further details of primers are given in Table 1.
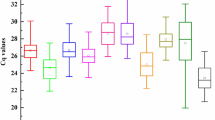
The expression levels of all candidate reference genes were determined by RT-qPCR under all of the following conditions: different tissues, H. robusta treatment, 4 °C treatment, MeJA treatment, and PEG6000 treatment. The expression levels of candidate genes were very different across the samples. The maximum cycle threshold (CT) value was 31.66, and the minimum was 13.18 (Fig. 1). Among them, PPIA26 showed the highest expression abundance, with the maximum, minimum, and median of the CT values being 31.66, 20.07, and 23.36, respectively. EF1 showed the lowest expression abundance, with the maximum, minimum, and median CT values being 22.16, 13.18, and 17.17, respectively. In addition, candidate genes exhibited significant variability in expression. MUB1 and UBC5B had a relatively narrow range of CT values compared with other genes, indicating that they are more stably expressed. Notably, these results show that none of the genes are expressed stably across all conditions, so it is necessary to screen reference genes for T. ciliata under specific conditions.

Distribution of threshold cycle (CT) values for 20 candidate reference genes across all samples. The middle line within each box represents the 50th percentile. The lower boundary and upper boundary of each box represent the 25th and 75th percentile respectively
Stability of expression of candidate reference genes
The software packages geNorm, NormFinder, and BestKeeper were used to evaluate the expression stability of the 20 candidate reference genes under different experimental conditions. The R software RankAggreg package was used for overall ranking [34].
GeNorm analysis
In geNorm analysis, M value is calculated for each pair of genes. The stability of gene expression is evaluated based on the M value; the genes with threshold M value below 1.5 are considered as stably expressed, and the gene with the lowest M value is regarded as the most stably expressed reference gene. The results of geNorm analysis of 20 candidate reference genes under different conditions are shown in Fig. 2a-h. The M values of all candidate genes from all the tested samples were below 1.5 (Fig. 3). Under H. robusta treatment, UBC17, PP2C59, and UBC5B were most stably expressed in leaves (Fig. 2a), while HIS1, UBC5B, and ACT7 exhibited few expression fluctuations in young stems (Fig. 2b). Data Analyses from the two tissues under H. robusta treatment showed that UBC5B, HIS1, and ACT7 were with the most stable expression as their M values are the lowest (Fig. 2c). The most stably expressed genes across different tissues were 18S and TUB-α, with M values around 0.2 (Fig. 2d). PPIA95 showed good stability under both 4 °C and MeJA treatments; 60S-18 and UBC17 were stably expressed only under 4 °C and MeJA treatment, respectively (Fig. 2e and f). The two genes with the lowest M values under drought stress which was simulated by PEG6000 treatment were PP2C57 and EF1 (Fig. 2g). And EF2 and EF1 had the highest stability with M value of 0.49 (Fig. 2h).

Average expression stability values (M) for 20 candidate reference genes calculated by geNorm

Pairwise variations (V) for the 20 candidate reference genes calculated by geNorm to determine the optimal number of reference genes for accurate normalization. The threshold used was 0.15
In general, it is more reliable to use multiple reference genes are more reliable than a single reference gene for quantitative gene analysis. Given this, geNorm calculates the pairwise variation (Vn/n + 1) of the normalization factor after the introduction of a new reference gene, and determines the optimal number of reference genes based on this ratio. The default Vn/n + 1 value for the software is 0.15. If the ratio is less than 0.15, the number of internal gene combinations that can meet the requirements for relative quantification is n, otherwise another reference gene needs to be introduced. In our study, the values of pairwise variation V2/3 under conditions with H. robusta treatment, 4 °C treatment, MeJA treatment, PEG6000 treatment, and different tissues, were all less than 0.15, indicating that the optimal number of reference gene combinations is two (Fig. 3). Across all samples, pairwise variation (V2/3) was 0.180, V3/4 was 0.15, and V4/5 was 0.126, indicating that the addition of the third and fourth reference genes has different impacts on the results. It always better to use fewer reference gene due to the time and cost economy consideration, hence the best reference gene combination was EF2, EF1, ACT7 and UBC5B for all the samples.
NormFinder analysis
In order to further determine the stability of candidate reference genes, NormFinder was used to re-analyze the data. The results are shown in Fig. 4a-h. Under H. robusta treatment, the top three genes with stable expression in leaves were TUB-α (stability value =0.038), HIS1 (0.105), and PP2C59 (0.161) (Fig. 4a), while the most stable genes were ACT7 (0.042), UBC5B (0.042), and TIP41 (0.109) in young stems (Fig. 4b). The top three reference candidates in two tissues (leaves and stems) were TUB-α (0.170), UBC5B (0.206), and PPIA95 (0.250) (Fig. 4c). The genes 18S (0.088) and TUB-α (0.112) had lower stability values across different tissues since they showed the most stable expression, which is consistent with the results of geNorm analysis (Fig. 4d). However, ACT7 (0.014), TUB-α (0.082), and PGK (0.099) were the most stable candidate genes under 4 °C treatment (Fig. 4e), which is inconsistent with the results of geNorm analysis; this may be due to the fact that the two software packages use different algorithms. Under MeJA treatment, the two most stable reference genes were 18S (0.078) and UBC17 (0.100) (Fig. 4f), while under PEG6000 treatment, the two most stable were 18S (0.055) and HIS6 (0.082) (Fig. 4g). TUB-α (0.281), 18S (0.316), and PGK (0.335) were the three genes with the lowest stability values for all samples (Fig. 4h). It is not consistent with geNorm analysis which showed that PPIA26, PP2C59 and HIS6 were the most unstable genes.

Expression stability of the 20 candidate reference genes as calculated by NormFinder
BestKeeper analysis
BestKeeper takes as input the CT data for each gene-primer pair combination and calculates the coefficient of variation (CV) and standard deviation (SD), as shown in Table 2. The stability of genes is evaluated by the value of CV ± SD. More stable genes have a lower value of CV ± SD. UBC17 (1.16 ± 0.23) and 18S (1.62 ± 0.36) were the most stable genes in leaves under H. robusta treatment, and the expression of HIS1 (1.40 ± 0.31) and UBC17 (1.70 ± 0.33) were the most stable in young stems. Genes with the most stable expression across two tissues were UBC17 (1.73 ± 0.34) and 18S (2.12 ± 0.47), as was the case in leaves. HIS6 (5.41 ± 1.38) and MUB1 (5.48 ± 1.16) were the most stably expressed genes in the different tissues, 18S (0.73 ± 0.16) and 60S-18 (0.83 ± 0.18) in 4 °C treatment, 60S-18 (0.67 ± 0.14) and EF1 (0.83 ± 0.14) in MeJA treatment, PPIA26 (3.89 ± 0.87) and 60S-18 (4.48 ± 1.02) in PEG6000 treatment. For all samples, MUB1 (3.95 ± 0.87) showed the highest value for expression stability.
RankAggreg analysis
In this study, three algorithms were used to analyze the expression stability of 20 candidate reference genes. The gene ranking tables generated by them are different because of their different algorithms. RankAggreg is an algorithm designed to aggregate large ranking lists. It performs aggregation of ordered lists based on the rankings via the Cross-Entropy Monte Carlo algorithm or a Genetic Algorithm [34]. To provide a consensus ranking, we used RankAggreg to calculate the overall gene ranking for each experimental condition, as shown in Table 3. The consensus for the top two genes in H. robusta treatment on leaves and under MeJA treatment was consistent with the results of geNorm analysis. HIS1 ranked first for young stem tissue under H. robusta treatment. The first-placed genes were 60S-18 and HIS6 under 4 °C treatment and PEG6000 treatment, respectively. TUB-α was the most stable gene in different tissues and all samples. The expression of PPIA26 was the most unstable under all experimental conditions except PEG6000 treatment.
Validation of reference genes
In order to verify the expression stability of the selected reference genes by the software, expression of the TcMYB3 gene was quantified using either the two most stable genes, alone and in combination, or the two most unstable genes in the consensus ranking. Under H. robusta treatment, the relative expression of TcMYB3 in leaves and young stems reached a peak at 12 h when the most stable genes and their combinations were used for standardized. But the relative expression of TcMYB3 was abnormally increased when standardized with the most unstable genes (Fig. 5a, b). As shown in Fig. 5c, when using the most stable genes as the reference genes, the expression level of TcMYB3 increased 1–1.5 times in young leaves compared with the expression level in mature leaves and the expression of other tissues (shoots, young stems, roots, and flowers) was down-regulated and the depression multiple was basically the same. But the expression level of TcMYB3 in the flower was the highest using the most unstable gene (PPIA26) as the reference gene. In addition, the expression level and trends were very similar when the most stable two reference genes and their combination were used for relative quantification under other stresses, including 4 °C treatment (Fig. 5d), MeJA treatment (Fig. 5e), and PEG6000 treatment (Fig. 5f). Whereas neither the expression level nor trend was consistent when the two most unstable internal reference genes were used for relative quantification. It is evident that the use of unstable references for gene expression analysis in T. ciliata can result in biased results.

Relative expression of TcMYB3 using the selected reference genes. The results were normalized using the selected stable reference genes (alone or in combination) and the unstable genes in sample sets across treatment with a H. robusta treatment in leaves, b H. robusta treatment in young stems, c different tissues, d 4 °C treatment, e MeJA treatment, f PEG6000 treatment. The bars indicate the standard error (±SE) evaluated from three biological replicates
Discussion
It is ideal that reference genes are stably expressed under all experimental conditions and show stable expression levels across various tissues and growth stages of the organism, but such genes are almost non-existent [35]. More and more studies are showing that the genes that are stably expressed in different species and under different conditions change [36,37,38,39]. Selection of the most suitable reference gene for specific conditions using RT-PCR is therefore very important. This study was dedicated to discovering the best reference genes for gene expression analysis in T. ciliata under different conditions. There were 20 candidate genes from the T. ciliata transcriptome database screened and analyzed by RT-qPCR. It was found that the best reference genes were not consistent across different conditions. For examples, PP2C59 and UBC5B were most suitable for leaves under H. robusta treatment, whereas HIS1 and ACT7 were more optimal for young stems under H. robusta treatment, TUB-α and PPIA95 for comparing different tissues, and 60S-18 and TUB-α for leaves under 4 °C treatment.
In this study, we used four methods, geNorm, NormFinder, BestKeeper, and RankAggreg, to evaluate the expression stability of 20 candidate genes. The first three algorithms were used to evaluate the expression stability of candidate genes. Our results demonstrated that reference values and calculation methods used by the three algorithms were very different [40]. NormFinder calculates stability values based on intra- and inter-group differences [25], while geNorm compares a reference gene with all genes in a given sample to evaluate the best reference gene [24]. In BestKeeper, CV and SD values determine the ranking of stability of candidate genes [41]. Due to the difference of the algorithms among the three software packages, they generated different rankings for the same set of experimental data, although the results of analysis with geNorm and NormFinder had few variations in this study. For example, 18S was the best reference gene to use across different tissue conditions according to geNorm and NormFinder, whereas the best reference gene was HIS6 identified by BestKeeper analysis. For young stem tissue exposed to H. robusta stress, ACT7 and UBC5B were put forward by geNorm and NormFinder, but ranked low in BestKeeper results. In order to consolidate the results from the three algorithms, RankAggreg was used for overall ranking [34]. Many researchers use ReFinder to calculate the final ranking [42,43,44]. ReFinder assigns an appropriate weight to each gene and calculates the geometric mean of its weights to give the final ranking [45]. RankAggreg uses a cross-entropy Monte Carlo algorithm or genetic algorithm to produce aggregated ordered lists based on rankings [34]. Both tools play very important roles in the consolidation of the screening results for internal reference genes from other softwares.
Other researchers have studied the best reference genes for plants under pest stress, and STP4 was found to be the best reference gene for use in Brassica juncea under biotic stress caused by aphid infestation [46]. ABCT and FBOX were found to be the most stable in soybean under soybean aphid (SBA) stress; TUB4 and TUA4 were stable under two-spotted spider mite (TSSM) stress [47]. Miranda indicated that both GmELF1A and GmTUA5 were stable reference genes for normalization of expression data obtained from soybean roots infected with Meloidogyne incognita, and GmCYP2 and GmELF1A were the best reference genes in soybean leaves infested by Anticarsia gemmatalis [48]. Under H. robusta stress, the reference genes that performed best in leaves and young stem tissues were different in our study. PP2C59 and UBC5B showed high stability of expression in leaves, while only PP2C59 ranked high for young stems. Once again, the appropriate reference genes for different species under different conditions and in different tissues vary. Hence, it is necessary to identify the best reference genes for specific conditions via RT-qPCR. Protein phosphatase can reverse the phosphorylation of protein kinases, thereby dynamically controlling protein phosphorylation and protein phosphatase 2Cs (PP2Cs) is the most abundant type of phosphatase in plants [49]. Although it not often has been used as a candidate internal reference gene, it is stable in our study under pest stress and in different tissues for GA treatment of Santalum album [50]. Therefore, when screening reference genes in other species, PP2Cs can be considered as a candidate reference gene.
Under most experimental conditions in this study (all except for PEG6000 treatment), the reference gene with the worst performance was PPIA26, which was the best reference gene recommended by BestKeeper for use under PEG6000 stress. Another gene in this family, PPIA95, ranked first in geNorm analysis for both 4 °C cold stress and MeJA treatment. For leaves and young stems under H. robusta treatment and MeJA stress, PPIA95 ranked third in NormFinder analysis. In BestKeeper analysis, PPIA95 ranked third under 4 °C cold stress and PEG6000-induced drought stress, and among all samples it ranked second. Overall, the PPIA gene family is a promising reference gene set in T. ciliata. The PPIA gene family encodes proteins with functions in immune responses, as well as resistance to cancer, autoimmune diseases, protozoan, and viral infections [51]. In plants, genes of the PPIA family are rarely used as internal reference genes, but they are abundantly expressed in the T. ciliata transcriptome data and the expression level in each sample is very similar, which is the main reason for choosing them. As reference genes, they are also stably expressed in animals. For example, in different heart and disease conditions, PPIA is recognized by ReFinder as the best reference gene in different skeletal muscles of mice, and it ranked first for human endometrial cancer [52, 53]. PPIB is believed as the optimal reference gene in analyzing the blood of Machado-Joseph disease (MJD) patients [54].
Conclusion
This study is the first report about screening and verification of expression stability analysis of a series of reference genes under different conditions in T. ciliata, showing that the optimal reference genes were TUB-α and PGK across all samples; PP2C59 and UBC5B in leaves and HIS1 and ACT7 in young stems under H. robusta treatment; TUB-α and PPIA95 in different tissues; 60S-18 and TUB-α under 4 °C treatment; UBC17 and PPIA95 under MeJA treatment; HIS1 and MUB1 under PEG6000 treatment, respectively. We believe this research is important for accurate quantification and expression analysis of genes under different conditions in T. ciliata. It will play a vital role in the molecular breeding work of T. ciliata, such as the research on the H. robusta-resistant and drought-resistant varieties, as well as the research on the metabolic pathways of precious compounds in plants in the future.
Methods
Plant materials
Five different experiments were conducted for data collection (Table 4). Experimental samples were all collected from one-year old T. ciliata, grown in a greenhouse in South China Agricultural University (SCAU). For samples from different tissues, mature leaves, young leaves, flowers, shoots, and young stems were collected at 9:00 am on August 25, 2019. Before treatment with the H. robusta, PEG6000, 4 °C, and MeJA, all the seedlings were pre-incubated in incubator for 7 days with 16 h of light at 28 °C and 8 h of dark at 22 °C to mimic the wild environment. For H. robusta treatment, seedlings were exposed to herbivores, and leaves and young stems were harvested after 0, 6, 12, 24, and 36 h. After seedlings were treated with 0, 5, 10, 20, and 30% (w/v) of PEG6000 for 7 days, leaves were collected. For 4 °C treatment, seedlings were placed at 4 °C, and samples (leaves) were taken at 0, 6, 12, 24, and 36 h. Seedlings sprayed with MeJA (100 μM) were sealed in plastic bags and leaves were collected at 0, 6, 12, 24, and 36 h. Three biological replicates were taken for each sampling point, and all samples were immediately frozen in liquid nitrogen and stored at − 80 °C.
RNA extraction, quality assessment, and DNA synthesis
Total RNA was extracted from all samples using a HiPure HP Plant RNA Mini Kit (Magen) with DNase treatment to remove genomic DNA. The quality of the RNA was determined with NanoDrop ND1000 (Thermo Scientific). RNA samples with absorbance ratios of A260/A280 and A260/A230 both around 2.0 were selected for further analysis. To synthesize cDNA, 0.5 μg of total RNA was used according to the manufacturer’s instructions for the HiScript II Reverse Transcript kit (Vazyme). Five-fold diluted cDNA was used for subsequent RT-qPCR experiments.
Selection of candidate reference genes and primer design
Twenty candidate reference genes were selected from the T. ciliata leaf transcriptome database by reviewing previous literature: PGK, 60S-18, 60S-13, HIS1, HIS6, PP2C57, PP2C59, UBC5B, UBC17, ACT7, SAMDC, EF1, EF2, 18S, TUB-α, TUB-β, MUB1, PPIA26, PPIA95, TIP41. Since there is no genomic sequence data available for T. ciliata, we designed primers based on the sequences in the T. ciliata transcriptome database. Firstly, the Coding Sequence (CDS) and genomic DNA sequences (gDNA) of the candidate reference genes were amplified separately (Table S1), and then the introns and exons of the candidate reference genes were obtained by sequence alignment (NCBI-Blast), and finally primers for RT-qPCR analysis of each reference gene were designed using the web-based Primer-Blast tool from NCBI (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). Apart from 60S-18, SAMDC, TUB-β and PPIA26, RT-qPCR primers were designed across introns. Details of these genes and primers are shown in Table 1.
PCR and RT-qPCR analysis
The volume of each PCR amplification reaction mix was 20 μL, containing 10 μL of Phata Max Buffer, 2 μL of five-fold diluted cDNA, 2 μL of each primer (10 μM), 0.5 μL of dNTP, 0.5 μL of Phata Max Super-Fidelity DNA Polymerase, and 5 μL ddH2O. The PCR reaction procedure was as follows: 95 °C for 3 min, 35 cycles of 95 °C for 15 s, 55 °C for 15 s, 72 °C for 15 s, followed by 5 min extension at 72 °C. The RT-qPCR reaction mixture consisted of 10 μL ChamQ Universal SYBR qPCR Master Mix (Vazyme), 2 μL of cDNA, 0.4 μL of each primer (10 μM), and 7.2 μL ddH2O to a final volume of 20 μL, and it was performed on LightCycler480 (Roche Molecular Biochemicals, Mannheim, Germany) with optical 96-well plate. To test the specificity of the RT-qPCR primers, the products of PCR were analyzed by nucleic acid electrophoresis on a 2% (w/v) gel and the melting curve was included after amplification. All samples used for RT-qPCR analysis had three biological replicates, each containing three technical replicates. In order to calculate the gene-specific PCR efficiency (E) and correlation coefficient (R2) of each gene, a standard curve was generated from the mixed complementary DNA (cDNA) using a fivefold dilution series.
Analysis of stability of expression of candidate reference genes
CT values were obtained by RT-qPCR, and used to evaluate the expression levels of candidate genes in different experimental conditions and tissues. Three commonly employed algorithms, geNorm [24], NormFinder [25], and BestKeeper [26], were used to evaluate the stability of candidate reference genes in different experiments.
The package geNorm (Version3.5) screens stable reference genes by calculating the M value of the stability of each candidate gene, and the criterion is that the smaller the M value is, the higher the stability of the candidate gene is. It also calculates pairwise variations of the normalized factor after introducing a new internal reference gene, and determines the number of optimal internal reference genes based on the ratio Vn / Vn + 1. If the value of Vn / Vn + 1 is less than 0.15, the number of optimal internal reference genes is n. If the value of Vn / Vn + 1 is greater than 0.15, the number of optimal reference genes is n + 1. NormFinder selects the most suitable internal reference gene by calculating a stability value for gene expression of the candidates. The lower the stability value is, the more stable the gene is. Using BestKeeper, the SD and CV of expression of each candidate gene can be obtained. The CV ± SD values of different genes were then compared to determine the relative stability of expression of the candidates. Finally, in order to generate an overall ranking of candidate genes from the data generated by geNorm, NormFinder, and BestKeeper, we used the RankAggreg (version 0.6.5) software package in R as previously described [14, 55,56,57]. RankAggreg is an algorithmic package that can combine different ranking lists. Based on the size of the rankings list, we used the Cross-Entropy Monte Carlo algorithm [34]. The rankings list previously generated by the three packages were used as input with the following parameters: the distance was calculated using Spearman’s Footrule function, with rho set at 0.1, the seed at 100, and the “convIn” argument at 50.
Validation of reference genes
In order to verify the accuracy of the rankings and the stability of expression of the selected reference genes, the two most stable reference genes, alone and in combination, and the two most unstable reference genes, as recommended by RankAggreg were used to verify the relative expression of TcMYB3 under 4 °C, MeJA, PEG6000, in different tissues, and under H. robusta treatment (leaves and young stems). Finally, we used the 2-△△CT method to calculate the relative expression levels of the verified genes, where △CT = CT (target gene)-CT (reference gene), △△CT = △CT (treatment)-△ CT (control), 2-△△CT = relative expression. Three technical replicates were performed for each biological sample [58].
Availability of data and materials
The datasets generated and/or analyzed during the current study are available in the [GeneBank] repository, [the GeneBank accession number are form MW003991 to MW004010.]
Abbreviations
- RT-qPCR:
-
Quantitative real time PCR
- T. ciliata :
-
Toona ciliata Roem
- H. robusta :
-
Hypsipyla robusta
- MeJA:
-
Methyl jasmonate
- CT:
-
Cycle threshold
- TcMYB3 :
-
Transcription factor MYB3
- CV:
-
Coefficient of variation
- SD:
-
Standard deviation
- CDS:
-
Coding Sequence
- gDNA:
-
Genomic DNA
- cDNA:
-
Complementary DNA
References
Li P, Zhan X, Que QM, Qu WT, Liu MQ, Ouyang KX, Li JC, Deng XM, Zhang JJ, Liao BY, et al. Genetic diversity and population structure of Toona Ciliata Roem. Based on sequence-related amplified polymorphism (SRAP) markers. Forests. 2015;6(12):1094–106.
Li P, Shang YY, Zhou W, Hu XS, Mao WM, Li JJ, Li JC, Chen XY. Development of an efficient regeneration system for the precious and fast-growing timber tree Toona ciliata. Plant Biotechnol (Tokyo, Japan). 2018;35(1):51–8.
Zhu GL, Wan LS, Peng XR, Shi QQ, Li XN, Chen JC, Zhou L, Qiu MH. Cytotoxic limonoids from the twigs and leaves of Toona ciliata. J Nat Prod. 2019;82(9):2419–29.
Beseni BK, Matsebatlela TM, Bagla VP, Njanje I, Poopedi K, Mbazima V, Mampuru L, Mokgotho MP. Potential antiglycation and hypoglycaemic effects of Toona ciliata M. Roem. and Schkuhria pinnata Lam. Thell. crude extracts in differentiated C2C12 cells. Evid-Based Complement Alternat Med. 2019;2019:5406862.
Luo J, Huang WS, Hu SM, Zhang PP, Zhou XW, Wang X, Yang MH, Luo JG, Wang C, Liu C, et al. Rearranged limonoids with unique 6/5/6/5 tetracarbocyclic skeletons from Toona ciliata and biomimetic structure divergence. Org Chem Front. 2017;4:2417–21.
Veiga TA, Gonzalez-Vazquez R, Neto JO, Silva MF, King-Diaz B, Lotina-Hennsen B. Siderin from Toona ciliata (Meliaceae) as photosystem II inhibitor on spinach thylakoids. Arch Biochem Biophys. 2007;465(1):38–43.
Xia JJ, Li XY, Zhang SZ, Liu JQ, Zhang WM, Yan YX, Ding ZT, Qiu MH. An unusual 9,11-seco limonoid from Toona ciliata. Tetrahedron Lett. 2014;55(13):2104–6.
Malairajan P, Gopalakrishnan G, Narasimhan S, Veni KJ, Kavimani S. Anti-ulcer activity of crude alcoholic extract of Toona ciliata Roemer (heart wood). J Ethnopharmacol. 2007;110(2):348–51.
Atuahene SKN, Souto D. The rearing and biology of the mahogany shoot borer Hypsipyla robusta Moore (Lepidoptera: Pyralidae) on an artificial medium. Int J Trop Insect Sci. 1983;4(04):319–25.
Abraham J, Opunifrimpong E, Weissbecker B, Schütz S, Angeli S. Olfactory cues of mahogany trees to female Hypsipyla robusta. Bull Insectol. 2014;67(1):21–30.
Cunningham SA, Floyd RB. Leaf compositional differences predict variation in Hypsipyla robusta damage to Toona ciliata in field trials. Can J For Res. 2011;34(3):642–8.
Mo J, Tanton MT, Bygrave FL. Within-tree distribution of attack by Hypsipyla robusta Moore (Lepidoptera: Pyralidae) in Australian red cedar (Toona australis (F. Muell.) Harmes). Forest Ecol Manag. 1997;96(1–2):139–46.
Mo J, Tanton MT, Bygrave FL. An improved technique for rearing the red cedar tip moth, Hypsipyla robusta Moore (Lepidoptera: Pyralidae). Austral Entomol. 2014;37(1):64–9.
Guo CS, Chi ZX, Jiang DL, Xu T, Yu WW, Wang Z, Chen S, Zhang L, Liu QY, Guo XC, et al. Cholesterol homeostatic regulator SCAP-SREBP2 integrates NLRP3 inflammasome activation and cholesterol biosynthetic signaling in macrophages. Immunity. 2018;49(5):842–856.e847.
Kundu A. Vanillin biosynthetic pathways in plants. Planta. 2017;245(6):1069–78.
Han B, Yang Z, Samma MK, Wang R, Shen W. Systematic validation of candidate reference genes for qRT-PCR normalization under iron deficiency in Arabidopsis. Biometals. 2013;26(3):403–13.
Reddy DS, Bhatnagar-Mathur P, Cindhuri KS, Sharma KK. Evaluation and validation of reference genes for normalization of quantitative real-time PCR based gene expression studies in peanut. PLoS One. 2013;8(10):e78555.
Warzybok A, Migocka M. Reliable reference genes for normalization of gene expression in cucumber grown under different nitrogen nutrition. PLoS One. 2013;8(9):e72887.
Gao MM, Liu YP, Ma X, Shuai Q, Gai JY, Li Y. Evaluation of reference genes for normalization of gene expression using quantitative RT-PCR under aluminum, cadmium, and heat stresses in soybean. PLoS One. 2017;12(1):e0168965.
Deng LT, Wu YL, Li JC, Ouyang KX, Ding MM, Zhang JJ, Li SQ, Lin MF, Chen HB, Hu XS, et al. Screening reliable reference genes for RT-qPCR analysis of gene expression in Moringa oleifera. PLoS One. 2016;11(8):e0159458.
Huang T, Long JM, Liu SW, Yang ZW, Zhu QJ, Zhao XL, Peng CC. Selection and validation of reference genes for mRNA expression by quantitative real-time PCR analysis in Neolamarckia cadamba. Sci Rep. 2018;8(1):9311.
Qu RJ, Miao YJ, Cui YJ, Cao Y, Zhou YW, Tang XQ, Yang J, Wang FY. Selection of reference genes for the quantitative real-time PCR normalization of gene expression in Isatis indigotica fortune. BMC Mol Biol. 2019;20(1):9.
Yu YT, Zhang G, Chen YK, Bai QQ, Gao CS, Zeng LB, Li ZM, Cheng Y, Chen J, Sun XP, et al. Selection of reference genes for qPCR analyses of gene expression in ramie leaves and roots across eleven abiotic/biotic treatments. Sci Rep. 2019;9(1):20004.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):Research0034.
Andersen CL, Jensen JL, Orntoft TF. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res. 2004;64(15):5245–50.
Czechowski T, Stitt M, Altmann T, Udvardi MK, Scheible WR. Genome-wide identification and testing of superior reference genes for transcript normalization in Arabidopsis. Plant Physiol. 2005;139(1):5–17.
Smitha PK, Vishnupriyan K, Kar AS, Anil Kumar M, Bathula C, Chandrashekara KN, Dhar SK, Das M. Genome wide search to identify reference genes candidates for gene expression analysis in Gossypium hirsutum. BMC Plant Biol. 2019;19(1):405.
Song Y, Wang Y, Guo DD, Jing L. Selection of reference genes for quantitative real-time PCR normalization in the plant pathogen Puccinia helianthi Schw. BMC Plant Biol. 2019;19(1):20.
Chen XJ, Chen H, Yuan JS, Köllner TG, Chen YY, Guo YF, Zhuang XF, Chen XL, Zhang YJ, Fu JY, et al. The rice terpene synthase gene OsTPS19 functions as an (S)-limonene synthase in planta, and its overexpression leads to enhanced resistance to the blast fungus Magnaporthe oryzae. Plant Biotechnol J. 2018;16(10):1778–87.
Pu XJ, Yang LX, Liu LN, Dong XM, Chen SL, Chen ZX, Liu GJ, Jia YX, Yuan WY, Liu L. Genome-wide analysis of the MYB transcription factor superfamily in Physcomitrella patens. Int J Mol Sci. 2020;21(3):975.
Wang H, Zhang H, Yang Y, Li MF, Zhang YT, Liu JS, Dong J, Li J, Butelli E, Xue Z, et al. The control of red colour by a family of MYB transcription factors in octoploid strawberry (Fragaria x ananassa) fruits. Plant Biotechnol J. 2020;18:1169–84.
Zhang YL, Zhang CL, Wang GL, Wang YX, Qi CH, Zhao Q, You CX, Li YY, Hao YJ. The R2R3 MYB transcription factor MdMYB30 modulates plant resistance against pathogens by regulating cuticular wax biosynthesis. BMC Plant Biol. 2019;19(1):362.
Zhu L, Guo JS, Ma ZY, Wang JF, Zhou C. Arabidopsis transcription factor MYB102 increases plant susceptibility to aphids by substantial activation of ethylene biosynthesis. Biomolecules. 2018;8(2):39.
Pihur V, Datta S, Datta S. Weighted rank aggregation of cluster validation measures: a Monte Carlo cross-entropy approach. Bioinformatics (Oxford, England). 2007;23(13):1607–15.
Razavi SA, Afsharpad M, Modarressi MH, Zarkesh M, Yaghmaei P, Nasiri S, Tavangar SM, Gholami H, Daneshafrooz A, Hedayati M. Validation of reference genes for normalization of relative qRT-PCR studies in papillary thyroid carcinoma. Sci Rep. 2019;9(1):15241.
Poli M, Salvi S, Li M, Varotto C. Selection of reference genes suitable for normalization of qPCR data under abiotic stresses in bioenergy crop Arundo donax L. Sci Rep. 2017;7(1):10719.
Yang QP, Li Z, Cao JJ, Zhang SD, Zhang HJ, Wu XY, Zhang QW, Liu XX. Selection and assessment of reference genes for quantitative PCR normalization in migratory locust Locusta migratoria (Orthoptera: Acrididae). PLoS One. 2014;9(6):e98164.
Zhang K, Li MN, Cao SH, Sun Y, Long RC, Kang JM, Yan L, Cui HT. Selection and validation of reference genes for target gene analysis with quantitative real-time PCR in the leaves and roots of Carex rigescens under abiotic stress. Ecotoxicol Environ Saf. 2019;168:127–37.
Zhu XY, Li XP, Chen WX, Chen JY, Lu WJ, Chen L, Fu DW. Evaluation of new reference genes in papaya for accurate transcript normalization under different experimental conditions. PLoS One. 2012;7(8):e44405.
Li WG, Zhang LH, Zhang YD, Wang GD, Song DY, Zhang YW. Selection and validation of appropriate reference genes for quantitative real-time PCR normalization in staminate and perfect flowers of andromonoecious Taihangia rupestris. Front Plant Sci. 2017;8:729.
Pfaffl MW, Tichopad A, Prgomet C, Neuvians TP. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper--excel-based tool using pair-wise correlations. Biotechnol Lett. 2004;26(6):509–15.
Gao D, Kong FN, Sun PP, Bi GQ, Mao YX. Transcriptome-wide identification of optimal reference genes for expression analysis of Pyropia yezoensis responses to abiotic stress. BMC Genomics. 2018;19(1):251.
Li HB, Dai CG, Zhang CR, He YF, Ran HY, Chen SH. Screening potential reference genes for quantitative real-time PCR analysis in the oriental armyworm, Mythimna separata. PloS one. 2018;13(4):e0195096.
Perez LJ, Rios L, Trivedi P, D'Souza K, Cowie A, Nzirorera C, Webster D, Brunt K, Legare JF, Hassan A, et al. Validation of optimal reference genes for quantitative real time PCR in muscle and adipose tissue for obesity and diabetes research. Sci Rep. 2017;7(1):3612.
He SN, An T, Liu SA. Validation of reliable reference genes for RT-qPCR studies of target gene expression in Colletotrichum camelliae during spore germination and mycelial growth and interaction with host plants. Front Microbiol. 2019;10:2055.
Ram C, Koramutla MK, Bhattacharya R. Identification and comprehensive evaluation of reference genes for RT-qPCR analysis of host gene-expression in Brassica juncea-aphid interaction using microarray data. Plant Physiol Biochem. 2017;116:57–67.
Bansal R, Mittapelly P, Cassone BJ, Mamidala P, Redinbaugh MG, Michel A. Recommended reference genes for quantitative PCR analysis in soybean have variable stabilities during diverse biotic stresses. PLoS One. 2015;10(8):e0134890.
de Jesus Miranda V, Coelho RR, Viana AA, de Oliveira Neto OB, Carneiro RM, Rocha TL, de Sa MF, Fragoso RR. Validation of reference genes aiming accurate normalization of qPCR data in soybean upon nematode parasitism and insect attack. BMC Res Notes. 2013;6:196.
Bhaskara GB, Wong MM, Verslues PE. The flip side of phospho-signalling: regulation of protein dephosphorylation and the protein phosphatase 2Cs. Plant Cell Environ. 2019;42(10):2913–30.
Yan H, Zhang Y, Xiong Y, Chen Q, Liang H, Niu M, Guo B, Li M, Zhang X, Li Y, et al. Selection and validation of novel RT-qPCR reference genes under hormonal stimuli and in different tissues of Santalum album. Sci Rep. 2018;8(1):17511.
Ayakannu T, Taylor AH, Willets JM, Brown L, Lambert DG, McDonald J, Davies Q, Moss EL, Konje JC. Validation of endogenous control reference genes for normalizing gene expression studies in endometrial carcinoma. Mol Hum Reprod. 2015;21(9):723–35.
Molina CE, Jacquet E, Ponien P, Munoz-Guijosa C, Baczko I, Maier LS, Donzeau-Gouge P, Dobrev D, Fischmeister R, Garnier A. Identification of optimal reference genes for transcriptomic analyses in normal and diseased human heart. Cardiovasc Res. 2018;114(2):247–58.
Nakao R, Okauchi H, Hashimoto C, Wada N, Oishi K. Determination of reference genes that are independent of feeding rhythms for circadian studies of mouse metabolic tissues. Mol Genet Metab. 2017;121(2):190–7.
Ferreira AF, Raposo M, Vasconcelos J, Costa MDC, Lima M. Selection of reference genes for normalization of gene expression data in blood of Machado-Joseph disease/Spinocerebellar Ataxia type 3 (MJD/SCA3) subjects. J Mol Neurosci. 2019;69(3):450–5.
Ambroise V, Legay S, Guerriero G, Hausman JF, Cuypers A, Sergeant K. Selection of appropriate reference genes for gene expression analysis under abiotic stresses in Salix viminalis. Int J Mol Sci. 2019;20(17):4210.
Mallona I, Lischewski S, Weiss J, Hause B, Egea-Cortines M. Validation of reference genes for quantitative real-time PCR during leaf and flower development in Petunia hybrida. BMC Plant Biol. 2010;10:4.
Ponton F, Chapuis MP, Pernice M, Sword GA, Simpson SJ. Evaluation of potential reference genes for reverse transcription-qPCR studies of physiological responses in Drosophila melanogaster. J Insect Physiol. 2011;57(6):840–50.
Wong ML, Biotechniques MJFJ. Real-time PCR for mRNA quantitation. Bio Tech. 2005;39(1):75–85.
Acknowledgments
We thank Guangdong Key Laboratory for Innovative Development and Utilization of Forest Plant Germplasm, for providing laboratory apparatus.
Funding
This research is supported by grants from the National Key Research Projects, Forestry Resource Cultivation and Utilization Technology Innovation (Grant No.2016YFD0600606), the Natural Science Foundation of Guangdong Province of China (Grant No.2018A030313798) and Characteristic innovation projects of department of education of Guangdong province (Grant No.2019KTSCX017).
Author information
Authors and Affiliations
Contributions
PL and XC designed the research; HS and WM conducted the experiment; QQ, ZD and WZ analyzed the data; PL XC and HS wrote the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1 Figure S1
: Amplification products of the twenty candidate reference genes and TcMYB3. Figure S2: Melting curves of candidate reference genes and TcMYB3.
Additional file 2 Table S1.
Coding sequences and genomic DNA sequences of candidate reference gene.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Song, H., Mao, W., Duan, Z. et al. Selection and validation of reference genes for measuring gene expression in Toona ciliata under different experimental conditions by quantitative real-time PCR analysis. BMC Plant Biol 20, 450 (2020). https://doi.org/10.1186/s12870-020-02670-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-020-02670-3