
Abstract
Background
Ferula sinkiangensis is an increasingly endangered medicinal plant. Arbuscular mycorrhiza fungi (AMF) are symbiotic microorganisms that live in the soil wherein they enhance nutrient uptake, stress resistance, and pathogen defense in host plants. While such AMF have the potential to contribute to the cultivation of Ferula sinkiangensis, the composition of AMF communities associated with Ferula sinkiangensis and the relationship between these fungi and other pertinent abiotic factors still remains to be clarified.
Results
Herein, we collected rhizosphere and surrounding soil samples at a range of depths (0–20, 20–40, and 40–60 cm) and a range of slope positions (bottom, middle, top). These samples were then subjected to analyses of soil physicochemical properties and high-throughput sequencing (Illumina MiSeq). We determined that Glomus and Diversispora species were highly enriched in all samples. We further found that AMF diversity and richness varied significantly as a function of slope position, with this variation primarily being tied to differences in relative Glomus and Diversispora abundance. In contrast, no significant relationship was observed between soil depth and overall AMF composition, although some AMF species were found to be sensitive to soil depth. Many factors significantly affected AMF community composition, including organic matter content, total nitrogen, total potassium, ammonium nitrogen, nitrate nitrogen, available potassium, total dissolvable salt levels, pH, soil water content, and slope position. We further determined that Shannon diversity index values in these communities were positively correlated with total phosphorus, nitrate-nitrogen levels, and pH values (P < 0.05), whereas total phosphorus, total dissolvable salt levels, and pH were positively correlated with Chao1 values (P < 0.05).
Conclusion
In summary, our data revealed that Glomus and Diversispora are key AMF genera found within Ferula sinkiangensis rhizosphere soil. These fungi are closely associated with specific environmental and soil physicochemical properties, and these soil sample properties also differed significantly as a function of slope position (P < 0.05). Together, our results provide new insights regarding the relationship between AMF species and Ferula sinkiangensis, offering a theoretical basis for further studies of their development.
Similar content being viewed by others

Background
Ferula sinkiangensis is a perennial plant found only in the Yining region of Xinjiang province, China that blooms only once, and that produces seedlings each March [1, 2]. In years when these plants do not reach the flowering stage, their root systems instead gradually expand and tufted basal leaves develop. After entering the flowering stage, these plants bloom at the beginning of May, bear fruit by mid-late May, and die at the end of June [3, 4]. Ferula sinkiangensis exhibits potent anticancer [5], antibiotic [6], antiviral [7], antioxidant [8], and anti-inflammatory [9] properties, and was thus included in the Pharmacopoeia of the People’s Republic of China in 1977 [4]. Extensive Ferula sinkiangensis harvesting in recent years, however, has caused serious habitat damage [10]. This, coupled with its low reproductive rate and the prevalence of pests, diseases, and poor environmental conditions, has led Ferula sinkiangensis to become increasingly endangered [11]. Efforts to conserve this valuable medicinal herb are thus essential.
Slope position is an important topographical factor that governs microenvironmental heterogeneity by impacting the temperature, light, soil physicochemical properties, and water levels to which plants are exposed [12, 13]. Abiotic and biotic factors additionally vary with soil depth [14]. Soil nutrient content and environmental gradients observed as a function of depth may influence the abundance, composition, and function of soil microbial communities [15]. Although slope position and depth are not direct ecological factors that govern microbial survival, they can influence microbe distributions by controlling the spatiotemporal distribution of a range of ecological factors and combinations thereof. However, the impacts of slope position and soil depth on the rhizosphere arbuscular mycorrhizal fungi (AMF) community composition associated with Ferula sinkiangensis remain poorly understood.
Soil microorganisms have been an increasingly important focus of ecological research [16], as they have been shown to be key regulators of plant growth, development, and overall ecosystem stability [17]. AMF species represent a particularly important subset of soil microbes that are able to form mutualistic symbiotic relationships with the roots of > 80% of all land plants [18,19,20]. Mycorrhizal symbionts are key sources of plant nutrients, and these AMF species can also enhance host plant resistance to environmental stressors [21, 22] and to pathogen infections [23, 24]. Few studies to date, however, have conducted comprehensive evaluations of AMF in the rhizosphere soil associated with Ferula sinkiangensis, and current understanding of the diversity and distribution of these microbial communities remains limited. In addition, the association between these microbes and soil physicochemical properties remains uncertain. As such, it is important that field studies of these AMF communities be conducted.
AMF exhibit rich species diversity, and approaches to studying these fungi have historically included both spore identification efforts and molecular analyses [25, 26]. Spore identification, however, is a time- and energy-intensive task that is susceptible to variability in spore morphology as a function of regional variations, host species, and microbial age, making it challenging to differentiate between the spores of similar species [27]. In contrast, high-throughput sequencing [28, 29] represents an increasingly robust and common approach to reliably studying AMF community structure and diversity. High-throughput approaches have been widely used in studies of AMF in the context of forestry [13], agriculture [30, 31], and environmental remediation [32], while they have also been used to explore the relationship between slope position and soil depth on AMF community composition associated with specific plants.
While prior studies have leveraged high-throughput sequencing to evaluate rhizosphere microorganisms associated with Ferula sinkiangensis [33, 34], no studies have comprehensively assessed the diversity, community composition, or distribution patterns of AMF species associated with this plant. As such, in the present study, we employed an Illumina MiSeq sequencing approach [29] to assess the diversity and structure of rhizosphere AMF communities associated with Ferula sinkiangensis. The goals of this study were as follows: (1) to research the rhizosphere AMF diversity associated with Ferula sinkiangensis; (2) to evaluate AMF community composition and distribution patterns as a function of slope position and soil depth; and (3) to discover the important topographic and edaphic factors affecting AMF diversity, community composition, and distribution patterns.
Results
AMF species diversity
We identified 77 total AMF OTUs in our 27 soil samples, which were separated into 9 groups. Dilution curves generated for these 9 groups were flat, indicating that sequencing depth was sufficient and that additional sequencing depth would have revealed only a small number of additional species (Supplementary Fig. S1).
We next conducted taxonomic analyses of these representative OTUs, leading us to determine that these fungi were associated with 1 Class, 4 Orders, 4 Families, 4 Genera, and 20 Species, with additional unidentified species having been detected at various taxonomic levels. No significant differences in Alpha diversity, Sobs, Shannon, Chao1, or Simpson index values were observed among samples as a function of soil depth, whereas these index values did vary significantly as a function of slope position. Phylogenetic diversity (PD) was unrelated to soil depth or slope position. Greater than 99.98% coverage was achieved in this sequencing analysis, confirming that these data met with the targeted sequencing depth requirements (Table 1).
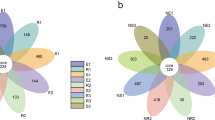
Intermediate junctions were used to identify OTUs common to all samples as well as OTUs unique to specific samples. These analyses revealed that there were 12 core OTUs in our samples, with two of these OTUs belonging to the Diversispora genus and all others belonging to the Glomus genus (Fig. 1).

Operational taxonomic units (OTU)-based petal maps. Each petal corresponds to a sample group, with the shared overlapping region representing OTUs common to all samples, and the numbers on individual petals representing the number of OTUs unique to a given sample group. SW, ZW, and XW respectively correspond to the top, middle, and bottom slope positions. 1, 2, and 3 respectively represent samples collected at a soil depth of 0-20 cm, 20-40 cm, and 40-60 cm
AMF community composition and distribution
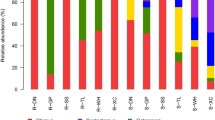
Clear differences in AMF community composition in different rhizosphere soil samples were observed in this study. Glomus species accounted for 83.46% of total AMF species, while Diversispora accounted for 14.73%, Ambispora for 1.21%, and Paraglomus species were only detected in a few samples (Fig. 2a).

The proportion of AMF genera in soil. All the soil samples (a) and soil samples at different slope positions (b). SW, ZW, and XW respectively correspond to the top, middle, and bottom slope positions
In our constructed species relationship diagrams, Glomus distributions in individual samples ranged from 7.7–13%. Of these Glomus species, 31, 28, and 39% were detected in the soil samples from the upper, middle, and lower slope positions, respectively, while 31, 28, and 39% of Glomus species were detected in samples collected at respective soil depths of 0–20 cm, 20–40 cm, and 40–60 cm. Diversispora species were primarily distributed in the upper and middle slope positions, with 52, 47.8, and 0.2% of these species being found in the upper, middle, and lower slope positions, respectively. In addition, 30, 31, and 39% of Diversispora species were detected in samples collected at 0–20 cm, 20–40 cm, and 40–60 cm depths, respectively (Fig. 2b). Glomus and Diversispora species were present at all soil depths and slope positions, with Glomus species composing the largest proportion of all samples. Ambispora species were only detected in soil samples collected at depths of 0–20 cm and 40–60 cm from the middle slope position (Supplementary Fig. S2).
A cluster analysis conducted according to Unweighted UniFrac distance values revealed that samples were separable into three primary categories based upon environmental variables. Clusters of samples collected at the same slope site indicated that AMF community composition was more similar at a given slope site, whereas this composition differed substantially among slope sites (Fig. 3).

A UPGMA tree based on Unweighted Unifrac Distances at the OTU level. The UPGMA cluster tree structure is shown on the left, while on the right is the relative abundance distributions at the OTU level for each sample. SW, ZW, and XW respectively correspond to the top, middle, and bottom slope positions. 1, 2, and 3 respectively represent samples collected at a soil depth of 0-20 cm, 20-40 cm, and 40-60 cm
A Bray-Curtis PCoA analysis indicated that there was no difference in AMF community composition at different soil depths, whereas these communities did differ significantly as a function of sample slope position. While there were no differences in AMF community composition in the middle or upper slope positions, the composition of these samples did differ significantly with respect to the composition of AMF communities in soil samples collected from lower slope positions (R = 0.332, P = 0.001) (Fig. 4a).

A Bray-Curtis PCoA analysis on OTU levels (a). CCA analysis based on OTU levels and sample slope positions (b). Blue triangles, red circles, and green diamonds correspond to samples collected at the middle (ZW), top (SW), and bottom (XW) of the slope, respectively. The light blue circles correspond to the top 20 OTUs by abundance
LEFse analyses were also used to identify biomarkers associated with AMF diversity in rhizosphere soil. At different soil depths, only Glomus species were significantly enriched at the species and OTU levels. The most abundant biomarkers were detected in soil samples collected at a depth of 0–20 cm, with decreasing levels of these biomarkers as soil depth increased (Fig. 5b). With respect to slope position, biomarker abundance was highest in the lower slope position. However, Glomus species were significantly enriched in samples collected from the lower slope position, whereas Diversispora species were significantly enriched in samples collected from the upper slope position (Fig. 5a).

A linear discriminant analysis effect size (Lefse) analysis of differences in AMF community composition as a function of soil depth and slope position. Differently colored nodes correspond to microbial groups that were significantly enriched in the corresponding groups and that significantly influenced the differences between groups. Light yellow nodes correspond to microbial groups with no significant differences in different groups or that had no significant influence on the difference between groups. SW, ZW, and XW respectively correspond to the top, middle, and bottom slope positions. 1, 2, and 3 respectively represent samples collected at a soil depth of 0-20 cm, 20-40 cm, and 40-60 cm
Relationships between soil AMF diversity and soil properties
Significant differences in pH and TP were observed between soil samples as a function of soil depth (Table 2; P < 0.05), but these differences may be a function of different slope positions, as soil from the same slope position did not differ as a function of depth. When comparing soil samples collected from different slope positions, there were significant differences in OM, TN, TP, TK, AN, NN, TDS, pH, and SM values (P < 0.05).
Spearman correlation analyses revealed that TP, AN, and soil pH were significantly positively correlated with Shannon index values (P < 0.05), whereas TP, TDS, pH, AE, and Chao 1 values were positively correlated with one another (P < 0.05) (Supplementary Table S1).
CCA analysis revealed that OM (R2 = 0.3939, P = 0.002), TN (R2 = 0.2701, P = 0.021), TK (R2 = 0.3209, P = 0.012), NN (R2 = 0.3003, P = 0.012), AN (R2 = 0.2692, P = 0.03), AK (R2 = 0.3803, P = 0.001), TDS (R2 = 0.5328, P = 0.001), pH (R2 = 0.4834, P = 0.001), SM (R2 = 0.4108, P = 0.004), and AE (R2 = 0.335, P = 0.011) all had a significant impact on AMF community composition, explaining 24.07% of the overall variability in this composition. In addition, AE was positively correlated with TDS and depth, whereas it was negatively correlated with other environmental factors (Fig. 4b).
The top 20 most abundant OTUs were identified and used to assess relationships between species and environmental factors. Among these OTUs were OTU33, which corresponded to an Ambispora species and OTU61, OTU12, and OTU24 which corresponded to Diversispora species, with all other top OTUs corresponding to Glomus species. OM, NN, AN, TP, AE, TDS, and pH were all significantly correlated with three or more OTUs (Fig. 6).

A correlation heat map of the relationship between the top 20 OTUs and soil properties. The X- and Y-axes correspond to environmental factors and OTUs, respectively. Different colors are used to indicate R values corresponding to the correlations between individual variables. *** P < 0.001 level, ** P < 0.01 level, * P < 0.05 level
Discussion
In this study, the relative diversity of Glomus, Diversispora, and Ambispora fungi varied significantly, with these genera accounting for 83.46, 14.73, and 1.21% of the fungi in analyzed samples, respectively (Fig. 2a). Owing to their adaptability, Glomus species are abundant in many ecosystems, consistent with our findings. As Glomus and Diversispora were the two dominant genera detected in soil samples in the present study, this suggests that these fungi are better adapted to the desert environment at this study site. In addition, we were unable to identify certain AMF species, and Paraglomus species were detected in only a few samples. Some molecular studies of AMF communities have reported difficulties in the detection of Paraglomerales and Archaeosporales species [35, 36], potentially due to PCR primer-related issues [37], owing to the use of different target genes or genomic regions and primer combinations that exhibit differences in specificity and efficacy across fungal genera [38, 39]. Maarten et al. demonstrated the complementary specificity of AMV4.5NF–AMDGR with AML1–AML2 primer sets, and found that a greater number of high-quality AMF sequences were obtained for the AMV4.5NF–AMDGR primers when evaluating six primer pairs targeting the nuclear rRNA operon as a means of characterizing AMF communities [38]. However, their results also suggested that this primer pair favored the amplification of Glomeraceae sequences at the expense of Ambisporaceae, Claroideoglomeraceae, and Paraglomeraceae sequences. As such, future efforts to identify more reliable primer pairs may be warranted.
Our petal chart analysis identified 12 core OTUs shared among different soil sample groups (Fig. 1), including 10 Glomus OTUs and 2 Diversispora OTUs that were present at all soil depths and slope positions. Given their universality among collected samples, we hypothesize that these fungi are closely associated with Ferula sinkiangensis growth, potentially suggesting that further study of these fungi may offer key insights into soil microbiology that can support artificial Ferula sinkiangensis cultivation.
In LEFse analyses, we determined that biomarkers [40,41,42] differed significantly as a function of soil depth and slope position, with decreasing biomarker levels as soil depth increased, suggesting that certain AMF species are sensitive to soil depth (Fig. 5b). We also found that most of these soil depth-sensitive AMF biomarkers were located in the lower slope position. This finding, together with the data shown in Fig. 5a, indicated that most AMF biomarkers were enriched at a soil depth of 0–20 cm in samples collected from the bottom of the slope, which may be a consequence of the fact that plant residues typically accumulate on the soil surface [33, 41, 42], particularly on relatively flat regions like those found at the bottom of a given slope. Such residues are associated with high soil nutrient contents, good ventilation, and favorable hydrothermal conditions that are conducive to the growth of soil microorganisms. Moreover, microorganisms can function synergistically with other AMF species [43, 44] to promote Ferula sinkiangensis growth.
Spearman correlation analyses revealed that soil physicochemical properties were significantly associated with AMF alpha diversity indices, with TP and pH being positively correlated with Shannon and Chao1 index values (P < 0.05). Soil phosphorus levels are one of the most important factors regulating AMF community diversity [44, 45], with certain studies having found AMF diversity to be significantly negatively correlated with AP levels [45, 46]. Herein, we found AMF diversity to be significantly positively correlated with soil TP (P < 0.05), whereas it was not significantly related to levels of AP. This may be due to the low levels of AP in these soil samples (1.67–6.85 mg/kg). It has been shown that the function whereby AMF species provide phosphorus to their host plants is phylogenetically conserved [47], such that different AMF phylogenetic groups would exhibit significant differences in availability. For example, the diversity of AMF communities associated with soybean roots was significantly influenced by P application [48], whereas such application did not affect AMF root colonization or the diversity/structure of AMF communities associated with tomato plants [49]. As such, it is possible that low P availability may select for functionally similar AMF species exhibiting highly efficient P uptake. In contrast, TP contents varied from 0.49–0.85 g/kg in the soil samples in the present study, suggesting a high potential phosphorus abundance in these soil samples. Ferula sinkiangensis growth is dependent upon the absorption of available soil phosphorus, and AMF species can facilitate such phosphorus uptake [46, 47]. This thus explains the increase in TP content, which was consistent with the substantial enrichment of AMF species adapted to low AP levels within the rhizosphere.
Soil pH is another key parameter that influences AMF community diversity, with AMF diversity often being significantly negatively correlated with pH [50]. In contrast, in the present study, we found that AMF community diversity was significantly positively correlated with soil pH, potentially due to unique local environmental factors. Some studies have shown that the tolerance of different AMF species to soil pH varies greatly [50, 51], and that the diversity and community composition of AMF species in soils with different pH values were significantly different [52,53,54]. The soil pH range in the present study was from 7.80–8.81, with only certain Glomus and Diversispora AMF species being able to survive in this pH range. As pH values rose, we found that the richness and diversity values corresponding to these AMFs also increased.
We detected significant differences in AMF diversity and richness as a function of slope position but not as a function of soil depth. This may be because the physical and chemical properties of soil at different slope locations differed significantly, whereas these properties did not vary as a function of soil depth. Cluster analyses (Fig. 3) clearly separated soil samples into three categories, which revealed that AMF community composition at a given slope level was similar, whereas this composition varied significantly as a function of slope level. We additionally observed no significant differences in soil properties as a function of soil depth, whereas these properties did differ at different slope positions, with significant differences being observed in OM, TP, TK, AN, pH, and SM (P < 0.05, Table 2). AMF diversity and richness were closely associated with environmental factors, and CCA analyses revealed that OM, TN, TK, NN, AN, AK, TDS, pH, SM, and AE all had a significant impact on AMF community composition (Fig. 4b). AE was found to be positively correlated with TDS and DE, and to be negatively correlated with other environmental factors (Fig. 4b). These factors were also correlated with AMF community composition, with OM, TN, TP, TK, AN, NN, TDS, pH, and SM all being positively correlated with the abundance of many OTUs, whereas AE and TDS were negatively correlated with the abundance of several OTUs (Fig. 6). Soil composition thus differed significantly as a function of slope position, in turn affecting AMF community diversity and richness.
Conclusion
In summary, our results provide new insights regarding the composition and diversity of rhizosphere AMF communities associated with Ferula sinkiangensis. We found that Glomus and Diversispora were enriched in our samples, and that rhizosphere AMF diversity and richness varied significantly among slope positions, as evidenced by differences in Glomus and Diversispora abundance. In contrast, rhizosphere soil depth did not significantly affect overall AMF diversity, although certain AMF species were found to be sensitive to depth. In addition, the physical and chemical properties of soil varied significantly as a function of slope position (P < 0.05), potentially explaining the differences in AMF community composition across these slope positions. Together, we hope that our results will help guide efforts to improve soil structure and AMF communities associated with Ferula sinkiangensis in order to improve the protection and cultivation of this valuable medicinal plant.
Methods
Site description and experimental design
The chosen experimental site was located in the Yining region of Xinjiang Province, China (82°7′E, 43°44′N) [1]. We selected three 10 m × 5 m rectangular plots at the top (altitude:1121 m), middle (altitude:1087 m), and bottom (altitude:1053 m) of a slope in a region containing Ferula sinkiangensis plants in May 2019. We then randomly selected 9 Ferula plants at each of these slope positions and collected rhizosphere soil samples at depths of 0–20 cm, 20–40 cm, and 40–60 cm [55] as per the methods previously described by Riley and Barber [56, 57]. We also collected samples of the surrounding soil within 5 cm of the root system. Next, we pooled together three of the nine plants, and we similarly pooled the rhizosphere soil samples from these three plants at each depth level, as well as the surrounding soil samples. Rhizosphere soil was stored in liquid nitrogen and was sent to Shanghai Maso Bio-Pharmaceutical Techno-logy Co., Ltd. for high-throughput sequencing. Samples of surrounding soil were air-dried and were then used to assess soil physicochemical properties [58, 59]. Soil samples from the top, middle, and bottom slope positions were respectively denoted with the SW, ZW, and XW designations, while samples collected at depths of 0–20 cm, 20–40 cm, and 40–60 cm were respectively designated with the numbers 1, 2, and 3.
Library preparation
Microbial DNA [60] was extracted from 0.5 g aliquots of each sample using the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, GA, USA) according to the manufacturer’s protocols. Final DNA concentrations and purity were determined by NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, DE, USA), and DNA quality was assessed via 1% agarose gel electrophoresis. The genomic DNA pellet was stored at − 30 °C prior to use.
The partial small subunit (SSU) region of the 18S rRNA gene was amplified via nested PCR [61, 62]. AML1F (forward primer) (5′-ATCAACTTTCGATGGTAGGATAGA-3′) and AML2R (reverse primer) (5′-GAACCCAAACACTTTGGTTTCCTTGGTTTCC-3 ‘) [38, 39] were used as the primers in the first round of amplification using a thermocycler PCR system (GeneAmp 9700, ABI, USA), whereas AMV4.5NF (forward primer) (5’-AAGCTCGTAG-TTGAATTTCG-3′) and AMDGR (reverse primer) (5′-CCCAACTATCCCTATTAATCAT-3′) [38, 61, 63] were used as the primers in the second amplification step. The first-round PCR reactions were conducted using the following thermocycler settings: 3 min at 95 °C, followed by 32 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, followed by a final extension at 72 °C for 10 min. PCR reactions were performed in triplicate, with each reaction being conducted in a 20 μL volume containing 4 μL of 5 × FastPfu Buffer, 2 μL of 2.5 mM dNTPs, 0.8 μL of each primer (5 μM), 0.4 μL of FastPfu Polymerase, and 10 ng of template DNA. After amplification, replicates from each sample were pooled and separated via 2% agarose gel electrophoresis. These first-round PCR products were diluted 10-fold and used as templates for the second-round PCR amplification step using the following thermocycler settings: 3 min at 95 °C, followed by 30 cycles of 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 45 s, followed by a final extension at 72 °C for 10 min. Reaction mixtures were prepared identically to those for the first round of PCR. The resultant PCR products were purified via 2% agarose gel electrophoresis with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, CA, USA), and DNA levels were quantified using QuantiFluor™-ST (Promega, USA) according to the manufacturer’s instructions.
Illumina MiSeq sequencing
Purified amplicons were pooled in equimolar amounts, and paired-end sequencing (2 × 300) was conducted using an Illumina MiSeq platform (Illumina, CA, USA) according to the standard protocols produced by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). Sequence read processing was performed using QIIME v.1.9.1.
Processing of sequencing data
Raw fastq files were demultiplexed, quality-filtered using Trimmomatic, and merged via FLASH with the following criteria: (i) The reads were truncated at any site receiving an average quality score < 20 over a 50 bp sliding window. (ii) Primers were exactly matched allowing for 2 nucleotide mismatching, and reads containing ambiguous bases were removed. (iii) Sequences with > 10 bp overlap were merged based upon the overlapping sequence.
Operational taxonomic units (OTUs) were clustered with a 97% similarity cutoff using UPARSE (v.7.1 http://drive5.com/uparse/), and chimeric sequences were identified and removed using UCHIME. The taxonomy of each 18S rRNA gene sequence was analyzed using RDP Classifier (v.2.2 http://sourceforge.net/projects/rdp-classifier/) and the MaarjAM (https://www.maarjam.botany.ut.ee/) 18S rRNA database at a confidence threshold of 70%.
Soil physicochemical properties
Soil properties were assessed as described in prior studies [59, 64]. Analyzed soil physicochemical properties included gravimetric soil water content (assessed via by oven drying at 105 °C) [65, 66], organic matter content (assessed using the KCr2O7 method) [66], total nitrogen (assessed using the HClO4-H2SO4 digestion method) [67, 68], total phosphorus (assessed using a Mo-Sb colorimetric method) [68, 69], total potassium (as measured via atomic absorption spectrometry) [69, 70], nitrate nitrogen, ammonium nitrogen (as assessed via a 0.01 M calcium chloride extraction method using a BRAN+LUEBBE flow analyzer) [67, 68], available phosphorus (using NaH-CO3 extracts and analyzed via the Mo-Sb colorimetric method) [68, 70], available potassium (measured using NH4OAc extracts and analyzed via atomic absorption spectrometry), pH (as measured with a Mettler Tolido FiveEasy Plus pH meter) [70], and total dissolvable salts (as assessed via atomic absorption spectrometry and titration) [59, 70].
Data analysis
In the diversity indices, all the rarefaction and diversity analysis for community analysis was performed at the lowest number of reads (14991) per sample. The Mothur (version v.1.30.1 http://www.mothur.org/wiki/Schloss_SOP#Alpha_diversity) was used to calculate Sobs (the observed richness) as well as the Chao1 (the Chao1 estimator), Shannon (the Shannon diversity index), Simpson (the Simpson diversity index), coverage (Good’s coverage indices), and Phylogenetic diversity (PD), which were respectively used to assess sample richness, diversity, and coverage. Since the data were not normally distributed, Kruskal-Wallis test was used to detect whether there was a significant difference in index values between the groups. The dilution curve was plotted with R (v3.6.1) to count the Alpha diversity index of the corresponding samples of these sequences by randomly selecting 14,991 sequences from the samples. The extracted data volume was taken as the x-coordinate and the Alpha diversity index as the y-coordinate.
The Venn diagram diagram were drawn using R (v3.6.1) to count the number of common and unique species (such as OTU).
The community column were drawn using R (v3.6.1) to count the species composition of different groups (or samples) at each level of classification.
The Circos sample relationship diagram is a visual circle diagram describing the correspondence between samples (or groups) and species, which was drawn by Circos-0.67-7 (http://circos.ca/). The abundance of samples in the group was calculated using mean values. In all samples, species with less than 0.01 abundance were combined as others.
The Unweighted Pair-group Method with Arithmetic Means (UPGMA) clustering was performed as a type of hierarchical clustering method to interpret the distance matrix using average linkage. The UPGMA clustering based on Unweighted Unifrac Distances at the OTU level, which was conducted by QIIME (Version 1.9.1) and drawn by R (v3.6.1).
Principal co-ordinate analysis (PCoA) based on bray-curtis at OUT level analysis the community of AMF by R (v3.6.1). The Analysis of similarities (ANOSIM) in “vegan” R package is used to examine differences between groups. The test for significance is 999 permutations.
Differences between groups were assessed based upon linear discriminant analysis (LDA) effect size (LEfSe) (http://huttenhower.sph.harvard.edu/galaxy/root?tool_id=lefse_upload). First, the non-parametric factorial Kruskal-Wallis (KW) sum-rank test is applied to detect the significant difference of abundance and find out the group with significant difference. Finally, linear discriminant analysis (LDA) estimate the impact of abundance of each component (species) on the difference effect, the microbes of LDA (> 2) are considered to be significant groups of microbes. The multi-group comparison strategy is that species can only be considered as differential species if there are differences in multiple groups.
Since the axis length > 4 in the DCA results, canonical correlation analysis (CCA) was used to test the relationship among environmental factors, samples and microbes. The CCA was estimated using the “vegan” package in R (v3.6.1).
Correlations between soil properties and dominant OTUs were assessed via Spearman correlation analyses, while relationships between rhizosphere composition, soil properties. The Correlation Heatmap was drawn using the “pheatmap” package in R (v3.6.1).
Since the data were not normally distributed, Kruskal-Wallis test was used to detect whether there was a significant difference in the soil physicochemical factors between the groups. Spearman correlation analyses were also run among the soil physicochemical factors with Alpha diversity index values. The Statistical analysis was carried out with SPSS 19.0 (IBM Inc., Armonk, USA).
Availability of data and materials
The datasets generated and/or analysed during the current study are not publicly available due the datasets also forms part of an ongoing study, but they are available from the corresponding author on reasonable request.
Abbreviations
- OM:
-
Organic matter content
- TN:
-
Total nitrogen
- TP:
-
Total phosphorus
- TK:
-
Total potassium
- NN:
-
Nitrate nitrogen
- AN:
-
Ammonium nitrogen
- AP:
-
Available phosphorus
- AK:
-
Available potassium
- TDS:
-
Total dissolvable salt
- pH:
-
Hydrogen ion concentration
- SM:
-
Soil water content
- AE:
-
Slope position
- DE:
-
Depth
References
Liu Y, Guo J, Li L, Asem MD, Zhang Y, Mohamad OA, et al. Endophytic bacteria associated with endangered plant Ferula sinkiangensis K. M. Shen in an arid land: diversity and plant growth-promoting traits. J Arid Land. 2017;9(3):432–45. https://doi.org/10.1007/s40333-017-0015-5.
Tian C, Xie L, Li G. Germination characters of the seeds of the Ferula sinkiangensis KM Shen. Seed. 2008;27(5):88–90.
Meng L, Tan D-y. An observation on pollinating insects and their flower-visiting behavior on Ferula sinkiangensis (Umbelliferae). Acta Botanica Boreali-occidentalia Sinica. 2003;23(3):488.
Fang L, Wenbin Z, Yuhuai C. A study on biological characters of wild Ferula sinkiangensis K.M.SHENin. Xinjiang. Lishizhen Medicine and Materia Medica Research. 2008;19(2):396–7.
Alkhatib R, Hennebelle T, Joha S, Idziorek T, Preudhomme C, Quesnel B, et al. Activity of elaeochytrin A from Ferula elaeochytris on leukemia cell lines. Phytochemistry. 2008;69(17):2979–83. https://doi.org/10.1016/j.phytochem.2008.09.019.
Xing Y, Li N, Zhou D, Chen G, Jiao K, Wang W, et al. Sesquiterpene Coumarins from Ferula sinkiangensis act as Neuroinflammation inhibitors. Planta Med. 2016;83(01/02):135–42. https://doi.org/10.1055/s-0042-109271.
Lee C-L, Chiang L-C, Cheng L-H, Liaw C-C, Abd El-Razek MH, Chang F-R, et al. Influenza A (H1N1) antiviral and cytotoxic agents fromFerula Assa-foetida. J Nat Prod. 2009;72(9):1568–72. https://doi.org/10.1021/np900158f.
Kartal N, Sokmen M, Tepe B, Daferera D, Polissiou M, Sokmen A. Investigation of the antioxidant properties of Ferula orientalis L. using a suitable extraction procedure. Food Chem. 2007;100(2):584–9. https://doi.org/10.1016/j.foodchem.2005.09.084.
Iranshahi M, Kalategi F, Rezaee R, Shahverdi A, Ito C, Furukawa H, et al. Cancer Chemopreventive activity of Terpenoid Coumarins from Ferula species. Planta Med. 2008;74(2):147–50. https://doi.org/10.1055/s-2008-1034293.
Aybek R, Nilufar M, Keyser S. Determination of Volatile Oil and Ferulic Acid in Different Parts of Wild Ferula sinkiangensis KM Shen and Ferula fukanensis KM Shen Cultivars. Medicinal Plant. 2018;9(1):36–8. https://doi.org/10.19600/j.cnki.issn2152-3924.2018.01.009.
Sun L, Shi S, Zhu J, Li X. Traditional use and modern research review of Ferula in China. Mod Chinese Med. 2013;15(7):620–6. https://doi.org/10.13313/j.issn.1673-4890 2013.07.024.
Birhane E, Aregawi K, Giday K. Changes in arbuscular mycorrhiza fungi spore density and root colonization of woody plants in response to exclosure age and slope position in the highlands of Tigray, Northern Ethiopia. J Arid Environ. 2017;142:1–10. https://doi.org/10.1016/j.jaridenv.2017.03.002.
Xu X, Wang X, Cleary M, et al. Slope position rather than thinning intensity affects arbuscular mycorrhizal fungi (AMF) community in Chinese fir plantations. 2020;11(3):273. https://doi.org/10.3390/f11030273.
Schreiner RP. Depth structures the community of arbuscular mycorrhizal fungi amplified from grapevine (Vitis vinifera L.) roots. Mycorrhiza. 2020;30:149–60. https://doi.org/10.1007/s00572-020-00930-6.
Higo M, Isobe K, Yamaguchi M, Drijber RA, Jeske ES, Ishii R. Diversity and vertical distribution of indigenous arbuscular mycorrhizal fungi under two soybean rotational systems. Biol Fertil Soils. 2013;49(8):1085–96. https://doi.org/10.1007/s00374-013-0807-5.
Singh JS, Pandey VC, Singh DP. Efficient soil microorganisms: A new dimension for sustainable agriculture and environmental development. Agric Ecosyst Environ. 2011;140(3–4):339–53. https://doi.org/10.1016/j.agee.2011.01.017.
Carney KM, Matson PA. Plant communities, soil microorganisms, and soil carbon cycling: does altering the world belowground matter to ecosystem functioning? Ecosystems. 2005;8(8):928–40. https://doi.org/10.1007/s10021-005-0047-0.
Smith JE. Mycorrhizal Symbiosis (Third Edition). Soil Sci Soc Am J. 2009;73(2):694. https://doi.org/10.2136/sssaj2008.0015br.
Spatafora JW, Chan Y, Benny GL, Lazarus K, Smith ME, Berbee ML, et al. A phylum-level phylogenetic classification of zygomycete fungi based on genome-scale data. Mycologia. 2016;108(5):1028–46. https://doi.org/10.3852/16-042.
Tedersoo L, Sánchez-Ramírez S, Kljalg U, Bahram M, Dring M, Schigel D, et al. High-level classification of the fungi and a tool for evolutionary ecological analyses. Fungal Divers. 2018;90(1):135–59. https://doi.org/10.1007/s13225-018-0401-0.
Kurth F, Zeitler K, Feldhahn L, Neu TR, Weber T, Krištůfek V, et al. Detection and quantification of a mycorrhization helper bacterium and a mycorrhizal fungus in plant-soil microcosms at different levels of complexity. BMC Microbiol. 2013;13:205. https://doi.org/10.1186/1471-2180-13-205.
Mathur S, Tomar RS, Jajoo A. Arbuscular Mycorrhizal fungi (AMF) protects photosynthetic apparatus of wheat under drought stress. Photosynth Res. 2018;139(1–3):227–38. https://doi.org/10.1007/s11120-018-0538-4.
Vigo C, Norman JR, Hooker JE. Biocontrol of the pathogen Phytophthora parasitica by arbuscular mycorrhizal fungi is a consequence of effects on infection loci. Plant Pathol. 2000;49(4):509–14. https://doi.org/10.1046/j.1365-3059.2000.00473.x.
Wang Y-Y, Yin Q-S, Qu Y, Li G-Z, Hao L. Arbuscular mycorrhiza-mediated resistance in tomato against Cladosporium fulvum -induced mould disease. J Phytopathol. 2017;166(1):67–74. https://doi.org/10.1111/jph.12662.
Alguacil MM, Torrecillas E, Roldán A, Díaz G, Torres MP. Perennial plant species from semiarid gypsum soils support higher AMF diversity in roots than the annual Bromus rubens. Soil Biol Biochem. 2012;49:132–8. https://doi.org/10.1016/j.soilbio.2012.02.024.
BHAT BA, SHEIKH MA, TIWARI A. Impact of various edaphic factors on AMF spore population and diversity in Catharanthus roseus at Gwalior. Int J Plant Sci. 2014;9(1):1–6.
Rodríguez-Echeverría S, Teixeira H, Correia M, Timóteo S, Heleno R, Öpik M, et al. Arbuscular mycorrhizal fungi communities from tropical Africa reveal strong ecological structure. New Phytol. 2016;213(1):380–90. https://doi.org/10.1111/nph.14122.
Faggioli V, Menoyo E, Geml J, Kemppainen M, Pardo A, Salazar MJ, et al. Soil lead pollution modifies the structure of arbuscular mycorrhizal fungal communities. Mycorrhiza. 2019;29(4):363–73. https://doi.org/10.1007/s00572-019-00895-1.
Rodríguez-Caballero G, Caravaca F, Roldán A. The unspecificity of the relationships between the invasive Pennisetum setaceum and mycorrhizal fungi may provide advantages during its establishment at semiarid Mediterranean sites. Sci Total Environ. 2018;630:1464–71. https://doi.org/10.1016/j.scitotenv.2018.02.321.
Higo M, Tatewaki Y, Gunji K, Kaseda A, Isobe K. Cover cropping can be a stronger determinant than host crop identity for arbuscular mycorrhizal fungal communities colonizing maize and soybean. PeerJ. 2019;7(2):e6403. https://doi.org/10.7717/peerj.6403.
Higo M, Tatewaki Y, Iida K, et al. Amplicon sequencing analysis of arbuscular mycorrhizal fungal communities colonizing maize roots in different cover cropping and tillage systems. Sci Rep. 2020;10(1):6039. https://doi.org/10.1038/s41598-020-58942-3.
Higo M, Kang D, Isobe K. First report of community dynamics of arbuscular mycorrhizal fungi in radiocesium degradation lands after the Fukushima-Daiichi nuclear disaster in Japan. Sci Rep. 2019;9(1):8240. https://doi.org/10.1038/s41598-019-44665-7.
Zhang T, Wang Z, Lv X, Li Y, Zhuang L. High-throughput sequencing reveals the diversity and community structure of rhizosphere fungi of Ferula Sinkiangensis at different soil depths. Sci Rep. 2019;9(1):6558. https://doi.org/10.1038/s41598-019-43110-z.
Wang X, Wang Z, Jiang P, He Y, Mu Y, Lv X, et al. Bacterial diversity and community structure in the rhizosphere of four Ferula species. Sci Rep. 2018;8(1):5345. https://doi.org/10.1038/s41598-018-22802-y.
Hijri I, SÝKorovÁ Z, Oehl F, Ineichen K, MÄDer P, Wiemken A, et al. Communities of arbuscular mycorrhizal fungi in arable soils are not necessarily low in diversity. Mol Ecol. 2006;15(8):2277–89. https://doi.org/10.1111/j.1365-294x.2006.02921.x.
Hempel S, Renker C, Buscot F. Differences in the species composition of arbuscular mycorrhizal fungi in spore, root and soil communities in a grassland ecosystem. Environ Microbiol. 2007;9(8):1930–8. https://doi.org/10.1111/j.1462-2920.2007.01309.x.
Santos JC, Finlay RD, Tehler A. Molecular analysis of arbuscular mycorrhizal fungi colonising a semi-natural grassland along a fertilisation gradient. New Phytol. 2006;172(1):159–68. https://doi.org/10.1111/j.1469-8137.2006.01799.x.
Van Geel M, Busschaert P, Honnay O, et al. Evaluation of six primer pairs targeting the nuclear rRNA operon for characterization of arbuscular mycorrhizal fungal (AMF) communities using 454 pyrosequencing. J Microbiol Methods. 2014;106:93–100. https://doi.org/10.1016/j.mimet.2014.08.006.
Petr K, Radka, Sudová, Martina, Janoušková, et al. Comparison of commonly used primer sets for evaluating arbuscular mycorrhizal fungal communities: is there a universal solution? Soil Biol Biochem. 2014;68:482–93. https://doi.org/10.1016/j.soilbio.2013.08.027.
Bouffaud ML, Bragalini C, Berruti A, Peyret-Guzzon M, Voyron S, Stockinger H, et al. Arbuscular mycorrhizal fungal community differences among European long-term observatories. Mycorrhiza. 2016;27(4):331–43. https://doi.org/10.1007/s00572-016-0753-9.
Olibone D, Rosolem CA. Phosphate fertilization and phosphorus forms in an Oxisol under no-till. Sci Agric. 2010;67(4):465–71. https://doi.org/10.1590/s0103-90162010000400014.
Casazza G, Lumini E, Ercole E, Dovana F, Guerrina M, Arnulfo A, et al. The abundance and diversity of arbuscular mycorrhizal fungi are linked to the soil chemistry of screes and to slope in the Alpic paleo-endemic Berardia subacaulis. PLoS One. 2017;12(2):e0171866–e. https://doi.org/10.1371/journal.pone.0171866.
Y-h T, Y-b L, Zheng Y-l, Z-q C. Synergistic effect of colonization with arbuscular mycorrhizal fungi improves growth and drought tolerance of Plukenetia volubilis seedlings. Acta Physiol Plant. 2012;35(3):687–96. https://doi.org/10.1007/s11738-012-1109-5.
Liang Y, He X, Chen C, Feng S, Liu L, Chen X, et al. Influence of plant communities and soil properties during natural vegetation restoration on arbuscular mycorrhizal fungal communities in a karst region. Ecol Eng. 2015;82:57–65. https://doi.org/10.1016/j.ecoleng.2015.04.089.
Jayachandran K, Shetty KG. Growth response and phosphorus uptake by arbuscular mycorrhizae of wet prairie sawgrass. Aquat Bot. 2003;76(4):281–90. https://doi.org/10.1016/s0304-3770(03)00075-5.
Alguacil MM, Lozano Z, Campoy MJ, Roldán A. Phosphorus fertilisation management modifies the biodiversity of AM fungi in a tropical savanna forage system. Soil Biol Biochem. 2010;42(7):1114–22. https://doi.org/10.1016/j.soilbio.2010.03.012.
Powell JR, Parrent JL, Hart MM, Klironomos JN, Rillig MC, Maherali H. Phylogenetic trait conservatism and the evolution of functional trade-offs in arbuscular mycorrhizal fungi. Proc Biol Sci. 2009;276(1676):4237–45. https://doi.org/10.1098/rspb.2009.1015.
Higo M, Sato R, Serizawa A, Takahashi Y, Gunji K, Tatewaki Y, Isobe K. Can phosphorus application and cover cropping alter arbuscular mycorrhizal fungal communities and soybean performance after a five-year phosphorus-unfertilized crop rotational system? PeerJ. 2018;6:e4606. https://doi.org/10.7717/peerj.4606.
Higo M, Azuma M, Kamiyoshihara Y, Kanda A, Tatewaki Y, Isobe K. Impact of phosphorus fertilization on tomato growth and Arbuscular Mycorrhizal fungal communities. Microorganisms. 2020;8(2):178. https://doi.org/10.3390/microorganisms8020178.
Sidhu O, Behl H. Response of three Glomus species on growth of Prosopis juliflora Swartz at high pH levels. Symbiosis; 1997.
Helgason T, Fitter AH. Natural selection and the evolutionary ecology of the arbuscular mycorrhizal fungi (phylum Glomeromycota). J Exp Bot. 2009;60(9):2465–80. https://doi.org/10.1093/jxb/erp144.
Higo M, Isobe K, Kang D, et al. Molecular diversity and spore density of indigenous arbuscular mycorrhizal fungi in acid sulfate soil in Thailand. Ann Microbiol. 2011;61:383–9. https://doi.org/10.1007/s13213-010-0142-0.
Yao L, Wang D, Kang L, Wan D, Guo Y. Effects of fertilizations on soil bacteria and fungi communities in a degraded arid steppe revealed by high through-put sequencing. PeerJ. 2018;6(2):e4623. https://doi.org/10.7717/peerj.4623.
De Pontes JS, Oehl F, Pereira C, Torres DTMC, Coyne D, Da Silva DKA, et al. Diversity of arbuscular mycorrhizal fungi in the brazilian's cerrado and in soybean under conservation and conventional tillage. Appl Soil Ecol. 2017;117-118:178–89. https://doi.org/10.1016/j.apsoil.2017.04.023.
Montero Sommerfeld H, Díaz LM, Alvarez M, Añazco Villanueva C, Matus F, Boon N, et al. High winter diversity of arbuscular mycorrhizal fungal communities in shallow and deep grassland soils. Soil Biol Biochem. 2013;65:236–44. https://doi.org/10.1016/j.soilbio.2013.06.002.
Riley D, Barber SA. Bicarbonate accumulation and pH changes at the soybean (Glycine max (L.) Merr.) root-soil Interface. Soil Sci Soc Am J. 1969;33(6):905–8. https://doi.org/10.2136/sssaj1969.03615995003300060031x.
Riley D, Barber SA. Salt accumulation at the soybean (glycine max. (L.) Merr.) root-soil Interface. Soil Sci Soc Am J. 1970;34(1):154–5. https://doi.org/10.2136/sssaj1970.03615995003400010042x.
Liu W, Jiang S, Zhang Y, Yue S, Christie P, Murray PJ, et al. Spatiotemporal changes in arbuscular mycorrhizal fungal communities under different nitrogen inputs over a 5-year period in intensive agricultural ecosystems on the North China Plain. FEMS Microbiol Ecol. 2014. https://doi.org/10.1111/1574-6941.12405.
Naga Raju M, Golla N, Vengatampalli R. Soil physicochemical properties. In: SpringerBriefs in Environmental Science. Springer International Publishing; 2016. p. 5–10.
Awoyemi OM, Adeleke EO, Dzantor EK. Arbuscular mycorrhizal fungi and exogenous glutathione mitigate coal fly ash (CFA)-induced phytotoxicity in CFA-contaminated soil. J Environ Manag. 2019;237:449–56. https://doi.org/10.1016/j.jenvman.2019.02.103.
Heid CA, Stevens J, Livak KJ, Williams PM. Real time quantitative PCR. Genome Res. 1996;6(10):986–94. https://doi.org/10.1101/gr.6.10.986.
Rumsby P. PCR protocols—A guide to methods and applications. Food Chem Toxicol. 1991;29(11):788–9. https://doi.org/10.1016/0278-6915(91)90192-a.
Suzuki K, Takahashi K, Harada N. Evaluation of primer pairs for studying arbuscular mycorrhizal fungal community compositions using a MiSeq platform. Biol Fertil Soils. 2020;56(6):853–8. https://doi.org/10.1007/s00374-020-01431-6.
Melo CD, Walker C, Krüger C, Borges PAV, Luna S, Mendonça D, et al. Environmental factors driving arbuscular mycorrhizal fungal communities associated with endemic woody plant Picconiaazorica on native forest of Azores. Ann Microbiol. 2019;69(13):1309–27. https://doi.org/10.1007/s13213-019-01535-x.
Chandrasekaran M, Boughattas S, Hu S, Oh S-H, Sa T. A meta-analysis of arbuscular mycorrhizal effects on plants grown under salt stress. Mycorrhiza. 2014;24(8):611–25. https://doi.org/10.1007/s00572-014-0582-7.
Ren C, Chen J, Deng J, et al. Response of microbial diversity to C:N:P stoichiometry in fine root and microbial biomass following afforestation. Biol Fertil Soils. 2017;53:457–68. https://doi.org/10.1007/s00374-017-1197-x.
Zhao F, Feng X, Guo Y, et al. Elevation gradients affect the differences of arbuscular mycorrhizal fungi diversity between root and rhizosphere soil. Agric For Meteorol. 2020;284:107894. https://doi.org/10.1016/j.agrformet.2019.107894.
Tang X, Xia M, Guan F, et al. Spatial distribution of soil nitrogen, phosphorus and potassium stocks in Moso bamboo forests in subtropical China. Forests. 2016;7(11):267. https://doi.org/10.3390/f7110267.
Moreira H, Pereira SIA, Vega A, et al. Synergistic effects of arbuscular mycorrhizal fungi and plant growth-promoting bacteria benefit maize growth under increasing soil salinity. J Environ Management. 2020;257(Mar.1):109982.1–109982.12. https://doi.org/10.1016/j.jenvman.2019.109982.
Zhang W, Cao J, Zhang S, et al. Effect of earthworms and arbuscular mycorrhizal fungi on the microbial community and maize growth under salt stress. Appl Soil Ecol. 2016;107:214–23. https://doi.org/10.1016/j.apsoil.2016.06.005.
Acknowledgements
We would like to thank Shanghai Meiji biomedical technology co., ltd. for its assistance in the analysis of the Illumina genome. We would like to thank Professor Ping Yan who is from the College of life Sciences, Shihezi University to identify the voucher specimens.
Research involving plants
The collection of plant material complied with local and national guidelines. Voucher specimens are are identified by Professor Ping Yan from the College of life Sciences, Shihezi University and deposited in the herbarium of the College of life Sciences, Shihezi University.
Funding
The work was funded by the national natural science foundation of China regional project (project number: 41561010; 2016–2019 and 31560177; 2016–2019). The funding agency has no role in the design, data collection, analysis or interpretation of the research or in the writing of the manuscript.
Author information
Authors and Affiliations
Contributions
Y.L. coordinated the study, collected data, conducted data analysis, interpreted data, curated the metagenomic data, drafts the manuscript and writes the manuscripts. Y.H., G.L., and X.L. coordinated the study, conducted data analysis, interpreted data. L.Z. and Z.W. allocate funds, collect data and jointly analyze data, curated the metagenomic data, and critically analyze manuscripts. X.L., G.L. allocate funds and co-analysis the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
All authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Table S1.
The relationships between alpha diversity indices and abiotic factors. Spearman correlation analyses of the relationships between alpha diversity indices and abiotic factors in all samples. Only variables with significant relationships are shown. ** P < 0.01 level (two-tailed). * P < 0.05 level (two-tailed). Figure S1. Rhizosphere soil sample dilution curves. SW, ZW, and XW respectively correspond to the top, middle, and bottom slope positions. 1, 2, and 3 respectively represent samples collected at a soil depth of 0-20 cm, 20-40 cm, and 40-60 cm. Figure S2. A Circos sample relationship diagram. The small left semi-circle corresponds to the species composition within a given sample, while the color of the outer ribbon corresponds to the group, the color of the inner ribbon corresponds to the species, and the length of the ribbons correspond to the relative abundance of these species within the indicated sample. The right semi-circle corresponds to the distributions of species within different samples at the taxonomic level, the outer band represents species, the inner band represents different groups, and the length corresponds to the distribution proportion of the sample in a given species. SW, ZW, and XW respectively correspond to the top, middle, and bottom slope positions. 1, 2, and 3 respectively represent samples collected at a soil depth of 0-20 cm, 20-40 cm, and 40-60 cm.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Luo, Y., Wang, Z., He, Y. et al. High-throughput sequencing analysis of the rhizosphere arbuscular mycorrhizal fungi (AMF) community composition associated with Ferula sinkiangensis. BMC Microbiol 20, 335 (2020). https://doi.org/10.1186/s12866-020-02024-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-02024-x