
Abstract
Background
Regulation of lignin biosynthesis is known to occur at the level of transcription factors (TFs), of which R2R3-MYB family members have been proposed to play a central role via the AC cis-elements. Despite the important roles of TFs in lignin biosynthesis, the post-translational regulation of these TFs, particularly their ubiquitination regulation, has not been thoroughly explored.
Results
We describe the discovery of a Populus tomentosa E2 ubiquitin-conjugating enzyme 34 (PtoUBC34), which is involved in the post-translational regulation of transactivation activity of lignin-associated transcriptional repressors PtoMYB221 and PtoMYB156. PtoUBC34 is localized at the endoplasmic reticulum (ER) membrane where it interacts with transcriptional repressors PtoMYB221 and PtoMYB156. This specific interaction allows for the translocation of TFs PtoMYB221 and PtoMYB156 to the ER and reduces their repression activity in a PtoUBC34 abundance-dependent manner. By taking a molecular biology approach with quantitative real-time polymerase chain reaction (qRT-PCR) analysis, we found that PtoUBC34 is expressed in all aboveground tissues of trees in P. tomentosa, and in particular, it is ubiquitous in all distinct differentiation stages across wood formation, including phloem differentiation, cambium maintaining, early and developing xylem differentiation, secondary cell wall thickening, and programmed cell death. Additionally, we discovered that PtoUBC34 is induced by treatment with sodium chloride and heat shock.
Conclusions
Our data suggest a possible mechanism by which lignin biosynthesis is regulated by ER-localized PtoUBC34 in poplar, probably through the ER-associated degradation (ERAD) of lignin-associated repressors PtoMYB221 and PtoMYB156.
Similar content being viewed by others


Background
Lignin is an aromatic heteropolymer that deposits mostly in the secondary cell walls of tracheophytes and helps large upright land plants to survive by providing mechanical support and efficient water conductance [1]. It is the second most abundant polymer in wood and has been estimated to represent almost 30% of the total biomass produced in the biosphere [2]. However, lignin limits the efficient utilization of lignocellulose as feedstock in pulping, papermaking, and biofuel industry, in which lignin removal is a necessary and costly step [3,4,5]. A preferred strategy to optimize lignocellulosic feedstock with improved performance in various applications is to utilize biotechnology to engineer the desired amount and structure of lignin [5,6,7]. Elucidation of the mechanisms underlying lignin biosynthesis and regulation is critical in plant biology and likely will guide the lignin bioengineering design.
For more than 20 years, the lignin biosynthesis field has been focused on identifying the monolignol biosynthetic genes and their transcriptional regulators. Many steps of the biochemical pathway and transcriptional control of the monolignol biosynthesis already have been discovered [2, 8,9,10]. It is well known that regulation of lignin biosynthesis occurs at the level of transcription factors (TFs) [8,9,10]. Among these, R2R3-MYB TFs are key players [11,12,13,14] which coordinate the gene expression of the complex monolignol biosynthetic pathway by binding to the AC cis-elements present in the promoter regions of most monolignol biosynthetic genes [15,16,17,18]. The identified R2R3-MYB key players in lignin biosynthesis include secondary cell wall master activators AtMYB46 and AtMY83 from Arabidopsis, and their orthologs in other plant species [19,20,21,22,23,24,25,26]; and lignin-specific activators MYB58, MYB63, MYB85 in Arabidopsis, and PtMYB1 in pine [17, 18, 27, 28]; and lignin transcriptional repressors, most of which belong to subgroup 4 of the R2R3-MYB TF family [29,30,31,32,33,34,35,36,37] (Table 1). The unique TF-directed hierarchical gene regulatory networks [38] in lignin biosynthesis integrate transcriptional activators and repressors to maintain transcriptional homeostasis of lignin pathway genes [8, 13]. Failure to maintain the transcriptional homeostasis results in limited growth and ectopic lignin deposition, as demonstrated by overexpression of the MYB lignin activators AtMYB46, AtMYB85, AtMYB58, and AtMYB63 [17, 19, 27]. Indeed, transcriptional repression through repressors is a useful strategy used to maintain transcriptional homeostasis [39].
Although lignin biosynthesis and transcriptional regulation have been studied extensively, the post-translational regulation of lignin complex biosynthetic pathway, particularly ubiquitination, was not studied in detail until recent years. Ubiquitination of a substrate, which has been shown to play crucial roles in plant senescence, xylogenesis, embryogenesis, floral development, and stress responses [40,41,42], is performed by a set of enzymes: ubiquitin activation enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin ligase (E3) [43]. In Arabidopsis, a WD40-repeat adaptor protein AtULCS1, a subunit of a cullin-RING E3 ligase, was shown to function in cell wall modification and lignin-specific deposition in anther cell wall, and control anther dehiscence [44]. In rice, an F-box protein OsFBK1, a component of an SCF E3 ligase, has been shown to play crucial roles in the site-specific deposition of lignin in the anther endothecium, which mediates the turnover of a cinnamoyl-CoA reductase. Importantly, this specific lignin deposition is believed to regulate anther dehiscence and plant fertility [45]. The ubiquitination regulation of lignin deposition in poplar has not yet been reported, even though lignin accounts for approximately 25% of the total lignocellulose in poplar.
Given the crucial roles that lignin repressors play in maintaining transcriptional homeostasis of complex lignin biosynthetic pathway [39] and activating lignin deposition in plant defense [8] and because their conditional expression responds to the developmental signals and environmental cues [32, 33, 46], we propose that fine-tuned mechanisms post-translationally regulate transactivation activity of subgroup 4 MYB lignin repressors MYB221 and MYB156 in poplar, which likely mediate the regulation of secondary wall formation and lignin biosynthesis. Because protein-protein interactions are a common strategy to modify the activity of TFs post-translationally [47], it is essential to isolate proteins that interact with poplar subgroup 4 MYB221 and MYB156 repressors.
In this study, we screened P. tomentosa differentiating secondary vascular tissue cDNA library with PtoMYB221 and PtoMYB156 as baits, and isolated a partial sequence of PtoUBC34, a specific ubiquitin-conjugating enzyme. Further characterization through co-localization, translocation, bimolecular fluorescence complementation (BiFC), coimmunoprecipitation, and transactivation assay in poplar mesophyll protoplasts suggested that PtoUBC34 was involved in the lignin biosynthesis through the regulation of transactivity of lignin transcriptional repressors, probably by the ER-associated degradation (ERAD) pathway in P. tomentosa.
Results
Isolation of proteins that interact with PtoMYB221 and PtoMYB156
To isolate proteins that potentially interact with MYB subgroup 4 transcriptional repressors PtoMYB221 and PtoMYB156 during wood formation, we used the yeast two-hybridization (Y2H) system based on split-ubiquitin to screen normalized cDNA library from P. tomentosa differentiating secondary vascular tissue (including vascular cambium, developing phloem, and xylem). In this library vector, N-terminal half of the ubiquitin (Nub) is modified at position 3 of the protein, where isoleucine is exchanged with glycine (NubI → NubG), to abolish the strong affinity between wild-type Nub and C-terminal half of the ubiquitin (Cub). This system can limit self-activation by TF proteins in the classical protein interaction assay [48], in which transcriptional repressor PtoMYB156 has been shown to significantly activate the transcription of his3 and β-galactosidase (β-Gal) reporter genes (Additional file 1: Figure S1). To prevent leaky expression of the his3 reporter gene, we added 3-aminotriazole (1 mM) to the selection medium. We fused the entire PtoMYB221 or PtoMYB156 protein to the Cub and used them as baits. We transformed 28 μg of library DNA into yeast strain NMY51 containing PtoMYB221 or PtoMYB156 bait vector as recommended in the user manual. We rescued approximately 5 × 106 independent transformants, covering the cDNA library about 2.5 times, whose complexity is 1.92 × 106 independent clones. We screened the transformants by growth selection in medium lacking histidine and then by the activity of β-Gal. A protein designated N35 was isolated and was capable of activating transcription of the two reporter genes in the presence of PtoMYB221. We confirmed the specific interaction between N35 and PtoMYB221 by the re-introduction of the corresponding plasmids into yeast NMY51 (Fig. 1).

The specific interaction between PtoMYB221 and the N35 protein in a split-ubiquitin Y2H system. Bait protein PtoMYB221 fused to Cub was co-expressed in yeast NMY51 with prey proteins, NubI, NubG, or the NubG:N35 protein. NubI is a positive prey control with strong affinity between wild-type NubI and Cub, whereas NubG is a negative prey control with weak affinity between mutant NubG and Cub. Interaction of the bait was shown with positive control NubI and the test NubG:N35 fusion protein but not with the negative control NubG, as indicated by growth of the transformed strain on synthetic defined medium without Trp, Leu, and His and by the activity of β-Gal
N35 represents a partial sequence of an ER-localized E2 ubiquitin-conjugating enzyme
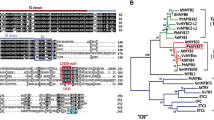
Sequence analysis revealed that N35 is a partial sequence of an E2 ubiquitin-conjugating enzyme using a BLASTP search against the P. trichocarpa genome (http://phytozome.jgi.doe.gov/pz/portal.html), with the best hit for Potri.001G162200. The corresponding coding sequence (CDS) of the N35 protein was missing the first 72 nucleotides. Then we obtained the full-length cDNA sequence from P. tomentosa by reverse transcription polymerase chain reaction (RT-PCR) with primers designed according to the homologous sequence of Potri.001G162200 and deposited the sequence into the GenBank with accession number MH708242. It shares 98.7 and 98.30% sequence identity with orthologs from P. trichocarpa (Potri.001G162200) and Populus euphratica (PeUBC34-like) in amino acid, respectively, and was named after PtoUBC34. In comparison, the truncated UBC34 of the N35 protein was designated as PtoUBC34s. PtoUBC34 is homologous with the yeast Ubc6 and mammalian UBE2J1 and UBE2J2, which are involved in the ERAD process [49,50,51,52]. A whole-genome BLAST search (https://phytozome.jgi.doe.gov) indicated that three other genes are homologous with Potri.001G162200 in P. trichocarpa: Potri.003G073100, Potri.008G106300 and Potri.010G143900. All four genes are homologous with yeast Ubc6 and mammalian UBE2J1 and UBE2J2. Similarly to Arabidopsis and humans [52,53,54,55], P. trichocarpa Ubc6 homologs evolved into two subfamilies, and Potri.001G162200 and Potri.003G073100 are both similar to UBE2J2, whereas Potri.008G106300 and Potri.010G143900 belong to the UBE2J1 subfamily (Fig. 2a).

Characteristics of PtoUBC34. a. Phylogenetic analysis of PtoUBC34 and other ERAD-related UBC proteins in yeast (UBC6 and UBC7), humans (UBE2J1, UBE2J2, and UBE2G2), poplar, and Arabidopsis. The phylogenetic tree was established with full-length protein sequences by the neighbor-joining method. The bootstrap values out of 500 retrials are indicated at each branch. The scale (0.1) represents a 10% change in sequences. b. Schematic of PtoUBC34. The UBCc domain (blue) and a transmembrane domain (orange) are predicted in PtoUBC34 at the N-terminus and the C-terminus, respectively. c. Subcellular localizatin of PtoUBC34. PtoUBC34 was fused with YFP and co-transformed with the ER marker BiP:RFP [57] into P. tomentosa protoplasts. d. Multiple sequence alignment of yeast UBC6, human UBE2J2, Arabidopsis UBC34 and P. tomentosa UBC34. The conserved active Cys residue with red star, E3 interaction residues with black stars and Ub thioester intermediate interaction residues with blue stars are indicated. The transmembrane regions are shown in boxes. Bar in (c) = 10 μm
PtoUBC34 encodes a protein composed of 235 amino acid with a predicted molecular mass of ~ 27 kDa using Compute pI/Mw (https://web.expasy.org/compute_pi/). InterPro program (http://www.ebi.ac.uk/interpro/) [56] predicted the presence of an E2 ubiqutin-conjugating enzyme catalytic (UBCc) domain located at the N-terminus and a transmembrane domain at the C-terminus of the PtoUBC34 protein. The UBCc domain includes a conserved active Cys at position 87, which forms a thiol-ester with ubiquitin and serves as a point of attachment for ubiquitin before transfer to the cellular target, and several E3 interaction residues 8 K, 65P, 66Y, 100P, 101 M, and many Ub thioester intermediate interaction residues 79R, 80F, 82 T, 86I, 87C, 88 L, 90 M, 91S, 98 W, 99 N, 102 W, 107I, 110G, 113S, 118 N, 119S, 121 T, 123G, and 124S, most of which are well conserved among yeast, humans, and plants (Fig. 2b and d). To investigate the subcellular localization of the PtoUBC34 protein, the yellow fluorescent protein (YFP)-UBC34 fusion protein (YFP:PtoUBC34) driven by the cauliflower mosaic virus 35S (CaMV35S) promoter was co-transformed into P. tometosa mesophyll protoplasts with a fusion protein (BiP:RFP) between red fluorescent protein (RFP) and portions of the chaperone binding protein (BiP). BiP is an ER resident and is used widely as an ER marker [57]. Confocal examination revealed that the fluorescent signals of YFP and RFP clearly overlapped in the protoplasts (Fig. 2c). These results suggest that PtoUBC34 is localized to the ER compartment.
UBC34 is expressed ubiquitously and responds to stress factors in poplar
We analyzed the expression pattern of PtoUBC34 by quantitative real-time polymerase chain reaction (qRT-PCR) in P. tomentosa and detected PtoUBC34 transcripts in all examined tissues, including mature leaf, stem, shoot tip, phloem, and developing xylem (Fig. 3a). To investigate the differential expression level of PtoUBC34 under heat shock, we treated the plant at 37 °C for 1 h, allowed it to recover for 2 h at 24 °C, and then subjected it to 42 °C for 2.5 h. The accumulation of PtoUBC34 transcripts was significantly higher after initial heat shock treatment at 37 °C and then reversed after a 2-h recovery from the heat shock. The subsequent heat stress at 42 °C also caused significant increase of PtoUBC34 transcripts in the leaves (Fig. 3b). Furthermore, we found PtoUBC34 to be induced significantly by salt (NaCl) treatment (Fig. 3c). During wood formation, UBC34 in P. trichocarpa also has been shown to have a high and constitutive expression in all five traditionally recognized distinct differentiation stages, that is, cambial cell division, cell expansion, secondary wall deposition, lignification, and cell death, according to the research by Sundell et al. [58] (Fig. 3d). This result is in contrast to most of the well-characterized genes during wood formation, which generally are expressed at a high level in a specific wood differentiation stage. For example, poplar CesA8-B, a marker gene for secondary cell wall (SCW) formation [59], was highly expressed in SCW-thickening xylem (Fig. 3d). These results suggest that poplar UBC34 might play important roles in plant development and physiology.

PtoUBC34 is expressed ubiquitously and responds to stress factors. a. qRT-PCR analysis of PtoUBC34 expression in various poplar tissues, using Actin6 (Potri.001G309500), eIF-5A (Potri.018G107300), and UBQ (Potri.014G115100) genes as internal controls, according to Wang et al. [94]. The expression level of PtoUBC34 in leaves was set as 1. b. The expression levels of PtoUBC34 in mature leaves in response to heat shock treatment by qRT-PCR, using Actin6 (Potri.001G309500) and EF1-beta (Potri.009G018600) genes as internal controls, which were validated in an evaluation assay of eight reference genes during heat shock treatment in P. tomentosa (unpublished data). Plants were treated at 37 °C for 1 h (HS1), allowed to recover for 2 h at 24 °C (HSR), and then were subjected to 42 °C for 2.5 h (HS2). The fold-expression was normalized relative to the 24 °C control (CK). c. The expression levels of PtoUBC34 in mature leaves in response to 300 mM sodium chloride (NaCl) treatment were analyzed by qRT-PCR, using UBQ (Potri.014G115100) and TUB (Potri.003G126800) genes as internal controls, which were validated in an evaluation assay of eight reference genes during salt stress in P. tomentosa (unpublished data). The fold-expression was normalized relative to the non-NaCl-treatment control. Data are mean of three biological samples from three plants, respectively, with three technical replicates for each one. Error bar represents standard deviation of the three biological samples. Significance was tested with one-way ANOVA to evaluate the effect of heat shock and NaCl treatment on UBC34 expression. The different alphabets above the bar indicates statistically significant differences with p value < 0.05, while the same alphabet mean no significant differences. d. UBC34 from P. trichocarpa was expressed constitutively in distinct differentiation stages across wood formation. The expression data are derived from the ASPWOOD project (http://aspwood.popgenie.org/aspwood-v3.0/) [58]. Phloem, cambium, developing xylem, and mature xylem were collected from stems through longitudinal continuous sections using a cryo-microtome. The tissue type was characterized by examining the images of cross-sections during sampling. All of the samples were collected during the current growth year. Poplar CesA8-B, specifically expressed during SCW thickening, was used as a reference
Characterization of the interaction between PtoMYB221/PtoMYB156 and PtoUBC34
According to Tuskan et al. [60] and Castanet-Tolosan [13], the poplar genome contains two putative subgroup 4 R2R3-MYB members, MYB221 and MYB156, which are close homologs of Arabidopsis MYB4. Analysis by the Y2H assay demonstrated that each of the two subgroup 4 R2R3-MYB members interacted with full-length PtoUBC34 in yeast (Fig. 4).

The Y2H demonstrated that full-length PtoUBC34 interacted with both the PtoMYB221 and PtoMYB156 protein in a split-ubiquitin Y2H system. Co-expression with NubI and NubG was used as positive and negative controls, respectively
We performed subcellular co-localization to further investigate the interaction between the proteins. To co-localize each of the subgroup 4 MYB members with PtoUBC34, fusion gene construct either 35S-PtoMYB221:YFP or 35S-PtoMYB156:YFP was co-expressed with 35S-RFP:PtoUBC34 in poplar mesophyll protoplasts. Because both 35S-PtoMYB221:YFP and 35S-PtoMYB156:YFP vectors showed very weak fluorescent signals in the ER and the degradation of PtoMYB221 and PtoMYB156 protein was suggested under the presence of entire PtoUBC34 protein in plant cells, the PtoUBC34s fused with RFP was used for the subcellular co-localization. Importantly, in the presence of PtoUBC34s, TFs PtoMYB221 and PtoMYB156 were translocated from the nucleus (Fig. 5a and c) to the ER, showing significant overlap with PtoUBC34s and a typical punctate pattern indicative of the ER (Fig. 5b and d). This translocation of PtoMYB221 or PtoMYB156 was exclusive to all co-transfected protoplasts. Overall, these results suggest that transcriptional repressors PtoMYB221 and PtoMBY156 are retained in the ER through interaction with the ER-localized ubiquitin-conjugating enzyme, PtoUBC34.

Protein co-localization in poplar mesophyll protoplasts. a and b. Protein co-localization of PtoUBC34 and PtoMYB221. c and d. Protein co-localization of PtoUBC34 and PtoMYB156. Both the PtoMYB221 and PtoMYB156 were fused with YFP and were co-transferred with the nuclear marker CBF1:RFP or RFP:PtoUBC34s into poplar protoplasts. The co-localization showed that TFs PtoMYB221 and PtoMY156 were translocated from the nucleus (a and c) to the ER (b and d) by PtoUBC34. Bar = 10 μm
The co-localization of PtoUBC34 with either PtoMYB221 or PtoMYB156 in the ER of poplar protoplasts and the interaction in yeast provided strong evidence that the pairs of proteins interact with each other in vivo. To further examine this interaction, we performed BiFC assay in poplar mesophyll protoplasts. Only combinations of the eYFPN:PtoUBC34s and PtoMYB221:eYFPC pair and the eYFPN:PtoUBC34s and PtoMYB156:eYFPC pair—that is, PtoUBC34 fused at the C-terminus to eYFPN (aa 1–173)—gave a positive BiFC signal. The BiFC signal was shown to be co-localized with the Bip:RFP marker in the ER (Fig. 6a), suggesting that PtoUBC34 interacts with both PtoMYB221 and PtoMYB156 in the ER of the plant cell. Furthermore, coimmunoprecipitation assays showed that PtoUBC34s:Flag coimmunoprecipitated with each of PtoMYB221:Myc and PtoMYB156:Myc in poplar mesophyll protoplasts (Fig. 6b), further confirming the interaction between PtoUBC34 and each of PtoMYB221 and PtoMYB156 in vivo.

PtoUBC34 interacts with both PtoMYB221 and PtoMYB156 in vivo. a. BiFC in poplar protoplasts demonstrated that PtoUBC34 interacts with both the PtoMYB221 and PtoMYB156 in the ER. The BiFC signals co-localized with the marker Bip:RFP in the ER, suggesting that PtoUBC34 and each of PtoMYB221 and PtoMYB156 were co-localized in the same BiFC complex in the ER. Bar = 10 μm. b. Coimmunoprecipitation analysis showed an interaction of PtoUBC34s with both the PtoMYB221 and PtoMYB156. PtoUBC34s:Flag was co-expressed with each of MYB221:Myc and MYB156:Myc in poplar protoplasts. Protein extracts were incubated with anti-Flag coupled agarose. Immunoprecipitates (IP) and input proteins were analyzed by immunoblotting using anti-Flag and anti-Myc antibodies as indicated
PtoUBC34 reduces the repression activity of PtoMYB221 in a dose-dependent manner
Both poplar MYB221 and MYB156 members from subfamily 4 of R2-R3 MYB proteins displayed activity of transcriptional repression, which significantly decreased the level of expression of the reporter gene (Fig. 7a and b) and functioned in the transcriptional downregulation of phenylpropanoid and lignin genes [36, 37]. By using electrophoretic mobility shift assay, PdMYB221 was found to bind to the promoter regions of the cell wall biosynthetic genes PdCESA8, PdGT47C, and PdCOMT2, which contain AC element consensus sequences ACC(A/T)A(C/A) C [36]. Therefore, the possible AC elements for PtoMYB221 can be each of the following: ACCTACC (AC-I), ACCAACC (AC-II), ACCTAAC (AC-III), and ACCAAAC (AC-IV), which also are target cis-elements for several other subfamily 4 MYB transcriptional repressors [30, 61, 62]. To identify the specific AC element that has the highest affinity for PtoMYB221, we set up a transient expression assay in poplar mesophyll protoplasts in which the reporter plasmid containing the luciferase (LUC) reporter gene under the control of CaMV35S promoter with three copies of each of the four AC elements inserted immediately upstream of the TATA box (CaMV35S-3xACx-TATA-LUC-NOS), and the effector plasmid encoding the PtoMYB221 protein (CaMV35S-PtoMYB221) was co-transfected into protoplasts (Fig. 8a and b). As shown in Fig. 8c, PtoMYB221 effectively repressed the expression of LUC reporter gene driven by each of the four AC-I, AC-II, AC-III, and AC-IV elements fused to the CaMV35S promoter with a reduction ranging from 56 to 86% relative to the corresponding control, indicating that PtoMYB221 can bind to all four AC elements. Moreover, PtoMYB221 blocked the expression of the LUC reporter gene most effectively (approximately 86%) with the AC-II element, suggesting that PtoMYB221 binds to the AC-II element with the highest affinity.

PtoMYB221 and PtoMYB156 are transcriptional repressors. a. Schematic representation of the constructs used for evaluation of the repression activity of PtoMYB221 and PtoMYB156 in dual LUC assay in poplar mesophyll protoplasts. The EAR-like motif repression domain of SUPERMAN (SUPRD) was used as a reference repressor [93]. b. Relative LUC activities after co-transformation with the reporter and effectors, where pUC18 was used as a control vector. Three independent experiments show similar results. Error bar represents standard deviation of three independent batches of P. tomentosa mesophyll protoplast transfections from one of the experiments. Significance was tested with one-way ANOVA to evaluate the roles of the effectors on the LUC expression. The different alphabets above the bar indicates statistically significant differences with p value < 0.05, while the same alphabet mean no significant differences

PtoMYB221 targets to the AC cis-elements of monolignol pathway genes and its repression activity is reduced with the involvement of PtoUBC34. a. Schematic representation of the effector and reporter plasmids used for evaluation of the affinity of PtoMYB221 to AC cis-elements in a dual LUC assay. b. Synthetic AC elements tested in dual LUC assay. c. LUC activity assay driven by PtoMYB221 binding to the AC elements in poplar mesophyll protoplasts. AC-I, AC-II, AC-III, and AC-IV are the reporters containing three copies of the ACx elements fused to the CaMV35S promoter and LUC reporter gene. d. PtoUBC34 reduced the repression activity of PtoMYB221. 2.4 μg of the effector plasmids PtoMYB221, 600 ng of PtoUBC34 or PtoUBC34m, along with the reporter plasmid and internal control plasmid, were used for co-transfection. Three independent experiments show similar results. Error bar represents standard deviation of three independent batches of P. tomentosa mesophyll protoplast transfections from one of the experiments. Significance was tested with one-way ANOVA to evaluate the roles of the effectors on the LUC expression. The different alphabets above the bar indicates statistically significant differences with p value < 0.05, while the same alphabet mean no significant differences
To analyze the in vivo function of PtoUBC34 on the transcriptional repression activity of PtoMYB221, we co-transformed a LUC reporter gene driven by three copies of the AC-II element fused to the CaMV35S promoter and effector genes under the control of CaMV35S promoter in poplar mesophyll protoplasts. The CaMV35S promoter with the AC-II element used here allowed the LUC reporter gene to reach a detectable expression level in the presence of the repressor PtoMYB221, which the tissue-specific promoters of lignin biosynthetic genes could not achieve. The effectors included PtoUBC34m in addition to PtoMYB221 and PtoUBC34. PtoUBC34m is a mutated version of PtoUBC34, in which the conserved active site cysteine residue at position 87 is replaced by an alanine residue. This mutant was expected to abolish the ubiquitin-conjugating activity of PtoUBC34 and its various biological functions, as reported in a yeast E2 ubiquitin-conjugating enzyme RAD6 and Pas2 [63, 64], as well as in Arabidopsis E2s UBC1 and UBC18 [65, 66]. As shown in Fig. 8d, the expression level of the reporter gene was not significantly affected by co-expression with PtoUBC34, PtoUBC34m, or the control vector pUC18, indicating that the expression of PtoUBC34 and PtoUBC34m did not change the general transcription. Co-expression of the effector PtoUBC34 with PtoMYB221 led to a significant increase in the LUC reporter gene activity compared with PtoMYB221 alone as an effector, whereas co-expression of PtoUBC34m with PtoMYB221 did not affect the LUC activity significantly. Because PtoUBC34 did not affect the basal LUC activity, this result suggests that PtoUBC34 reduces the repression activity of PtoMYB221 and likely is involved in the degradation of PtoMYB221.
One of our most significant findings regarding repression activity of PtoMYB221 regulated via PtoUBC34 was that the repression activity depends both on PtoMYB221 and PtoUBC34 protein abundance (Fig. 9). With the increased amount of PtoMYB221, the reporter LUC activity decreased significantly in a dose-dependent manner (Fig. 9). As predicted, the combined transient expression of PtoMYB221 and PtoUBC34 led to a significant decrease in the repression activity of PtoMYB221, compared with that of PtoMYB221 alone as the effector, due to the probable degradation of PtoMYB221 mediated by PtoUBC34 (Fig. 9). Moreover, we observed a lower level of repression activity in the co-transformation with a higher level of PtoUBC34. For example, 2.56 μg PtoMYB221 of the effector plasmid resulted in a 35.16% decrease in the LUC activity compared with the control (without PtoMYB221) in the presence of 1200 ng PtoUBC34, whereas it resulted in a 54.81% decrease with 600 ng PtoUBC34, and a 72.66% decrease when no exogenous PtoUBC34 was added. Indeed, the differential expression of both PtoMYB221 and PtoUBC34 might have occurred under the physiological conditions, as heat shock and high salinity significantly induced the transcription of PtoUBC34 (Fig. 3b and c), and moreover, high temperature, ultraviolet radiation, and cold and high salinity altered the transcriptional expression level of PtoMYB221 (unpublished data). Overall, these data suggest that the dose-dependent changes of the repression activity of PtoMYB221 on both PtoMYB221 and PtoUBC34 protein abundance might play a physiological role in regulating the transcription of downstream target genes in vivo.

PtoUBC34 reduced the repression activity of PtoMYB221 in a dose-dependent manner. All LUC activities are expressed relative to values obtained after transformation with only the reporter plasmid, which was set at 100%. Three independent experiments show similar results. Error bar represents standard deviation of three independent batches of P. tomentosa mesophyll protoplast transfections from one of the experiments. Significance was tested with one-way ANOVA to evaluate the roles of the effectors on the expression level of LUC reporter gene. The different alphabets above the bar indicates statistically significant differences with p value < 0.05, while the same alphabet mean no significant differences
Discussion
Lignin biosynthesis responds to many developmental and environmental cues, including plant hormones, light, sugar content, circadian rhythms, wounding, and pathogen attacks. To fine-tune the response to the various endogenous and exogenous signals, plants have developed a complex mechanism involving transcriptional hierarchical regulation of lignin pathway genes, which integrates many transcriptional activators and repressors [8, 13]. The repressors play equally important roles as activators in the transcriptional control of lignin biosynthesis, which is regulated by a variety of environmental cues and upstream TFs [8, 20, 46, 67]. In this study, we discovered another layer of the post-translational regulation of the subgroup 4 repressors of the R2R3-MYB family by an ER-localized ubiquitin-conjugating enzyme PtoUBC34 in poplar.
We reported several distinct mechanisms of regulation of the subgroup 4 repressors of the R2R3-MYB family by the ER-localized E2 PtoUBC34 in poplar. We showed that transcriptional repressors PtoMYB221 and PtoMYB156 interact with PtoUBC34 (Figs. 5 and 6), which decreases transrepression activity of PtoMYB221 (Figs. 8d and 9), through either the ERAD of the transcriptional repressors or sequestration in the ER. Typically, the ERAD process, as the primary means of quality control within the protein secretory pathway, targets misfolded or misassembled proteins [51, 53, 68,69,70,71]. However, the PtoUBC34-mediated regulation is likely linked with the quantity control of the substrate protein rather than quality control and is independent of the maturation status of substrate proteins. The transient expression assay in poplar mesophyll protoplasts indicated that the increased level of PtoMYB221 (from 640 ng to 2560 ng per transfection) failed to further reduce the reporter LUC activity, although no exogenous PtoUBC34 was added, suggesting that the native PtoUBC34 was involved in regulating the activity of PtoMYB221 in the poplar protoplasts (Fig. 9). Our conclusion of protein quantity control by the ER-localized E2 PtoUBC34 is supported by the best studied example of the protein abundance control of HMG-CoA-reductase (HMGR) by gp78 ubiquitin ligase complex [72, 73]. Moreover, PtoUBC34 decreased the transrepression activity of PtoMYB221 in a dose-dependent manner (Fig. 9), and lower transrepression activity of PtoMYB221 occurred with higher levels of PtoUBC34. This result is consistent with the data obtained in yeast and Arabidopsis in which case the elevated expression level of ERAD components increased the degradation rate of their substrates [50, 52, 74]. This strategy to regulate MYB protein abundance might have physiological significance in plants because the abundance of MYB TFs highly regulates their transactivation activity and the downstream target selectivity [46, 75].
Although PtoMYB221 and PtoMYB156 localize exclusively to the nucleus as a common TF (Fig. 5a and c), its complex with PtoUBC34 is ER specific (Fig. 5b and d). The reduced transrepression activity of MYB221 in protoplast when co-transfected with PtoUBC34 (Figs. 8d and 9) revealed that PtoUBC34 functioned to regulate the transrepression activity of MYB221 by redirecting MYB221 to ER instead of to the nucleus after the TF departed from ribosomal machinery. Whether or not the MYB221 protein is to be degraded in the ER needs further clarification. The interaction between PtoMYB34 and TF PtoMYB221 must occur in the cytosolic side of the ER membrane, in which PtoUBC34 might function in a similar manner as its yeast ortholog Ubc6p with its C-terminal hydrophobic tail embedded in the ER membrane and the catalytic domain facing the cytosol [76, 77]. Conversely, yeast TF Matalpha 2, a well-studied ERAD substrate, gained access in situ to the ERAD ubiquitin-protein ligase Doa10, which was transferred and co-localized at the inner nuclear membrane together with the other components of the ubiquitination machinery [78]. This suggests that substrate-enzyme interaction in the ubiquitination mechanism is regulated spatially, which is also a key aspect of the specificity of E2s and E3s [78, 79]. This mechanism provides real-time surveillance for proteins during the entire journey form ribosomes to their final destination. This notion is supported by the ubiquitination of PHOSPHATE TRANSPORTER1, which was regulated by ubiquitin-conjugating enzyme PHO2/UBC24 in post-endoplasmic reticulum [80] and was ubiquitinated by an E3 ubiquitin ligase NLA in plasma membranes [81]. The ER-localized E2 PtoUBC34 described here provides a potential mechanism for the temporal and spatial regulation of TFs in the compartment of ER prior to reaching the nucleus.
Generally, substrate specificity in ubiquitination is conferred by the E3 ubiquitin ligases [82, 83], which is consistent with the fact that there are many E3 component-associated genes (approximately 1300) in Arabidopsis genome [40] and only 37 members in the E2 family [84]. However, many E2 proteins have been shown to interact with the substrates and regulate their stability. For example, tobacco UBC2 interacts with NtERF3 and promotes its degradation [85]. Arabidopsis PHO2/UBC24 interacts with PHO1 and mediates the degradation of PHO1 to maintain Pi homeostasis [86]. Moreover, Arabidopsis UBC1–6, UBC19–20, and UBC22 have been shown to display variable levels of E3-independent ubiquitination activity [84]. In this study, we found that poplar UBC34 interacted with TF PtoMYB221 in the ER and regulated its transrepression activity (Figs. 6 and 8d). Whether or not PtoUBC34 functions on the ubiquitination and degradation of TF PtoMYB221 as a bonafide E2 and/or an E3-independent E2 needs to be further clarified by additional protein stability and ubiquitination assays.
Poplar TFs PtoMYB221 and PtoMYB156 function as transcriptional repressors in the lignin pathway (Fig. 7), leading to downregulation of lignin pathway genes [36, 37]. However, their physiological roles in plants remain unclear. The contradictory facts appear between the gene expression specificity and their biological function. As transcriptional repressors for lignin biosynthesis, both PtoMYB221 and PtoMYB156 are expressed highly in xylem in which lignin biosynthesis predominantly takes place [36, 37].
We proposed that the activity of PtoMYB221 and PtoMYB156 was fine-tuned by PtoUBC34 and that the translocation of PtoMYB221 and PtoMYB156 to the ER could relieve their repressor activity on phenylpropanoid and lignin pathway. This fine-tuning mechanism might modulate lignin deposition in specific cells under specific physiological conditions by responding to various developmental and environmental cues, such as light, sugar content, circadian rhythms, plant hormones, and wounding (Fig. 10). Further studies will focus on evaluating genetic impact on the transcriptional activity and stability of PtoMYB221 and PtoMYB156 mediated by PtoUBC34 and the influence of environmental cues on the transcriptional activity and stability of the PtoMYB221 protein. This investigation might help to elucidate the fine-tuning mechanism modulating the transcriptional activity of PtoMYB221 and PtoMYB156 and to further elucidate the cross-talk between the lignin biosynthesis pathway and other physiological processes.

Model of the roles of PtoUBC34 on the regulation of lignin biosynthesis. a. Schematic of cell with on-duty UBC34, and the gray part indicated PtoMYB221 absence in nucleus. b. Schematic of cell with off-duty UBC34, and the gray part indicated PtoMYB221 absence in ER compartments
Conclusions
This study has contributed to increase the knowledge about the molecular regulation mechanisms in lignin biosynthesis. To address the role of post-regulation in lignin biosynthesis we screened a Y2H cDNA library of poplar developing vascular tissues against transcriptional repressors PtoMYB221 and PtoMYB156, which was shown to play important roles in lignin biosynthetic pathway. We identified and characterized a Populus tomentosa ER-localized E2 ubiquitin-conjugating enzyme, PtoUBC34. We showed that it interacts with transcriptional repressors PtoMYB221 and PtoMYB156. This interaction leads to translocation of PtoMYB221 and PtoMYB156 from the nucleus to the ER and reduces their repression activity in a PtoUBC34 abundance-dependent manner. Further studies are required to evaluate genetic impact on the transcriptional activity and stability of PtoMYB221 and PtoMYB156 mediated by PtoUBC34. Even so, our data here indicate a mechanism by which lignin biosynthesis is regulated by an ER-localized E2 ubiquitin-conjugating enzyme in plants, which probably links the ERAD process with lignin biosynthesis.
Methods
Plant materials
P. tomentosa ‘BJHR01’ was maintained as described [87]. We used four-week-old plants cultured on MS (Murashig and Skoog) medium with 3% sucrose and 0.6% agar for isolation of mesophyll protoplasts. These plants were multiplied by nodal segments in vitro every 6–8 weeks. We selected five healthy, rapidly growing, two-year-old trees at a field site in Beijing, China, for collection of developing vascular tissues samples for cDNA library construction. The sample collection was done in the morning on a clear day in May 2012. We used a 10 cm region starting from the ground level to collect the differentiating phloem and xylem, and vascular calcium after the stem bark was peeled. All samples were pooled. We used clonally propagated, two-month-old plants grown in a greenhouse under a 16 h/8 h day/night photoperiod at 24 °C for qRT-PCR analysis of the PtoUBC34 transcript level in various tissues. The following samples were harvested from three well-grown plants: 1 cm segment of shoot tip (shoot tip), the first fully expanded leaf without petiole and main vein (leaf), the stem segment of internode 9 which was undergoing secondary growth (stem), the bottom 10 cm of the stem which was used to collect differentiating xylem and phloem. By peeling off the bark, differentiating xylem was sampled by scraping the surface of the exposed wood, and differentiating phloem was collected by scaping the inner-surface of the exposed bark. The plants that were used for either NaCl or heat shock treatment were subjected to aerated hydroculture with Hoagland nutrient solution in a plant incubator (MLR-352H, Panasonic, Japan) under a 16 h/8 h day/night photoperiod at 24 °C for two months. For heat shock tests, plants were preheated at 37 °C for 1 h and allowed to recover at 24 °C for 2 h prior to 42 °C for 2.5 h. For salt treatments, the medium was supplemented with 300 mM NaCl. Mature leaves with the leaf plastochron index 5 of three plants were harvested either at each time point during the heat shock treatment, or at 0, 1, and 6 h of NaCl treatment. All collected plant materials were flash frozen in liquid nitrogen upon harvesting and stored at − 80 °C until RNA was extracted.
Library construction and screening
We conducted the Y2H cDNA library construction for P. tomentosa developing vascular tissues according to the instructions provided with the normalized cDNA library construction package (#P01011, Dualsystems Biotech AG, Schlieren, Switzerland). The library was screened using PtoMYB221 and PtoMYB156 as the bait in Saccharomyces cerevisiae strain NMY51, in accordance with the recommended procedures in the DUALhunter system (#P01601-P01629, Dualsystems Biotech AG, Schlieren, Switzerland). We identified the final isolated positive clones by sequencing.
qRT-PCR and RT-PCR
We isolated total RNA from mature leaf, stem, shoot tip, phloem, and xylem using RNeasy Plant Mini Kit (#74903, QIAGEN Inc., Valencia, CA, USA) and treated it with DNase I (#79254, QIAGEN Inc., Valencia, CA, USA) to remove DNA. We performed qTR-PCR as previously described [87] using specific primers (Additional file 2: Table S1) with three biological replicates (three biological samples from three plants, respectively). Each reaction was repeated three times. The full-length CDS of PtoUBC34 was amplified using a primer set PtoUBC34-fF1/−fR1(Additional file 2: Table S1).
Y2H assay and constructs
We performed split-ubiquitin Y2H assay according to the instructions provided in the DUALhunter starter kit (#P01601-P01629, Dualsystems Biotech AG, Schlieren, Switzerland) [48]. The coding sequences of PtoMYB221 and PtoMYB156 were amplified using primer sets PtoMYB221-yF1/−yR1, PtoMYB156-yF1/−yR1 (Additional file 2: Table S1), respectively, and then were cloned into the two SfiI sites of the bait vector pDHB1 and fused at the N-terminus to the Cub fragment. The full-length PtoUBC34 coding sequence was amplified with primer set PtoUBC34-yF1/−yR1, then cloned into the two SfiI sites of the prey vector pPR3-N, and fused at the C-terminus to Nub fragment. Yeast strain NMY51 cells were co-transformed with the resulting constructs and plated onto synthetic medium lacking Leu, Trp, and His. We confirmed the specificity of protein-protein interactions by the activity of β-Gal.
P. tomentosa protoplast isolation and transfection
We isolated P. tomentosa mesophyll protoplasts from fully extended leaves of the four-week-old plants grown on MS medium and transfected them with plasmid DNA as described by Guo et al. [88].
Subcellular localization and co-localization
To construct the fusion proteins YFP:PtoUBC34, C-repeat-binding factor 1:RFP (CBF1:RFP) [89], RFP:PtoUBC34, and RFP:PtoUBC34s, the coding regions of the fluorescent protein (YFP or RFP) and the target protein (PtoUBC34, PtoUBC34s, or CBF1) were amplified using the corresponding primer sets shown in Additional file 2: Table S1, and then were cloned into linearized pGreen0029-35S [90] digested with HindIII and SacI by homologous recombination, as described (Trelief™ SoSoo Cloning Kit Ver.2, Beijing TsingKe Biotech Co., Ltd.), producing pGreen0029-35S-YFP:PtoUBC34, pGreen0029-35S-RFP:PtoUBC34, pGreen0029-35S-RFP:PtoUBC34s, and the nuclear marker vector pGreen0029-35S-CBF1:RFP. To prepare YFP-tagged PtoMYB221 and PtoMYB156 protein, the coding region of YFP was amplified using primer sets YFP-F2/−R2, with a BamHI restriction site included in the forward primer and a SacI site in the reverse primer (Additional file 2: Table S1), cloned into pGreen0029-35S, producing pGreen0029-35S-YFP. Then we cloned the polymerase chain reaction (PCR) -amplified product of PtoMYB221 and PtoMYB156 coding regions with primer sets PtoMYB221-F1/−R1 and PtoMYB156-F1/−R1 into the KpnI and BamHI sites of pGreen0029-35S-YFP, producing pGreen0029-35S-PtoMYB221:YFP and pGreen0029-35S-PtoMYB156:YFP. We prepared the ER marker vector pGreen0029-35S-Bip:RFP according to the description by Kim et al. [57]. For subcellular localization, YFP:PtoUBC34 fusion was co-transformed with ER marker Bip:RFP, and each of the PtoMYB221:YFP and PtoMYB156:YFP was co-transformed with nuclear marker CBF1:RFP into poplar mesophyll protoplasts. For co-localization, each of the PtoMYB221:YFP and PtoMYB156: YFP was co-transformed with either RFP:PtoUBC34 or RFP:PtoUBC34s into the mesophyll protoplasts. After incubation for 12 h, we observed the fluorescence under a Nikon inverted fluorescence microscope TE2000-E equipped with a Nikon D-Eclipse A1 spectral confocal laser scanning system (Nikon, Tokyo, Japan). The excitation wavelength was 488 nm for YFP and 543 nm for RFP. We selected the images under a confocal microscope to be green for YFP and red for RFP. When these two fluorescent signals overlapped, the resulting merged image was yellow.
BiFC
The coding regions of PtoMYB221, PtoMYB156, and PtoUBC34s were amplified using primer sets PtoMYB221-F2/−R2, PtoMYB156-F2/−R2, and PtoUBC34s-F2/−R2, respectively, with a BamHI restriction site included in the forward primer and a SmaI site in the reverse primer, and then cloned into pUC-pSPYCE(M) [91], resulting in pUC-35S-PtoMYB221:eYFPC, pUC-35S-PtoMYB156:eYFPC, and pUC-35S-PtoUBC34s:eYFPC. Similarly, the coding regions of PtoMYB221, PtoMYB156, and PtoUBC34s were amplified using primer sets PtoMYB221-F2/−R3, PtoMYB156-F2/−R3, and PtoUBC34s-F2/−R3, respectively, with introduction of BamHI and SmaI restriction sites, and cloned into pUC-pSPYNE(R)173 [91], resulting in pUC-35S-eYFPN:PtoMYB221, pUC-35S-eYFPN:PtoMYB156, and pUC-35S- eYFPN:PtoUBC34s. Each of the four pairs eYFPN:PtoUBC34s and PtoMYB221:eYFPC, eYFPN:PtoUBC34s and PtoMYB156:eYFPC, PtoUBC34s:eYFPC and eYFPN:PtoMYB221, and PtoUBC34s:eYFPC and eYFPN:PtoMYB156 together with Bip:RFP were co-transfected into the mesophyll protoplasts. We detected fluorescent signals as described earlier.
Coimmunoprecipitation experiments in poplar mesophyll protoplasts
Coimmunoprecipitation assays were conducted as described previously [92]. The coding sequence of truncated PtoUBC34 (PtoUBC34s) was cloned into the pCAMBIA1307-3flag vector yielding UBC34s-Flag. The coding sequence of PtoMYB156 and PtoMYB221 was cloned into the pCAMBIA1307-6myc vector yielding MYB156:Myc and MYB221:Myc. The UBC34s:Flag vector was co-transformed with MYB156:Myc or MYB221:Myc in poplar protoplasts. After a 16-h incubation, total proteins were extracted from the protoplasts. The protoplast protein extract was incubated with anti-Flag agarose (Sigma-Aldrich) at 4 °C for 2 h. The immunoprecipitates (IP) and input proteins were detected by immunoblotting with either anti-Myc (Abmart) or anti-Flag (MBL) antibodies.
Effector-reporter-based gene transactivation assays
The vectors CaMV35S-GAL4-TATA-Ω-LUC-Nos and CaMV35S-GAL4DB-SUPRD, gifts from Prof. Masaru Ohme-Takagi (National Institute of Advanced Industrial Technology and Science, Tokyo, Japan) [93], were used directly or modified to construct the corresponding reporter vectors CaMV35S-3xACx-TATA-Ω-LUC-Nos and effector constructs CaMV35S-GAL4DB-PtoMYB221, CaMV35S-GAL4DB-PtoMYB156. In brief, we chemically synthesized each of the 3xAC-I, 3x AC-II, 3x AC-III, and 3x AC-IV cis-elements of monolignol pathway genes combined with TATA-Ω sequences, which were flanked by HindIII and SalI sites. The synthesized fragments were digested with HindIII and SalI and then inserted into HindIII and SalI- linearized CaMV35S-GAL4-TATA-Ω-LUC-Nos, in which the elements GAL4-TATA-Ω were removed, resulting in four reporter constructs CaMV35S-3xAC-I-TATA-Ω-LUC-Nos, CaMV35S-3XAC-II-TATA-Ω-LUC-Nos, CaMV35S-3XAC-III-TATA-Ω-LUC-Nos, and CaMV35S-3XAC-IV-TATA-Ω-LUC-Nos. To prepare effector constructs shown in Fig. 7a, CaMV35S-GAL4DB-SUPRD was modified by using a Fast Mutagenesis System (TransGen Biotech, Beijing, China), to produce a SmaI restriction site before the element SUPRD, which then was linearized by SmaI and SalI. Next, we introduced the PCR products of PtoMYB221 and PtoMYB156 coding regions obtained with primer sets MYB221-F5/−R5 and PtoMYB156-F5/−R5 (Additional file 2: Table S1) into the linearized CaMV35S-GAL4DB-SUPRD (in which the element SUPRD was removed) by homologous recombination (Trelief™ SoSoo Cloning Kit Ver.2, Beijing TsingKe Biotech Co., Ltd.), to obtain effector constructs CaMV35S-GAL4DB-PtoMYB221 and CaMV35S-GAL4DB-PtoMYB156. To prepare effector constructs shown in Fig. 8a, PCR-amplified products of PtoMYB221 and PtoMYB156 coding regions with primer sets PtoMYB221-F4/−R4 and PtoMYB156-F4/−R4 (Additional file 2: Table S1) were cloned into the BamHI-EcoRI sites of pGreen0029-35S [90], generating pGreen0029-35S-PtoMYB221 and pGreen0029-35S-PtoMYB156. Mutated PtoUBC34 (PtoUBC34m), in which the cysteine residue at position 87 was replaced with an alanine residue, was made by a Fast Mutagenesis System (TransGen Biotech, Beijing, China). We obtained effector plasmids pGreen0029-35S-PtoUBC34 and pGreen0029-35S-PtoUBC34m by homologous recombination of PCR products of PtoUBC34 and PtoUBC34m coding regions with primer sets PtoUBC34-F3/−R (Additional file 2: Table S1) into the HindIII and SacI sites of linearized pGreen0029-35S [90]. Renilla luciferase gene (RLuc) under control of the CaMV35S promoter was used as an internal control for normalization of LUC expression values for each transfection. 3.2 μg of each of the reporter constructs was co-transformed with 0.8 μg of RLuc internal control construct and corresponding effector constructs into P. tomentosa mesophyll protoplasts [88]. After incubation for 12 h, we performed LUC assays with the Dual-Luciferase Reporter Assay System (#E1980, Promega, Madison, WI, USA) and a luminescence reader (GloMax®20/20; Promega, Madison, WI, USA). The LUC expression values of each transfection were normalized to RLuc values. We used pUC18 as the control vector, and expressed all LUC activities relative to values obtained after co-transformation with the reporter plasmid and pUC18 (with the value set at 100%). The experiments were repeated three times. We expressed the values of relative LUC activity as an average of three independent batches of P. tomentosa mesophyll protoplast transfections from one of the experiments.
Accession numbers
The A. thaliana and P. trichocarpa Genome Initiative locus identifiers or NCBI Protein IDs of the genes investigated in this study are as follows: UBC6 (NP_011026), UBC7 (NP_013735), UBE2J1 (NP_057105), UBE2J2 (NP_477515), UBE2G2 (NP_003334), AtUBC32 (AT3G17000), AtUBC33 (AT5G50430), AtUBC34 (AT1G17280), PtoUBC34 (MH708242), Potri.001G162200, Potri.003G073100, Potri.008G106300, Potri.010G143900, and PeUBC34-like (XP_011048632).
Abbreviations
- BiFC:
-
Bimolecular fluorescence complementation
- Bip:
-
Chaperone binding protein
- CaMV35S promoter:
-
Cauliflower mosaic virus 35S promoter
- CDS:
-
Coding sequence
- Cub:
-
C-terminal half of the ubiquitin
- ER:
-
Endoplasmic reticulum
- ERAD:
-
ER-associated protein degradation
- LUC:
-
Luciferase
- Nub:
-
N-terminal half of the ubiquitin
- PCR:
-
Polymerase chain reaction
- qRT-PCR:
-
Quantitative real-time polymerase chain reaction
- RFP:
-
Red fluorescent protein
- RLuc:
-
Renilla luciferase
- RT-PCR:
-
Reverse transcription polymerase chain reaction
- SCW:
-
Secondary cell wall
- TF:
-
Transcription factor
- UBCc:
-
Ubiqutin-conjugating enzyme catalytic
- Y2H:
-
Yeast two-hybridization
- YFP:
-
Yellow fluorescent protein
- β-Gal:
-
β-galactosidase
References
Weng JK, Chapple C. The origin and evolution of lignin biosynthesis. New Phytol. 2010;187(2):273–85.
Boerjan W, Ralph J, Baucher M. Lignin biosynthesis. Annu Rev Plant Biol. 2003;54:519–46.
Pilate G, Guiney E, Holt K, Petit-Conil M, Lapierre C, Leple JC, Pollet B, Mila I, Webster EA, Marstorp HG, et al. Field and pulping performances of transgenic trees with altered lignification. Nat Biotechnol. 2002;20(6):607–12.
Himmel ME, Ding SY, Johnson DK, Adney WS, Nimlos MR, Brady JW, Foust TD. Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science. 2007;315(5813):804–7.
Baucher M, Halpin C, Petit-Conil M, Boerjan W. Lignin: genetic engineering and impact on pulping. Crit Rev Biochem Mol Biol. 2003;38(4):305–50.
Huntley SK, Ellis D, Gilbert M, Chapple C, Mansfield SD. Significant increases in pulping efficiency in C4H-F5H-transformed poplars: improved chemical savings and reduced environmental toxins. J Agr Food Chem. 2003;51(21):6178–83.
Chapple C, Ladisch M, Meilan R. Loosening lignin's grip on biofuel production. Nat Biotechnol. 2007;25(7):746–8.
Zhao Q, Dixon RA. Transcriptional networks for lignin biosynthesis: more complex than we thought? Trends Plant Sci. 2011;16(4):227–33.
Zhong R, Lee C, Ye ZH. Evolutionary conservation of the transcriptional network regulating secondary cell wall biosynthesis. Trends Plant Sci. 2010;15(11):625–32.
Wang HZ, Dixon RA. On-off switches for secondary cell wall biosynthesis. Mol Plant. 2012;5(2):297–303.
Martin C, Paz-Ares J. MYB transcription factors in plants. Trends Genet. 1997;13(2):67–73.
Weisshaar B, Jenkins GI. Phenylpropanoid biosynthesis and its regulation. Curr Opin Plant Biol. 1998;1(3):251–7.
Castanet-Tolosan F. Transcriptional regulation of the lignin biosynthetic pathway revisited: new players and insights. Adv Bot Res. 2012;61:173–218.
Chezem WR, Clay NK. Regulation of plant secondary metabolism and associated specialized cell development by MYBs and bHLHs. Phytochemistry. 2016;131:26–43.
Raes J, Rohde A, Christensen JH, Van de Peer Y, Boerjan W. Genome-wide characterization of the lignification toolbox in Arabidopsis. Plant Physiol. 2003;133(3):1051–71.
Lacombe E, Van Doorsselaere J, Boerjan W, Boudet AM, Grima-Pettenati J. Characterization of cis-elements required for vascular expression of the cinnamoyl CoA reductase gene and for protein-DNA complex formation. Plant J. 2000;23(5):663–76.
Zhou J, Lee C, Zhong R, Ye ZH. MYB58 and MYB63 are transcriptional activators of the lignin biosynthetic pathway during secondary cell wall formation in Arabidopsis. Plant Cell. 2009;21(1):248–66.
Patzlaff A, Newman LJ, Dubos C, Whetten RW, Smith C, McInnis S, Bevan MW, Sederoff RR, Campbell MM. Characterisation of Pt MYB1, an R2R3-MYB from pine xylem. Plant Mol Biol. 2003;53(4):597–608.
Zhong R, Richardson EA, Ye ZH. The MYB46 transcription factor is a direct target of SND1 and regulates secondary wall biosynthesis in Arabidopsis. Plant Cell. 2007;19(9):2776–92.
Ko JH, Kim WC, Han KH. Ectopic expression of MYB46 identifies transcriptional regulatory genes involved in secondary wall biosynthesis in Arabidopsis. Plant J. 2009;60(4):649–65.
Kim WC, Ko JH, Kim JY, Kim J, Bae HJ, Han KH. MYB46 directly regulates the gene expression of secondary wall-associated cellulose synthases in Arabidopsis. Plant J. 2013;73(1):26–36.
McCarthy RL, Zhong R, Ye ZH. MYB83 is a direct target of SND1 and acts redundantly with MYB46 in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell Physiol. 2009;50(11):1950–64.
McCarthy RL, Zhong R, Fowler S, Lyskowski D, Piyasena H, Carleton K, Spicer C, Ye ZH. The poplar MYB transcription factors, PtrMYB3 and PtrMYB20, are involved in the regulation of secondary wall biosynthesis. Plant Cell Physiol. 2010;51(6):1084–90.
Patzlaff A, McInnis S, Courtenay A, Surman C, Newman LJ, Smith C, Bevan MW, Mansfield S, Whetten RW, Sederoff RR, et al. Characterisation of a pine MYB that regulates lignification. Plant J. 2003;36(6):743–54.
Goicoechea M, Lacombe E, Legay S, Mihaljevic S, Rech P, Jauneau A, Lapierre C, Pollet B, Verhaegen D, Chaubet-Gigot N, et al. EgMYB2, a new transcriptional activator from Eucalyptus xylem, regulates secondary cell wall formation and lignin biosynthesis. Plant J. 2005;43(4):553–67.
Zhong R, Lee C, McCarthy RL, Reeves CK, Jones EG, Ye ZH. Transcriptional activation of secondary wall biosynthesis by rice and maize NAC and MYB transcription factors. Plant Cell Physiol. 2011;52(10):1856–71.
Zhong R, Lee C, Zhou J, McCarthy RL, Ye ZH. A battery of transcription factors involved in the regulation of secondary cell wall biosynthesis in Arabidopsis. Plant Cell. 2008;20(10):2763–82.
Bomal C, Bedon F, Caron S, Mansfield SD, Levasseur C, Cooke JE, Blais S, Tremblay L, Morency MJ, Pavy N, et al. Involvement of Pinus taeda MYB1 and MYB8 in phenylpropanoid metabolism and secondary cell wall biogenesis: a comparative in planta analysis. J Exp Bot. 2008;59(14):3925–39.
Wang H, Zhao Q, Chen F, Wang M, Dixon RA. NAC domain function and transcriptional control of a secondary cell wall master switch. Plant J. 2011;68(6):1104–14.
Shen H, He X, Poovaiah CR, Wuddineh WA, Ma J, Mann DG, Wang H, Jackson L, Tang Y, Stewart CN Jr, et al. Functional characterization of the switchgrass (Panicum virgatum) R2R3-MYB transcription factor PvMYB4 for improvement of lignocellulosic feedstocks. New Phytol. 2012;193(1):121–36.
Fornalé S, Sonbol FM, Maes T, Capellades M, Puigdomènech P, Rigau J, Caparrós-Ruiz D. Down-regulation of the maize and Arabidopsis thaliana caffeic acid O-methyl-transferase genes by two new maize R2R3-MYB transcription factors. Plant Mol Biol. 2006;62(6):809–23.
Fornalé S, Shi X, Chai C, Encina A, Irar S, Capellades M, Fuguet E, Torres JL, Rovira P, Puigdomènech P, et al. ZmMYB31 directly represses maize lignin genes and redirects the phenylpropanoid metabolic flux. Plant J. 2010;64(4):633–44.
Sonbol FM, Fornalé S, Capellades M, Encina A, Touriño S, Torres JL, Rovira P, Ruel K, Puigdomènech P, Rigau J, et al. The maize ZmMYB42 represses the phenylpropanoid pathway and affects the cell wall structure, composition and degradability in Arabidopsis thaliana. Plant Mol Biol. 2009;70(3):283–96.
Karpinska B, Karlsson M, Srivastava M, Stenberg A, Schrader J, Sterky F, Bhalerao R, Wingsle G. MYB transcription factors are differentially expressed and regulated during secondary vascular tissue development in hybrid aspen. Plant Mol Biol. 2004;56(2):255–70.
Ma QH, Wang C, Zhu HH. TaMYB4 cloned from wheat regulates lignin biosynthesis through negatively controlling the transcripts of both cinnamyl alcohol dehydrogenase and cinnamoyl-CoA reductase genes. Biochimie. 2011;93(7):1179–86.
Tang X, Zhuang Y, Qi G, Wang D, Liu H, Wang K, Chai G, Zhou G. Poplar PdMYB221 is involved in the direct and indirect regulation of secondary wall biosynthesis during wood formation. Sci Rep. 2015;5:12240.
Yang L, Zhao X, Ran L, Li C, Fan D, Luo K. PtoMYB156 is involved in negative regulation of phenylpropanoid metabolism and secondary cell wall biosynthesis during wood formation in poplar. Sci Rep. 2017;7:41209.
Lin YC, Li W, Sun YH, Kumari S, Wei H, Li Q, Tunlaya-Anukit S, Sederoff RR, Chiang VL. SND1 transcription factor-directed quantitative functional hierarchical genetic regulatory network in wood formation in Populus trichocarpa. Plant Cell. 2013;25(11):4324–41.
Gaston K, Jayaraman PS. Transcriptional repression in eukaryotes: repressors and repression mechanisms. Cell Mol Life Sci. 2003;60(4):721–41.
Smalle J, Vierstra RD. The ubiquitin 26S proteasome proteolytic pathway. Annu Rev Plant Biol. 2004;55:555–90.
Petzold HE, Zhao M, Beers EP. Expression and functions of proteases in vascular tissues. Physiol Plant. 2012;145(1):121–9.
Vierstra RD. The ubiquitin-26S proteasome system at the nexus of plant biology. Nat Rev Mol Cell Biol. 2009;10(6):385–97.
Ciechanover A. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 1998;17(24):7151–60.
Beris D, Kapolas G, Livanos P, Roussis A, Milioni D, Haralampidis K. RNAi-mediated silencing of the Arabidopsis thaliana ULCS1 gene, encoding a WDR protein, results in cell wall modification impairment and plant infertility. Plant Sci. 2016;245:71–83.
Borah P, Khurana JP. The OsFBK1 E3 ligase subunit affects anther and root secondary Cell Wall thickenings by mediating turnover of a Cinnamoyl-CoA reductase. Plant Physiol. 2018;176(3):2148–65.
Jin H, Cominelli E, Bailey P, Parr A, Mehrtens F, Jones J, Tonelli C, Weisshaar B, Martin C. Transcriptional repression by AtMYB4 controls production of UV-protecting sunscreens in Arabidopsis. EMBO J. 2000;19(22):6150–61.
Schwechheimer C, Bevan M. The regulation of transcription factor activity in plants. Trends Plant Sci. 1998;3(10):378–83.
Möckli N, Deplazes A, Hassa PO, Zhang Z, Peter M, Hottiger MO, Stagljar I, Auerbach D. Yeast split-ubiquitin-based cytosolic screening system to detect interactions between transcriptionally active proteins. Biotechniques. 2007;42:725–30.
Sommer T, Jentsch S. A protein translocation defect linked to ubiquitin conjugation at the endoplasmic reticulum. Nature. 1993;365(6442):176–9.
Swanson R, Locher M, Hochstrasser M. A conserved ubiquitin ligase of the nuclear envelope/endoplasmic reticulum that functions in both ER-associated and Matalpha2 repressor degradation. Genes Dev. 2001;15(20):2660–74.
Ravid T, Kreft SG, Hochstrasser M. Membrane and soluble substrates of the Doa10 ubiquitin ligase are degraded by distinct pathways. EMBO J. 2006;25(3):533–43.
Lenk U, Yu H, Walter J, Gelman MS, Hartmann E, Kopito RR, Sommer T. A role for mammalian Ubc6 homologues in ER-associated protein degradation. J Cell Sci. 2002;115(Pt 14):3007–14.
Cui F, Liu L, Zhao Q, Zhang Z, Li Q, Lin B, Wu Y, Tang S, Xie Q. Arabidopsis ubiquitin conjugase UBC32 is an ERAD component that functions in brassinosteroid-mediated salt stress tolerance. Plant Cell. 2012;24(1):233–44.
Bachmair A, Novatchkova M, Potuschak T, Eisenhaber F. Ubiquitylation in plants: a post-genomic look at a post-translational modification. Trends Plant Sci. 2001;6(10):463–70.
Oh RS, Bai X, Rommens JM. Human homologs of Ubc6p ubiquitin-conjugating enzyme and phosphorylation of HsUbc6e in response to endoplasmic reticulum stress. J Biol Chem. 2006;281(30):21480–90.
Finn RD, Attwood TK, Babbitt PC, Bateman A, Bork P, Bridge AJ, Chang HY, Dosztanyi Z, El-Gebali S, Fraser M, et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45(D1):D190–9.
Kim DH, Eu YJ, Yoo CM, Kim YW, Pih KT, Jin JB, Kim SJ, Stenmark H, Hwang I. Trafficking of phosphatidylinositol 3-phosphate from the trans-Golgi network to the lumen of the central vacuole in plant cells. Plant Cell. 2001;13(2):287–301.
Sundell D, Street NR, Kumar M, Mellerowicz EJ, Kucukoglu M, Johnsson C, Kumar V, Mannapperuma C, Delhomme N, Nilsson O, et al. AspWood: high-spatial-resolution transcriptome profiles reveal uncharacterized modularity of wood formation in Populus tremula. Plant Cell. 2017;29(7):1585–604.
Kumar M, Thammannagowda S, Bulone V, Chiang V, Han KH, Joshi CP, Mansfield SD, Mellerowicz E, Sundberg B, Teeri T, et al. An update on the nomenclature for the cellulose synthase genes in Populus. Trends Plant Sci. 2009;14(5):248–54.
Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A. The genome of black cottonwood, Populus trichocarpa (Torr. & gray). Science. 2006;313(5793):1596–604.
Romero I, Fuertes A, Benito MJ, Malpica JM, Leyva A, Paz-Ares J. More than 80R2R3-MYB regulatory genes in the genome of Arabidopsis thaliana. Plant J. 1998;14(3):273–84.
Zhao J, Zhang W, Zhao Y, Gong X, Guo L, Zhu G, Wang X, Gong Z, Schumaker KS, Guo Y. SAD2, an importin -like protein, is required for UV-B response in Arabidopsis by mediating MYB4 nuclear trafficking. Plant Cell. 2007;19(11):3805–18.
Sung P, Prakash S, Prakash L. Mutation of cysteine-88 in the Saccharomyces cerevisiae RAD6 protein abolishes its ubiquitin-conjugating activity and its various biological functions. Proc Natl Acad Sci U S A. 1990;87(7):2695–9.
Wiebel FF, Kunau WH. The Pas2 protein essential for peroxisome biogenesis is related to ubiquitin-conjugating enzymes. Nature. 1992;359(6390):73–6.
Sullivan ML, Vierstra RD. Formation of a stable adduct between ubiquitin and the Arabidopsis ubiquitin-conjugating enzyme, AtUBC1. J Biol Chem. 1993;268(12):8777–80.
Cheng MC, Kuo WC, Wang YM, Chen HY, Lin TP. UBC18 mediates ERF1 degradation under light-dark cycles. New Phytol. 2017;213(3):1156–67.
Preston J, Wheeler J, Heazlewood J, Li SF, Parish RW. AtMYB32 is required for normal pollen development in Arabidopsis thaliana. Plant J. 2004;40(6):979–95.
Jin H, Yan Z, Nam KH, Li J. Allele-specific suppression of a defective brassinosteroid receptor reveals a physiological role of UGGT in ER quality control. Mol Cell. 2007;26(6):821–30.
Hong Z, Jin H, Tzfira T, Li J. Multiple mechanism-mediated retention of a defective brassinosteroid receptor in the endoplasmic reticulum of Arabidopsis. Plant Cell. 2008;20(12):3418–29.
Kostova Z, Tsai YC, Weissman AM. Ubiquitin ligases, critical mediators of endoplasmic reticulum-associated degradation. Semin Cell Dev Biol. 2007;18(6):770–9.
Muller J, Piffanelli P, Devoto A, Miklis M, Elliott C, Ortmann B, Schulze-Lefert P, Panstruga R. Conserved ERAD-like quality control of a plant polytopic membrane protein. Plant Cell. 2005;17(1):149–63.
DeBose-Boyd RA. Feedback regulation of cholesterol synthesis: sterol-accelerated ubiquitination and degradation of HMG CoA reductase. Cell Res. 2008;18(6):609–21.
Song BL, Sever N, DeBose-Boyd RA. Gp78, a membrane-anchored ubiquitin ligase, associates with Insig-1 and couples sterol-regulated ubiquitination to degradation of HMG CoA reductase. Mol Cell. 2005;19(6):829–40.
Gardner RG, Swarbrick GM, Bays NW, Cronin SR, Wilhovsky S, Seelig L, Kim C, Hampton RY. Endoplasmic reticulum degradation requires lumen to cytosol signaling. Transmembrane control of Hrd1p by Hrd3p. J Cell Biol. 2000;151(1):69–82.
Andersson KB, Berge T, Matre V, Gabrielsen OS. Sequence selectivity of c-Myb in vivo. Resolution of a DNA target specificity paradox. J Biol Chem. 1999;274(31):21986–94.
Hirsch C, Gauss R, Horn SC, Neuber O, Sommer T. The ubiquitylation machinery of the endoplasmic reticulum. Nature. 2009;458(7237):453–60.
Gilon T, Chomsky O, Kulka RG. Degradation signals recognized by the Ubc6p-Ubc7p ubiquitin-conjugating enzyme pair. Mol Cell Biol 2000;20(19):7214–7219.
Deng M, Hochstrasser M. Spatially regulated ubiquitin ligation by an ER/nuclear membrane ligase. Nature. 2006;443(7113):827–31.
Pines J, Lindon C. Proteolysis: anytime, any place, anywhere? Nat Cell Biol. 2005;7(8):731–5.
Huang TK, Han CL, Lin SI, Chen YJ, Tsai YC, Chen YR, Chen JW, Lin WY, Chen PM, Liu TY, et al. Identification of downstream components of ubiquitin-conjugating enzyme PHOSPHATE2 by quantitative membrane proteomics in Arabidopsis roots. Plant Cell. 2013;25(10):4044–60.
Lin WY, Huang TK, Chiou TJ. Nitrogen limitation adaptation, a target of microRNA827, mediates degradation of plasma membrane-localized phosphate transporters to maintain phosphate homeostasis in Arabidopsis. Plant Cell. 2013;25(10):4061–74.
Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev. 2002;82(2):373–428.
Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–33.
Kraft E, Stone SL, Ma L, Su N, Gao Y, Lau OS, Deng XW, Callis J. Genome analysis and functional characterization of the E2 and RING-type E3 ligase ubiquitination enzymes of Arabidopsis. Plant Physiol. 2005;139(4):1597–611.
Koyama T, Nii H, Mitsuda N, Ohta M, Kitajima S, Ohme-Takagi M, Sato F. A regulatory cascade involving class II ETHYLENE RESPONSE FACTOR transcriptional repressors operates in the progression of leaf senescence. Plant Physiol. 2013;162(2):991–1005.
Liu TY, Huang TK, Tseng CY, Lai YS, Lin SI, Lin WY, Chen JW, Chiou TJ. PHO2-dependent degradation of PHO1 modulates phosphate homeostasis in Arabidopsis. Plant Cell. 2012;24(5):2168–83.
Wang Y, Chen Y, Ding L, Zhang J, Wei J, Wang H. Validation of reference genes for gene expression by quantitative real-time RT-PCR in stem segments spanning primary to secondary growth in Populus tomentosa. PLoS One. 2016;11(6):e0157370.
Guo J, Morrell-Falvey JL, Labbe JL, Muchero W, Kalluri UC, Tuskan GA, Chen JG. Highly efficient isolation of Populus mesophyll protoplasts and its application in transient expression assays. PLoS One. 2012;7(9):e44908.
Stockinger EJ, Gilmour SJ, Thomashow MF. Arabidopsis thaliana CBF1 encodes an AP2 domain-containing transcriptional activator that binds to the C-repeat/DRE, a cis-acting DNA regulatory element that stimulates transcription in response to low temperature and water deficit. Proc Natl Acad Sci U S A. 1997;94(3):1035–40.
Hellens RP, Edwards EA, Leyland NR, Bean S, Mullineaux PM. pGreen: a versatile and flexible binary Ti vector for agrobacterium-mediated plant transformation. Plant Mol Biol. 2000;42(6):819–32.
Waadt R, Schmidt LK, Lohse M, Hashimoto K, Bock R, Kudla J. Multicolor bimolecular fluorescence complementation reveals simultaneous formation of alternative CBL/CIPK complexes in planta. Plant J. 2008;56(3):505–16.
Ye Q, Wang H, Su T, Wu WH, Chen YF. The ubiquitin E3 ligase PRU1 regulates WRKY6 degradation to modulate phosphate homeostasis in response to low-pi stress in Arabidopsis. Plant Cell. 2018;30(5):1062–76.
Hiratsu K, Mitsuda N, Matsui K, Ohme-Takagi M. Identification of the minimal repression domain of SUPERMAN shows that the DLELRL hexapeptide is both necessary and sufficient for repression of transcription in Arabidopsis. Biochem Bioph Res Co. 2004;321(1):172–8.
Wang Y, Chen YJ, Ding LP, Wei JH, Wang HZ. Validation of reference genes for gene expression analysis in different tissues of Populus tomentosa (in Chinese). Plant Physiol Commun. 2016;52:1312–20.
Acknowledgements
We acknowledge Prof. Masaru Ohme-Takagi (National Institute of Advanced Industrial Technology and Science, Japan) for providing the reporter vector CaMV35S-GAL4-TATA-Ω-LUC-Nos and effector vector CaMV35S-GAL4DB-SUPRD.
Funding
This work was supported by a grant to HW from the National Natural Science Foundation of China (Grant No. 31770708), a grant to JW from the National Key R&D Program of China (Grant No. 2016YFD0600104), grants to HW from the Science Foundation of the Beijing Academy of Agriculture and Forestry Sciences (Grant No. KJCX20170203) and the National Natural Science Foundation of China (Grant No. 31270593), and a grant to YC from the National Key Program on Transgenic Research of China (Grant No. 2018ZX0802002).
Availability of data and materials
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
HW and JW designed the research. DD, YC and LZ performed the research. YZ and LD prepared the plant materials. DD, YC and LZ analysed the data. HW, JW and LZ wrote the paper, LZ and HW revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
The plant materials were collected from the Research base of Beijing Academy of Agriculture and Forestry Sciences, Beijing, China. The experimental research on plants, including collection of plant material, was complied with the institutional, national, or international guidelines. No specific permits were required by the scientific research institute to use these materials.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Figure S1 Transactivation activity analysis of PtoMYB221 and PtoMYB156 in yeast. (DOCX 146 kb)
Additional file 2:
Table S1 Primer sequences. (DOCX 18 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zheng, L., Chen, Y., Ding, D. et al. Endoplasmic reticulum–localized UBC34 interaction with lignin repressors MYB221 and MYB156 regulates the transactivity of the transcription factors in Populus tomentosa. BMC Plant Biol 19, 97 (2019). https://doi.org/10.1186/s12870-019-1697-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12870-019-1697-y