
Abstract
Background
Several plant-pathogenic bacteria are transmitted by insect vector species that often also act as hosts. In this interface, these bacteria encounter plant endophytic, insect endosymbiotic and other microbes. Here, we used high throughput sequencing to examine the bacterial communities of five different psyllids associated with citrus and related plants of Rutaceae in Bhutan: Diaphorina citri, Diaphorina communis, Cornopsylla rotundiconis, Cacopsylla heterogena and an unidentified Cacopsylla sp.
Results
The microbiomes of the psyllids largely comprised their obligate P-endosymbiont ‘Candidatus Carsonella ruddii’, and one or two S-endosymbionts that are fixed and specific to each lineage. In addition, all contained Wolbachia strains; the Bhutanese accessions of D. citri were dominated by a Wolbachia strain first found in American isolates of D. citri, while D. communis accessions were dominated by the Wolbachia strain, wDi, first detected in D. citri from China. The S-endosymbionts from the five psyllids grouped with those from other psyllid taxa; all D. citri and D. communis individuals contained sequences matching ‘Candidatus Profftella armatura’ that has previously only been reported from other Diaphorina species, and the remaining psyllid species contained OTUs related to unclassified Enterobacteriaceae. The plant pathogenic ‘Candidatus Liberibacter asiaticus’ was found in D. citri but not in D. communis. Furthermore, an unidentified ‘Candidatus Liberibacter sp.’ occurred at low abundance in both Co. rotundiconis and the unidentified Cacopsylla sp. sampled from Zanthoxylum sp.; the status of this new liberibacter as a plant pathogen and its potential plant hosts are currently unknown. The bacterial communities of Co. rotundiconis also contained a range of OTUs with similarities to bacteria previously found in samples taken from various environmental sources.
Conclusions
The bacterial microbiota detected in these Bhutanese psyllids support the trends that have been seen in previous studies: psyllids have microbiomes largely comprising their obligate P-endosymbiont and one or two S-endosymbionts. In addition, the association with plant pathogens has been demonstrated, with the detection of liberibacters in a known host, D. citri, and identification of a putative new species of liberibacter in Co. rotundiconis and Cacopsylla sp.
Similar content being viewed by others

Background
Within the Psylloidae, three of the families proposed by Percy et al. [1], the Liviidae, Psyllidae and Triozidae, contain vectors of plant pathogenic bacteria. Vectors of liberibacters (Alphaproteobacteria) include species of Arytainilla, Bactericera, Cacospylla, Diaphorina and Trioza [[2] and references within]. Species of Cacopsylla transmit a range of phytoplasmas [3,4,5,6,7,8], and Bactericera trigonica Hodkinson vectors a phytoplasma to carrots [9]. Sometimes, individual psyllid species are able to vector a range of pathogens. Both Trioza erytreae Del Guercio (Triozidae) and Diaphorina citri Kuwayama (Liviidae) can transmit ‘Ca. Liberibacter asiaticus’ (hereafter CLas) and ‘Ca. L. africanus’ [10,11,12] and D. citri is also a vector for ‘Ca. L. americanus’ [13] and ‘Ca. Phytoplasma aurantifolia’ causing witches’ broom disease of lime [14].
These phytoplasmas and liberibacters are members of lineages that are ecologically specialized. Within their plant hosts, they are intracellular and restricted to the phloem, and within their insect hosts and vectors, they colonize various tissues and persist throughout the insects’ lifespan; hence, the insects are considered as alternative hosts rather than passive carriers [15, 16]. In addition to these two lineages, the bacterium, Erwinia amylovora (Burrill) Winslow et al. (Enterobacteriaceae), the causal agent of fireblight, has been found to be transmitted by Cacosylla pyricola (Förster) [17, 18] and, in South America, Russelliana solanicola Tuthill (Psyllidae) has been identified as the first known psyllid vector of a plant virus, the potato rugose stunting virus [19].
In addition to vectoring plant pathogens, psyllids, like other insects, harbour a diverse array of other bacteria [20] whose association with their host varies on a continuum from obligate to facultative [21]. The nitrogen content of phloem on which psyllids feed is low [22] particularly in concentrations of essential amino acids [23, 24]. To overcome these deficiencies, some insects have developed obligate associations with endosymbiotic bacteria (primary endosymbionts (P-endosymbionts)) [25] thereby allowing them to utilise nutrient-poor environments such as xylem and phloem [26, 27]. In psyllids, the P-endosymbiont is ‘Ca. Carsonella ruddii’ (hereafter Carsonella; Gammaproteobacteria) that is housed within specialised host cells called bacteriocytes that aggregate into bacteriomes [28,29,30]. These P-endosymbionts are vertically transmitted resulting in strict co-speciation with their psyllid hosts within families, genera and species [28,29,30]. The resulting clonality has induced a drastic reduction in genome size, through loss of up to 90% of ancestral genes including, in some cases, the loss of some genes or gene pathways involved in the production of essential amino acids [31].
Other endosymbiotic bacteria, secondary endosymbiont (S-endosymbionts), may be facultative or obligate, and a diverse group of S-endosymbionts, predominantly from the Enterobacteriaceae, are associated with psyllid species [32]. These may have positive or negative effects on the biology of their host, but unlike P-endosymbionts, do not show uniform signs of co-speciation with their hosts [29, 33]. Some P-endosymbionts have lost the ability to synthesise certain essential amino acids, and their synthesis has been subsumed by S-endosymbionts [30, 34, 35]. In addition to enabling feeding on nutrient poor food sources, S-endosymbionts may also have the potential to affect host plant range [30, 36]. Other benefits conferred by S-endosymbionts include resistance to specific biotic or abiotic stresses. For example, the pea aphid (Acyrthosiphon pisum Harris) (Aphididae) is protected from the hymenopteran parasitoid, Aphidius ervi Haliday (Braconidae) by two S-endosymbionts [37], from the entomopathogen, Pandora neoaphidis (Remaudière & Hennebert) by Regiella insecticola Moran et al. [38] and from heat stress by an S-endosymbiont belonging to the Gammaproteobacteria [39]. Another endosymbiont that can be found within the bacteriome and other tissues is Wolbachia [40]. This Alphaproteobacterium can be beneficial, and some strains have been shown to supply B vitamins to their hosts [41]. However, in many species it affects reproduction and can also interfere with the transmission of pathogens [42, 43].
In Bhutan, five species of psyllids have been found feeding on Rutaceae: D. citri, Diaphorina communis Mathur, Cornopsylla rotundiconis Lou, Li, Li & Cai, Cacopsylla heterogena Li and an unidentified species of Cacopsylla [44]. Diaphorina communis and Ca. heterogena [44, 45] and Ca. citrisuga (Yang & Li) have been shown to harbour CLas, and Ca. citrisuga (Yang & Li) can both acquire and transmit CLas [46, 47]. The microbiota of D. citri has been widely studied due to its ability to transmit the liberibacters associated with citrus, but little is known of the bacterial communities associated with Cacopsylla and Cornopsylla species. Given the potential of psyllids of these genera to harbour plant pathogens, for their host plants to also harbour plant pathogens [48], and for endosymbionts to be manipulated to manage vector-borne diseases [49,50,51], there is a need to examine their microbiomes. Therefore, this study has used high throughput 16S rRNA gene amplicon sequencing to examine the microbiome of these five species to: (1) identify the secondary endosymbionts and any other facultative endosymbionts in these psyllids; (2) to determine if these individuals carry plant pathogens such as liberibacters that are at low titre or novel and therefore difficult to detect by conventional screening.
Results
Psyllid microbiome analysis using 16S rRNA amplicon sequencing
A total of 48 individuals from five species were subject to 16S rRNA gene amplicon sequencing, and generated a total of 1,695,018 paired-end reads. After trimming, 92.9–96.7% of paired reads were joined, filtered (phred threshold > 19), and sequences were clustered into OTUs at 97% identity; OTUs comprising 2 or fewer reads were removed. For the remaining OTUs, 95 chimeras were identified and removed, leaving the total number of sequence reads per sample ranging from 5612 to 44,381, that clustered into 587 OTUs (Supplementary Table 2). Representative OTU sequence size ranged from 303 bp to 445 bp (mean +/− sd = 411.65 +/− 19.81). Rarefaction curves of observed OTUs per sample (Fig. S1) showed that read coverage was nearly complete. The Good’s coverage metric (Good’s coverage > 99.3%, mean 99.8%; Supplementary Table 4) showed that normalisation of read number to 5612 was sufficient for all samples, and based on rarefaction to 5612 sequences per sample (i.e., the sequence read number of the library with the overall lowest read number), collectively 416 OTUs remained, including 226 OTUs that were now only represented by a single read in the entire rarefied dataset (Supplementary Tables 3 & 4). All measures of alpha diversity showed low species richness with 9–60 detected OTUs (Supplementary Table 2), with individual psyllids only containing 2–6 OTUs at above 1% abundance.
Identity of primary and secondary endosymbionts
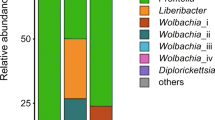
All psyllid individuals contained Carsonella (Figs. 1 & 2), the primary endosymbiont of psyllid species. Four OTUs were classified as Carsonella; D. citri, D. communis and Co. rotundiconis possessed one Carsonella strain each that was fixed in that species, and Ca. heterogena and the unclassified Cacopsylla species possessed the same Carsonella OTU in all individuals. The Carsonella sequence reads were present in individuals at up to 1.2%. Both Diaphorina species were dominated by ‘Ca. Profftella armatura’ (up to 96.2% of reads), but it was not present in the Cacopsylla or Cornopsylla species (Fig. 3).

Heatmap depicting abundance of OTUs within male and female psyllids of the five Bhutanese species. The heatmap was constructed using normalized log10 abundance of each OTU in each individual

Phylogenetic analysis of 16S rRNA gene sequences of potential P-endosymbionts of the five species of Bhutanese psyllids (emboldened). The evolutionary history was inferred using maximum likelihood based on the Hasegawa-Kishino-Yano model [52]. The tree with the highest log likelihood (− 1375.27) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.2691)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The tree is rooted with ‘Ca. Portiera aleyrodidarum’. (NB OTU numbers in bold classified according to Greengenes database, see Supplementary Table 3; OTU 25406 described in [53])

Phylogenetic analysis of 16S rRNA gene sequences of potential S-endosymbionts of the five species of Bhutanese psyllids (emboldened). The evolutionary history was inferred using maximum likelihood based on the Kimura 2-parameter model [54]. The tree with the highest log likelihood (− 1451.55) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 35.53% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The tree is unrooted. (NB OTU numbers classified according to Greengenes database; see Supplementary Table 3)
All Ca. heterogena possessed abundant Enterobacteriaceae from OTU 147629 (12.3–47.3%) and OTU12 (4.9–18.9%), which differed by 13 nucleotides; and Cacopsylla sp. harboured the same two OTUs, but OTU12 was more abundant (29.2–87.5%) (Fig. 1). All Co. rotundiconis individuals were dominated by two Enterobacteriaceae OTUs (OTU147629 at 63.5–67.4% abundance and OTU13 at 31.1–34.2% abundance), where the representative sequences for these OTUs differed by only a single nucleotide. For these symbionts classified to the Enterobacteriaceae, sequence searches using BLAST showed that the closest match to reference genomes in GenBank was Buchnera aphidicola Munson et al., but at only 91.6% match, these OTUs were not classified to genus and therefore, by convention, referred to as S-endosymbionts of its host. However, the sequences were closely related to other S-endosymbionts from other Psylloidae including other species of Cacopsylla (Fig. 3).
Identity of other endosymbionts
Wolbachia (Alphaproteobacteria: Rickettsiales) was prevalent in the five psyllid species (Figs. 1 & 4) at high titre in the Diaphorina and Cacopsylla species (in all individuals except a single D. citri male) but at low titre in all individuals of Co. rotundiconis. Wolbachia sequences from all five species were classified together into OTU 6118 from the Greengenes database; however, the representative sequence for this OTU was highly abundant only in Ca. heterogena and the unidentified Cacopsylla sp. This sequence was not identical to either of the highly prevalent Wolbachia strains previously reported in D. citri. Upon examination of the total reads, another sequence that differed by just one nucleotide, but still classified as OTU 6118, was found in D. citri samples as well as the Cornopsylla and Cacopsylla species, this sequence is identical to the Wolbachia sequences previously detected in Diaphorina citri from the USA. The Wolbachia strain from a Chinese isolate of D. citri, named wDi, was most abundant in D. communis, but this strain was not detected in the Bhutanese D. citri accessions screened here. These Wolbachia sequences were also confirmed in whole genome sequencing data derived from Bhutanese populations of D. communis, Ca. heterogena and the Cacopsylla sp. (data not shown). Other Wolbachia sequences were grouped into another five OTUs at low relative abundance levels (< 0.2% of reads) but the validity of these sequences could not be confirmed from whole genome sequence datasets.

Phylogenetic analysis of 16S rRNA gene sequences of potential Wolbachia endosymbionts within the five species of Bhutanese psyllids (emboldened). The evolutionary history was inferred using maximum likelihood based on the Kimura 2-parameter model [54]. The tree with the highest log likelihood (− 1128.97) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3912)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The tree was rooted using accession AB746416. (NB OTU numbers classified according to Greengenes database; see Supplementary Table 3)
One Rickettsia (Alphaproteobacteria: Rickettsiales) OTU that displayed similarity to the endosymbionts of a range of insect species, as well as environmental sources, was found at high concentrations in certain Co. rotundiconis individuals, at low concentrations in other individuals of this species and at low concentrations in two individuals of the unidentified Cacopsylla sp. (Figs. 1 & 5).

Phylogenetic analysis of 16S rRNA gene sequences of other bacteria found within the five species of Bhutanese psyllids (emboldened). The evolutionary history was inferred using maximum likelihood based on the Kimura 2-parameter model [54]. The tree with the highest log likelihood (− 1615.7) is shown. The percentage of trees in which the associated taxa clustered together is shown next to the branches. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.6415)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The tree is unrooted. (NB OTU numbers are reference sequences in the Greengenes database; see Supplementary Table 3)
Three individuals of the unidentified Cacopsylla sp. harboured a Rickettsiella-like OTU (Gammaproteobacteria: Legionellales) at up to 58% abundance and two individuals of the unidentified Cacopsylla sp. harboured the same OTU but at low concentrations, that showed high similarity to sequences of bacteria found in leaf hoppers and ixodid ticks, as well as other animal sources (Fig. S2).
The majority (6–9 out of 10) of individuals of D. commumis contained OTUs with similarity to Chelatococcus (Alphaproteobacteria: Rhizobiales); Geobacillus (Bacillis: Bacillales); Niabella (Sphingobacteria: Sphingobacteriales); Meiothermus (Deinococci: Thermales); Buchnera (Gammaproteobacteria: Enterobacteriales); and a member of the Comamonadaceae. These OTUs occurred at low abundance (0.02–0.77%). In addition, all individuals of D. citri harboured sequences of a single OTU at 0.6–3.8% abundance that shows high similarity to a range of Methylobacterium species (Alphaproteobacteria: Rhizobiales).
Individuals of both Co. rotundiconis and the unidentified Cacopsylla sp. contained OTUs with 100% identity with species of bacteria belonging in the genus Agrobacterium including A. pusense, A. salinitolerans and members of the A. tumefaciens complex. In addition, the majority of individuals (7 from 10) of the other species also contained low numbers of OTUs showing 100% identity with members of the genus Staphylococcus.
Plant pathogens
Nine of the 10 D. citri individuals tested using high throughput sequencing (HTS) contained OTUs with similarity to CLas found in other accessions of this psyllid species (Figs. 1 & 6). OTUs classified by the Greengenes data base as a species of ‘Ca. Liberibacter’ were detected in the Cacospylla and Cornospylla spp.; these sequences were not identical to other ‘Ca. Liberibacter’ sequences in GenBank. Five of the composite samples from Co. rotundicornis subjected to liberibacter-specific PCR and Sanger sequencing returned fragments of the expected sizes for one or both of the primer pairs. No differences in sequence were detected in the region of the 16S rRNA gene that overlaps between the HTS and specific-PCR data for OTU 2867020 and by only one base in OTU 3229. Phylogenetic analysis of the HTS and Sanger sequencing data (Fig. 6) showed that the bacterium did not group with any of the species of ‘Ca. Liberibacter’ found in GenBank and was sister to all accessions other than Liberibacter crescens Fagen et al.

Phylogenetic analysis of 16S rRNA gene sequences of the ‘Ca. Liberibacter sp.’ amplified from Cornopsylla rotundiconis by PCR (GenBank accession MN203627) and from Co. rotundiconis and the unidentified Cacopsylla sp. by HTS (OTU numbers classified according to Greengenes database, also see Supplementary Table 3). The evolutionary history was inferred by using the maximum likelihood method based on the Kimura 2-parameter model [54]. The tree with the highest log likelihood (− 1039.25) is shown. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.1544)). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. The tree is rooted with Agrobacterium tumefaciens
Bacterial community diversity
Measures of α-diversity are presented in Fig. 7. As judged by both the number of species and the Chao1 metric, species richness appears to be greater in Ca. heterogena than in the remainder of the species. Both the Chao1 metric and Good’s Coverage suggest that this is due to a higher number of OTUs with few reads. The number of species in the other taxa are similar to each other. The measures of heterogeneity (Simpson’s and Shannon’s Indices) suggest that Ca. heterogena is most diverse and that the two species of Diaphorina are less diverse than the other three. Males of the undescribed species of Cacopsylla and D. communis carry more bacterial species than females whereas the opposite is true for D. citri; all effects of sex are, however, small. With the exception of the two species of Diaphorina, principal coordinates analysis (PCoA) coordination of measures of β-diversity (Fig. 8) separate the taxa from each other; the Diaphorina species group together, a result of the dominance of Profftella reads in both Diaphorina species. Small effects of sex are also apparent, especially for D. citri and Ca. heterogena.

Measures of alpha-diversity, number of reads (A) Simpson’s (B), Shannon’s (C), Chao-1 (D), numbers of species (E) and Good’s (E) calculated from 16S rRNA gene amplicon sequencing of males and females of the five Bhutanese psyllid species. Error bars represent standard error of the means (n = 3–5). Means annotated with the same values are not significantly different from each other at P = 0.05 according to Student’s LSD tests

Principal coordinates analysis plot using a Bray-Curtis and b unweighted UniFrac distances generated from rarefied taxon abundances and depicting patterns of beta diversity among male and female psyllid of the five species found in Bhutan
Discussion
Given the importance of endophytic bacteria to crop protection and to the biology of psyllids, this study has looked at the microbiomes of five species of psyllids associated with Rutaceae in Bhutan. As has been found for a range of plant sap-feeding insects [32, 55], the microbiomes of the psyllids largely consisted of their P- and S-endosymbionts, and Wolbachia. All five species contained OTUs related to Carsonella, the P-endosymbiont present in all psyllid species [33]. The P-endosymbionts from the insects in this study grouped with those from closely related taxa showing the strong co-phylogeny between insects and their P-endosymbionts[28]. Cacopsylla heterogena and Cacopsylla sp. share the same Carsonella OTU, suggesting these two psyllid species are closely related [32]. Although there is no strict co-speciation between psyllids and their S-endosymbionts due to loss and acquisition [32, 56], the S-endosymbionts from the five psyllids grouped with those from closely related taxa. The accessions of D. citri and D. communis both contained sequences related to ‘Ca. Profftella armatura’, the defensive endosymbiont of some psyllids characterised by Nakabachi et al. [57]. So far, Diaphorina is the only genus found to harbour bacteria from this genus, and more work on the genomic and ecological features of the Profftella species associated with D. communis is needed to determine if it plays a similar defensive role as in D. citri. Previously, members of the genus Arsenophonus [58] were found in D. citri [56] as well as other psyllids [32, 56]; however, it was not detected in populations of the two Diaphorina species examined in this study. Arsenophonus sequences were also absent from D. citri sampled from Florida [59]; often invasive species suffer loss of bacterial endosymbionts in a new environment, and these Bhutanese populations may exemplify this scenario [32, 60].
The other three psyllid species, including Co. rotundiconis, all contained OTUs classified as Enterobacteriaceae. These bacteria are not currently able to be identified to the species level, and they represent novel S-endosymbionts that are a hallmark of the psyllid microbiome [32, 35]. These psyllid-associated S-endosymbionts appear fixed, or almost fixed, across individuals of a species, and are found in high relative abundance within an individual. It is likely that they share the attributes of the S-endosymbionts described in other psyllids in prior studies [32, 35], that is: (1) intracellular and non-culturable; (2) vertically transmitted; (3) evolving towards obligatory symbiosis within host; and (4) small, reduced genome size due to shedding genes during evolution to intracellular and obligate lifestyle. However, genome assemblies of these bacteria will be necessary to detect the signatures of genome reduction, and further tests performed to determine tissue localization and physiological effects in hosts. In addition, six of the populations of D. communis contained a relatively low abundance of sequences related to Buchnera aphidicola (99.8% identity), the P-endosymbiont of aphids [61]. Although Buchnera has been found in insects other than aphids [62], Buchnera have also been found in the plant, Solanum nigrum L. (Solanales: Solanaceae) [53] and in samples of human origin [63] and may be part of the group of OTUs representing environmentally-acquired bacteria.
Wolbachia was found in all five psyllid species and was abundant in all except Co. rotundiconis. Similar strains that all clustered as OTU6118 were detected in all species. Wolbachia is commonly found in arthropod species [64], and studies have found Wolbachia infection is fixed in D. citri populations in Brazil [65] but with differences detected in titres per individual [66]. The bacterium is a facultative, cytoplasmic symbiont that manipulates reproduction in its host [67]. The presence of Wolbachia proteins in the saliva of D. citri indicates that Wolbachia may play a role in psyllid–plant interactions [68]. Two prevalent Wolbachia strains that have been reported in D. citri, were also detected in this dataset at high abundance, with the Bhutanese D. citri dominated by a single Wolbachia strain first found in USA isolates of D. citri, while D. communis samples were dominated by the Wolbachia strain, wDi, first detected in D. citri from China. Due to the importance of D. citri as a vector of liberibacter, and the apparent lower competence of D. communis to transmit liberibacters, contrasting the potential impact of Wolbachia in both D. citri and D. communis adults and nymphs may prove insightful. The abundant Wolbachia strains in the two Cacopsylla species were most closely related to those from species of whitefly.
The unidentified Cacopsylla sp. was infected with a species of Rickettsia showing sequence identity to accessions of this genus from a range of insect species; however, the sequences were diverged from Rickettsia found in some other psyllids and the white fly, Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae) [32, 69, 70]. Rickettsia can be obligate, intracellular symbionts that are vertically transmitted but can also be acquired by whiteflies from a plant host inside which they become established in the phloem [71]. Like Wolbachia, some Rickettsia strains can affect host reproduction through male-killing [72] and parthenogenesis [73] and can also reduce microbiome diversity [74, 75]. They can also affect crop protection, as Rickettsia increase susceptibility to insecticides [76, 77], and Davis et al. [78] have shown that papaya bunchy top disease is caused by a Rickettsia vectored by Empoasca leafhoppers (Hemiptera: Cicadellidae). The role of this organism in Cacopsylla needs to be investigated.
CLas was found in D. citri but not in D. communis. The lack of CLas in D. communis is not surprising as, although the psyllid ‘feeds’ on Citrus spp. plus Murraya elongata A.DC. ex Hook. F. (Aurantieae), B. koenigii and Zanthoxylum spp., its development appears to be restricted to B. koenigii [44]; B. koenigii is not a host of CLas [44, 79]. The discovery of an unidentified ‘Ca. Liberibacter sp.’ in Co. rotundiconis and the unidentified Cacopsylla sp. parallels the discovery of ‘Ca. Liberibacter brunswickensis’ in Acizzia solanicola Kent and Taylor (Psyllidae) [80] and ‘Ca. Liberibacter ctenarytainae’ in Ctenarytaina fuchsiae (Maskell) (Aphalaridae) [81]. Both Co. rotundiconis and Cacopsylla sp. share a host plant that may enable acquisition of this liberibacter by both psyllids. Within D. citri, CLas is able to replicate and titres of this bacterium are relatively high [82]; however, in our study, the number of reads of the unidentified ‘Ca. Liberibacter sp.’ within individuals of Co. rotundiconis and the unidentified Cacopsylla sp. were low. This may suggest that the bacterium is unable to replicate and may have been passively acquired through feeding. Alternatively, the bacterium may replicate poorly within the psyllids. Therefore, the status of the new liberibacter as a plant pathogen, its transmission and its potential plant hosts are currently unknown.
The generic liberibacter primers used in this study proved useful to validate the liberibacter sequences obtained by HTS. Analysis of the primer pair 484F/803R in silico determined that they will amplify all known species and subspecies of liberibacter at the time they were designed. A single mismatch between the 803R reverse primer and the 16S rRNA sequence of ‘Ca. L. europaeus.’ did not prevent its amplification. Since their design, two new liberibacter species that have been identified also have a single mismatch in a noncritical region of the reverse primer. As with other generic primers, 484F/803R may occasionally amplify sequences from non-liberibacter species but the identity of the resulting amplicon is easily resolved by sequence analysis.
A Rickettsiella-like organism was detected in Co. rotundiconis and the unidentified Cacopsylla sp. This OTU (576671) has 99.5% sequence identity with Diplorickettsia massiliensis Mediannikov et al. (Legionellales: Coxiellaceae) from an Ixodes ticks and 98.6% identity from a Diplorickettsia sp. from a murine mucosa. Related organisms from the Coxiellaceae have been found in the psyllids, Cecidotrioza sozanica Boselli [83] and Glycaspis brimblecombei Moore [32]; however, these are more distantly related to OTU 576671. D. massiliensis is an obligate intracellular bacterium [84] and human pathogen [85] and is phylogenetically related to the genus Rickettsiella. Most Rickettsiella spp. are arthropod pathogens [86]; however, facultative symbionts belonging to the genus Rickettsiella have been identified in the aphid, Acyrthosiphon pisum Harris [87] and in the leafhoppers, Orosius albicinctus Distant [88], Macrosteles striifrons (Anufriev) and Macrosteles sexnotatus (Fallén) [89]. The Rickettsiella sp. within A. pisum causes changes in body color that, in turn, may influence predator-prey interactions [87], and any effects of OTU 576671 on the biology of its hosts need to be determined.
Diaphorina citri harboured bacteria belonging to the genus Methylobacterium that showed sequence homology with bacteria of this genus from a range of plant species. This xylem-associated bacterium has been isolated from citrus plants [90, 91], and it is possible that the psyllid acquired this bacterium during periods of xylem feeding. Diaphorina communis and to a lesser extent Co. rotundiconis and Ca. heterogena harboured a number of OTUs associated with environmental samples: Geobacillus and Chelatococcus, commonly found in straw products and composts; Niabella from soils; Meiothermus from biofilms from geothermal areas; Thermomonas from a fixed-bed reactor, hot springs, and kaolin slurry; and a member of the Comamonadaceae, other members of which are found ubiquitously in freshwater habitats and from activated sludge. Insect microbiomes can vary considerably in diversity with plant sap-feeding insects usually containing few gut bacteria [92, 93]. These bacteria in Co. rotundiconis may represent passive acquisition through consumption rather than active colonisation of the insect’s gut.
OTUs showing 100% identity with members of the Staphylococcus were found in the unidentified Cacopsylla sp. Staphylococci are commonly found in the microbiomes of insects [94] and have been shown to affect insect behaviour. For example, the velvetbean caterpillar (Anticarsia gemmatalis Hübner, Lepidoptera: Noctuidae) uses gut bacteria including staphylococci to overcome the activity of protease produced by its host plant [95] and the honey dew of Acyrthosiphon pisum contains a kairomone produced by Staphylococcus sciuri Kloos et al. (Bacillales: Staphylococcaceae) attracting ovipositing female hover flies (Syrphidae [96]). Further OTUs in the unidentified Cacopsylla sp. and in Co. rotundiconis showed 100% identity with members of the A. tumefaciens complex and with A. pusense. These bacteria have intimate associations with plants as gall-forming pathogens [97] or are involved in nitrogen fixation [98]. Further work is also required to determine whether the OTUs related to staphylococci and agrobacteria have biological roles associated with the psyllids or are again present due to consumption.
Conclusions
The bacterial microbiota detected in these Bhutanese psyllids support the trends that have been seen in previous studies: psyllids have microbiomes largely comprising their obligate P-endosymbiont Carsonella, and one or two S-endosymbionts that are fixed and specific to each lineage. In addition, the association with plant pathogens has been demonstrated, with the detection of liberibacters in a known host, D. citri, and identification of a putative new species of liberibacter in Co. rotundiconis and Cacopsylla sp. Further molecular and biological characterisation of the new liberibacter species will provide additional insights on the potential environmental and/or economic impacts of this organism. The significance of liberibacters to global horticultural industries will likely prompt testing to determine the distribution of the putative species detected in this study outside of Bhutan and its presence in other hosts.
Methods
Psyllid collection and characterisation
Five species of three genera within the Psylloidea were sampled from host plants in Bhutan (Table 1, Supplementary Table 2). Diaphorina citri was first described from adults found on citrus in Taiwan. Diaphorina communis was first recorded by Mathur [99] on curry leaf (Bergera koenigii L.) (Clauseneae) in the northern Indian foothills of the Himalayas, but was not described until 1975 [100]. In Bhutan, it is found on both curry leaf and mandarin (Citrus reticulata Blanco) (Aurantieae) at elevations ranging from 210 to 1223 m asl. Cacopsylla heterogena is closely related to Ca. citrisuga (Yang & Li); in Bhutan, it was found on mandarin at elevations 983–2444 m asl [44] and in China is known to occur on citrus [46, 101]. Cacopsylla sp. and Co. rotundiconis both feed on Zanthoxylum spp. (Amyridoideae).
Adult specimens for each species were used in high throughput bacterial microbiome analysis. Psyllids were collected by tapping branches of host plants onto a white enamel tray and aspirating the adults falling on the tray into a glass vial; the insects were then stored in absolute ethanol. For each species, the surfaces of five male and five female individuals were disinfested using 4% sodium hypochlorite as described in Morrow et al. [32] before DNA was extracted from whole, individual psyllids using QiaAmp Mini DNA Extraction kit (Qiagen) including an RNase treatment with 0.4 mg RNase (Sigma). The quantity and quality of the DNA was assessed by Nanodrop spectrophotometry, Qubit 2.0 Fluorometry and gel electrophoresis; all DNA samples were subject to a test amplification using general eubacterial 16S rRNA gene primers (63F and 1227R [102, 103]; Supplementary Table 1). DNA extracts were submitted to the HIE Next Generation Sequencing Facility (Hawkesbury Institute for the Environment, Western Sydney University) for HTS of the 16S rRNA gene using primers 341F and 805R, which cover the V3-V4 region of the 16S rRNA gene and produce a fragment of approximately 464 bp. Libraries were prepared using the Nextera XT kit (Illumina) with 7 ng of template DNA, and 2 × 300 bp paired end sequencing was run on a 384-multiplexed Illumina MiSeq lane.
Data analysis using QIIME
Raw sequencing reads (fastq format) from 48 individual libraries were examined using FastQC v0.11.4 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc). To increase the accuracy in these libraries [104], they were trimmed, then primer sequences were removed and paired reads joined as detailed in Morrow et al. [32].
QIIME 1.8.0 [105] was used to classify and analyse the bacterial communities associated with each sample using the procedures detailed in Morrow et al. [32]. Sequences were clustered into operational taxonomic units (OTUs) at 97% sequence similarity, and using uclust [106], classified according to the Greengenes 13_8-release database; OTUs comprising fewer than three sequences were removed. Chimeras were identified using the blast_fragments method [107] and then removed. Rarefaction graphs were generated in the vegan package within the R environment [108] to determine the optimal number of sequences for normalisation of samples. Metrics of alpha (Chao1, Shannon and Simpson indices and Good’s coverage) and beta diversity (Bray-Curtis similarities and weighted and unweighted UniFrac) were also determined as detailed in Morrow et al. [32]. Principal coordinate analyses were performed using the vegan package from within the R environment. Statistical tests of alpha and beta diversity comparisons by category (species, genus and sex) were performed using Statistica (Version 13.3; TIBCO Software Inc.).
16S rRNA gene phylogeny
DNA sequence alignments of 16S rRNA genes derived from the HTS and sequences sourced from GenBank were produced using Muscle, as implemented in MEGA7 (Version 7.0.26 [109]). Prior to analyses, the most appropriate evolutionary model for each data set was selected based on their Bayesian Information Criterion and phylogenetic relationships estimated using maximum likelihood. The percentages of trees in which the associated taxa clustered together were determined from 1000 bootstrap replications.
To confirm the novel liberibacter sequences discovered in the HTS data, DNA was extracted from 10 composite samples of five Co. rotundiconis adults. Almost the entire (1411 bp) 16S rRNA gene was amplified by PCR using the generic liberibacter primer pairs LG774F/LG1463R [80], 484F/1124R (Lia Liefting pers. comm.) and fD2 [110] /803R (Lia Liefting pers. comm.) (See Supplementary Table 1 for primer information). The fragments obtained were subjected to Sanger sequencing, and the data assembled and aligned using Geneious (Version 10; Biomatters Ltd.) together with relevant sequences from GenBank. The aligned data were subjected to phylogenetic analysis using MEGA7 as described above. The tree obtained was rooted using a sequence from Agrobacterium tumefaciens Smith & Townsend (Alphaproteobacteria: Rhizobiales). The sequence from Co. rotundiconis was submitted to GenBank (MN203627).
Availability of data and materials
The raw 16S rRNA genes amplicon reads are available from NCBI, SRA BioProject ID PRJNA563783. The assembled OTU representative sequences are found in fasta format in Supplementary Table S4. The DNA sequence for the Liberibacter sp. ex Cornopsylla rotundiconis 16S rRNA gene is available from GenBank, accession number MN203627.
Abbreviations
- OTU:
-
Operational taxonomic unit
- CLas:
-
‘Ca. Liberibacter asiaticus’
- P-endosymbiont:
-
Primary endosymbionts
- S-endosymbiont:
-
Secondary endosymbiont
- HTS:
-
High throughput sequencing
- PCR:
-
Polymerase chain reaction
- PCoA:
-
Principal coordinates analysis
- bp:
-
Base pair
References
Percy DM, Crampton-Platt A, Sveinsson S, Lemmon AR, Lemmon EM, Ouvrard D, Burckhardt D. Resolving the psyllid tree of life: Phylogenomic analyses of the superfamily Psylloidea (Hemiptera). Syst Entomol. 2018;43:762–76.
Tamborindeguy C, Huot OB, Ibanez F, Levy J. The influence of bacteria on multitrophic interactions among plants, psyllids, and pathogen. Insect Sci. 2017;24:961–74.
Davies DL, Guise CM, Clark MF, Adams AN. Parry’s disease of pears is similar to pear decline and is associated with mycoplasma-type organisms transmitted by Cacopsylla pyricola. Plant Pathol. 1992;41:195–203.
Carraro L, Loi N, Ermacora P, Gregoris A, Osler R. Transmission of pear decline using naturally infected Cacopsylla pyri L. Acta Hortic. 1998;472:665–8.
Frisinghelli C, Delaiti L, Grando MS, Forti D, Vindimian ME. Cacopsylla costalis (flor 1861), as a vector of apple proliferation in Trentino. J Phytopathol. 2000;148:425–31.
Carraro L, Ferrini F, Labonne G, Ermacora P, Loi N. Seasonal infectivity of Cacopsylla pruni, vector of European stone fruit yellows phytoplasma. Ann Appl Biol. 2004;144:191–5.
Jarausch B, Fuchs A, Schwind N, Krczal G, Jarausch W. Cacopsylla picta as most important vector for ‘Candidatus Phytoplasma Mali’ in Germany and neighbouring regions. Bull Insectol. 2007;60:189–90.
Malagnini V, Pedrazzoli F, Gualandri V, Forno F, Zasso R, Pozzebon A, Ioriatti C. A study of the effects of ‘Candidatus Phytoplasma Mali’ on the psyllid Cacopsylla melanoneura (Hemiptera: Psyllidae). J Invertebr Pathol. 2010;103:65–7.
Font I, Abad P, Albiñana M, Espino AI, Dally EL, Davis R, Jorda C. Amarilleos y enrojecimientos en zanahoria: Una enfermedad a diagnostico. Bol San Veg Plagas. 1999;25:405–15.
Catling HD. Notes on the biology of the south African citrus psylla, Trioza erytreae (Del Guercio) (Homoptera: Psyllidae). J Entomol Soc South Africa. 1973;36:299–306.
Massonié G, Garnier M, Bové JM. Transmission of Indian citrus decline by Trioza erytreae (Del Guercio), the vector of south African greening. In: Calavan EC, editor. Proceedings of the seventh conference of the International Organization of Citrus Virologists, Athens, Greece, 29 September-4 October 1975. Riverside: International Organization of Citrus Virologists. Riverside: University of California; 1976. p. 18–20.
Lallemand J, Fos A, Bové JM. Transmission de la bacterie associé à la forme africaine de la maladie du “greening” par le psylle asiatique Diaphorina citri Kuwayama. Fruits. 1986;41:341–3.
Yamamoto PT, Felippe MR, Garbim LF, Coelho JHC, Ximenes NL, Martins EC, Leite APR, Sousa MC, Abrahão DP, Braz JD. Diaphorina citri (Kuwayama) (Hemiptera: Psyllidae): Vector of the bacterium ‘Candidatus Liberibacter americanus’. Proceedings of the Huanglongbing-Greening International Workshop, Ribeirão Preto, São Paulo, Brazil, 16–20 July 2006. Araraquara: Fundecitrus; 2006. p. 96.
Queiroz RB, Donkersley P, Silva FN, Al-Mahmmoli IH, Al-Sadi AM, Carvalho CM, Elliot SL. Invasive mutualisms between a plant pathogen and insect vectors in the Middle East and Brazil. R Soc Open Sci. 2016;3:160557.
Weintraub PG, Beanland L. Insect vectors of phytoplasmas. Annu Rev Entomol. 2006;51:91–111.
Nadarasah G, Stavrinides J. Insects as alternative hosts for phytopathogenic bacteria. FEMS Microbiol Rev. 2011;35:555–75.
Wilde WHA, Carpenter J, Liberty J, Tunnicliffe J. Psylla pyricola (Hemiptera: Psyllidae) vector relationships with Erwinia amylovora. Can Entomol. 1971;103:1175–8.
Hildebrand M, Dickler E, Geider K. Occurrence of Erwinia amylovora on insects in a fire blight orchard. J Phytopathol. 2000;148:251–6.
Tenorio J, Chuquillanqui C, García A, Guillén M, Chávez R, Salazar LF. Sintomatología y efecto en el rendimiento de papa por el achaparramiento rugoso. Fitopatología. 2003;38:32–6.
Büchner P. Endosymbiose der Tiere mit Pflanzlichen Mikroorganismen. Basel: Gebundene Ausgabe; 1953.
Casteel CL, Hansen AK. Evaluating insect-microbiomes at the plant-insect interface. J Chem Ecol. 2014;40:836–47.
Mattson WJ Jr. Herbivory in relation to plant nitrogen content. Annu Rev Ecol Syst. 1980;11:119–61.
Sandström JP, Moran NA. How nutritionally unbalanced is phloem sap for aphids? Entomol Exp Appl. 1999;91:203–10.
Sandström JP, Pettersson J. Amino acid composition of phloem sap and the relation to intraspecific variation in pea aphid (Acyrthosiphon pisum) performance. J Insect Physiol. 1994;40:947–55.
Moran NA. Symbiosis as an adaptive process and source of phenotypic complexity. Proc Natl Acad Sci U S A. 2007;104:8627–33.
Baumann P. Biology of bacteriocyte-associated endosymbionts of plant sap-sucking insects. Annu Rev Microbiol. 2005;59:155–89.
Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–90.
Thao ML, Moran NA, Abbot P, Brennan EB, Burckhardt DH, Baumann P. Cospeciation of psyllids and their primary prokaryotic endosymbionts. Appl Environ Microbiol. 2000;66:2898–905.
Hall AA, Morrow JL, Fromont C, Steinbauer MJ, Taylor GS, Johnson SN, Cook JM, Riegler M. Codivergence of the primary bacterial endosymbiont of psyllids versus host switches and replacement of their secondary bacterial endosymbionts. Environ Microbiol. 2016;18:2591–603.
Fromont C, Riegler M, Cook JM. Phylogeographic analyses of bacterial endosymbionts in fig homotomids (Hemiptera: Psylloidea) reveal codiversification of both primary and secondary endosymbionts. FEMS Microbiol Ecol. 2016;92(12):fiw205.
Hansen AK, Moran NA. The impact of microbial symbionts on host plant utilization by herbivorous insects. Mol Ecol. 2014;23:1473–96.
Morrow JL, Hall AAG, Riegler M. Symbionts in waiting: the dynamics of incipient endosymbiont complementation and replacement in minimal bacterial communities of psyllids. Microbiome. 2017;5:58.
Thao ML, Clark MA, Baumann L, Brennan EB, Moran NA, Baumann P. Secondary endosymbionts of psyllids have been acquired multiple times. Curr Microbiol. 2000;41:300–4.
Pérez-Brocal V, Gil R, Ramos S, Lamelas A, Postigo M, Michelena JM, Silva FJ, Moya A, Latorre A. A small microbial genome: the end of a long symbiotic relationship? Science. 2006;314:312–3.
Sloan DB, Moran NA. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol Biol Evol. 2012;29:3781–92.
Tsuchida T, Koga R, Matsumoto S, Fukatsu T. Interspecific symbiont transfection confers a novel ecological trait to the recipient insect. Biol Lett. 2011;7:245–8.
Oliver KM, Degnan PH, Burke GR, Moran NA. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol. 2010;7:247–66.
Scarborough CL, Ferrari J, Godfray HC. Aphid protected from pathogen by endosymbiont. Science. 2005;310:1781.
Montllor CB, Maxmen A, Purcell AH. Facultative bacterial endosymbionts benefit pea aphids Acyrthosiphon pisum under heat stress. Ecol Entomol. 2002;27:189–95.
Kruse A, Fattah-Hosseini S, Saha S, Johnson R, Warwick E, Sturgeon K, Mueller L, MacCoss MJ, Shatters RG Jr, Heck MC. Combining ‘omics and microscopy to visualize interactions between the Asian citrus psyllid vector and the huanglongbing pathogen Candidatus Liberibacter asiaticus in the insect gut. PLoS One. 2017;12:e0179531.
Hosokawa T, Koga R, Kikuchi Y, Meng XY, Fukatsu T. Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc Natl Acad Sci U S A. 2010;107:769–74.
Moreira LA, Iturbe-Ormaetxe I, Jeffery JA, Lu G, Pyke AT, Hedges LM, Rocha BC, Hall-Mendelin S, Day A, Riegler M, Hugo LE. A Wolbachia symbiont in Aedes aegypti limits infection with dengue, Chikungunya, and plasmodium. Cell. 2009;139:1268–78.
Bian G, Xu Y, Lu P, Xie Y, Xi Z. The endosymbiotic bacterium Wolbachia induces resistance to dengue virus in Aedes aegypti. PLoS Pathog. 2010;6(4):e1000833.
Om N. The roles of psyllids, host plants and environment in the aetiology of huanglongbing in Bhutan. Dissertation: Western Sydney University; 2017.
Donovan NJ, Beattie GAC, Chambers GA, Holford P, Englezou A, Hardy S, Dorjee WP, Thinlay ON. First report of ‘Candidatus Liberibacter asisaticus’ in Diaphorina communis. Australas Plant Dis Notes. 2012;7:1–4.
Cen Y, Gao J, Deng X, Xia Y, Chen J, Zhang L, Guo J, Gao W, Zhou W, Wang Z. A new insect vector of ‘Candidatus Liberibacter asiaticus’ Cacopsylla (Psylla) citrisuga (Hemiptera: Psyllidae). Abstracts of the twelfth international Citriculture congress, Valencia, Spain 18–23, vol. 2012; 2012. p. 194.
Cen YJ, Zhang LN, Xia YL, Guo J, Deng XL, Zhou WJ, Sequeira GJ, Gao JY, Wang ZR, Yue JQ, Gao YQ. Detection of ‘Candidatus Liberibacter asiaticus’ in Cacopsylla (Psylla) citrisuga (Hemiptera: Psyllidae). Fla Entomol. 2012;95:304–11.
Ramadugu C, Keremane ML, Halbert SE, Duan YP, Roose ML, Stover E, Lee RF. Long-term field evaluation reveals huanglongbing resistance in citrus relatives. Plant Dis. 2016;100:1858–69.
Rio RVM, Hu Y, Aksoy S. Strategies of the home-team: symbioses exploited for vector-borne disease control. Trends Microbiol. 2004;12:325–36.
Douglas A. Symbiotic microorganisms: untapped resources for insect pest control. Trends Biotech Sci. 2007;25:338–42.
Crotti E, Balloi A, Hamdi C, Sansonno L, Marzorati M, Gonella E, Favia G, Cherif A, Bandi C, Alma A, Daffonchio D. Microbial symbionts: a resource for the management of insect-related problems. Microb Biotechnol. 2012;5:307–17.
Hasegawa M, Kishino H, Yano T. Dating the human-ape split by a molecular clock of mitochondrial DNA. J Mol Evol. 1985;22:160–74.
Chen L, Luo S, Chen J, Wan Y, Li X, Liu C, Liu F. A comparative analysis of endophytic bacterial communities associated with hyperaccumulators growing in mine soils. Environ Sci Pollut Res. 2014;21:7538–47.
Kimura M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Jing X, Wong AC, Chaston JM, Colvin J, McKenzie CL, Douglas AE. The bacterial communities in plant phloem-sap-feeding insects. Mol Ecol. 2014;23:1433–44.
Subandiyah S, Nikoh N, Tsuyumu S, Somowiyarjo S. Fukatsu T complex endosymbiotic microbiota of the citrus psyllid Diaphorina citri (Homoptera: Psylloidea). Zool Sci. 2000;17:983–90.
Nakabachi A, Ueoka R, Oshima K, Teta R, Mangoni A, Gurgui M, Oldham NJ, van Echten-Deckert G, Okamura K, Yamamoto K, Inoue H. Defensive bacteriome symbiont with a drastically reduced genome. Curr Biol. 2013;23:1478–84.
Nováková E, Hypša V, Moran NA. Arsenophonus, an emerging clade of intracellular symbionts with a broad host distribution. BMC Microbiol. 2009;9:143.
Meyer JM, Hoy MA. Molecular survey of endosymbionts in Florida populations of Diaphorina citri (Hemiptera: Psyllidae) and its parasitoids Tamarixia radiata (Hymenoptera: Eulophidae) and Diaphorencyrtus aligarhensis (Hymenoptera: Encyrtidae). Fla Entomol. 2008;91:294–304.
Nguyen DT, Spooner-Hart RN, Riegler M. Loss of Wolbachia but not Cardinium in the invasive range of the Australian thrips species, Pezothrips kellyanus. Biol Invasions. 2016;18:197–214.
Brinza L, Viñuelas J, Cottret L, Calevro F, Rahbé Y, Febvay G, Duport G, Colella S, Rabatel A, Gautier C, Fayard JM. Systemic analysis of the symbiotic function of Buchnera aphidicola, the primary endosymbiont of the pea aphid Acyrthosiphon pisum. Comptes Rendus Biologies. 2009;332:1034–49.
Ivens AB, Gadau A, Kiers ET, Kronauer DJ. Can social partnerships influence the microbiome? Insights from ant farmers and their trophobiont mutualists. Mol Ecol. 2018;27:1898–914.
Griffen AL, Beall CJ, Firestone ND, Gross EL, DiFranco JM, Hardman JH, Vriesendorp B, Faust RA, Janies DA, Leys EJ. CORE: a phylogenetically-curated 16S rDNA database of the core oral microbiome. PLoS One. 2011;6(4):e19051.
Zug R, Hammerstein P. Still a host of hosts for Wolbachia: analysis of recent data suggests that 40% of terrestrial arthropod species are infected. PLoS One. 2012;7(6):e38544.
Guidolin AS, Cônsoli FL. Molecular characterization of Wolbachia strains associated with the invasive Asian citrus psyllid Diaphorina citri in Brazil. Microb Ecol. 2013;65:475–86.
Hoffmann M, Coy MR, Gibbard HK, Pelz-Stelinski KS. Wolbachia infection density in populations of the Asian citrus psyllid (Hemiptera: Liviidae). Environ Entomol. 2014;43:1215–22.
Werren JH, Baldo L. Clark ME Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741.
Yu X, Killiny N. The secreted salivary proteome of Asian citrus psyllid Diaphorina citri. Physiol Entomol. 2018;43:324–33.
Perlman SJ, Hunter MS, Zchori-Fein E. The emerging diversity of Rickettsia. P Roy Soc B-Biol Sci. 2006;273:2097–106.
Weinert LA, Werren JH, Aebi A, Stone GN, Jiggins FM. Evolution and diversity of Rickettsia bacteria. BMC Biol. 2009;7(1):6.
Caspi-Fluger A, Inbar M, Mozes-Daube N, Katzir N, Portnoy V, Belausov E, Hunter MS, Zchori-Fein E. Horizontal transmission of the insect symbiont Rickettsia is plant-mediated. P Roy Soc B-Biol Sci. 2011;279:1791–6.
von der Schulenburg JH, Habig M, Sloggett JJ, Webberley KM, Bertrand D, Hurst GD, Majerus ME. Incidence of male-killing Rickettsia spp. (α-Proteobacteria) in the ten-spot ladybird beetle Adalia decempunctata L. (Coleoptera: Coccinellidae). Appl Environ Microbiol. 2001;67:270–7.
Hagimori T, Abe Y, Date S, Miura K. The first finding of a Rickettsia bacterium associated with parthenogenesis induction among insects. Curr Microbiol. 2006;52:97–101.
Sakurai M, Koga R, Tsuchida T, Meng XY, Fukatsu T. Rickettsia symbiont in the pea aphid Acyrthosiphon pisum: novel cellular tropism, effect on host fitness, and interaction with the essential symbiont Buchnera. Appl Environ Microbiol. 2005;71:4069–75.
van Treuren W, Ponnusamy L, Brinkerhoff RJ, Gonzalez A, Parobek CM, Juliano JJ, Andreadis TG, Falco RC, Ziegler LB, Hathaway N, Keeler C. Variation in the microbiota of Ixodes ticks with regard to geography, species, and sex. Appl Environ Microbiol. 2015;81:6200–9.
Kontsedalov S, Zchori-Fein E, Chiel E, Gottlieb Y, Inbar M, Ghanim M. The presence of Rickettsia is associated with increased susceptibility of Bemisia tabaci (Homoptera: Aleyrodidae) to insecticides. Pest Manag Sci. 2008;64:789–92.
Ghanim M, Kontsedalov S. Susceptibility to insecticides in the Q biotype of Bemisia tabaci is correlated with bacterial symbiont densities. Pest Manag Sci. 2009;65:939–42.
Davis MJ, Ying ZT, Brunner BR, Pantoja A, Ferwerda FH. Rickettsial relative associated with papaya bunchy top disease. Curr Microbiol. 1998;36:80–4.
Beloti VH, Alves GR, Coletta-Filho HD, Yamamoto PT. The Asian citrus psyllid host Murraya koenigii is immune to citrus huanglongbing pathogen ‘Candidatus Liberibacter asiaticus’. Phytopathology. 2018;108:1089–94.
Morris J, Shiller J, Mann R, Smith G, Yen A, Rodoni B. Novel ‘Candidatus Liberibacter’ species identified in the Australian eggplant psyllid, Acizzia solanicola. Microb Biotechnol. 2017;10:833–44.
Thompson S, Jorgensen N, Bulman S, Smith G. A novel Candidatus Liberibacter species associated with Ctenarytaina fuchsiae, the New Zealand native fuchsia psyllid. Abstract 4187. Australia: Science Protecting Plant Health 2017, Brisbane; 2017. p. 26–8.
Wu F, Huang J, Xu M, Fox EG, Beattie GA, Holford P, Cen Y, Deng X. Host and environmental factors influencing ‘Candidatus Liberibacter asiaticus’ acquisition in Diaphorina citri. Pest Manag Sci. 2018;74:2738–46.
Spaulding AW, von Dohlen CD. Psyllid endosymbionts exhibit patterns of co-speciation with hosts and destabilizing substitutions in ribosomal RNA. Insect Mol Biol. 2001;10:57–67.
Mediannikov O, Sekeyová Z, Birg ML, Raoult D. A novel obligate intracellular gamma-proteobacterium associated with ixodid ticks, Diplorickettsia massiliensis, Gen. Nov., Sp. Nov. PLoS One. 2010;5(7):e11478.
Subramanian G, Mediannikov O, Angelakis E, Socolovschi C, Kaplanski G, Martzolff L, Raoult D. Diplorickettsia massiliensis as a human pathogen. Eur J Clin Microbiol Infect Dis. 2012;31:365–9.
Bouchon D, Cordaux R, Grève P. Rickettsiella, intracellular pathogens of arthropods. In: Zchori-Fein E, Bourtzis K, editors. Manipulative tenants: bacteria associated with arthropods. Boca Raton: CRC Press; 2011. p. 127–48.
Tsuchida T, Koga R, Horikawa M, Tsunoda T, Maoka T, Matsumoto S, Simon J-C, Fukatsu T. Symbiotic bacterium modifies aphid body color. Science. 2010;330:1102–4.
Iasur-Kruh L, Weintraub PG, Mozes-Daube N, Robinson WE, Perlman SJ, Zchori-Fein E. Novel Rickettsiella bacterium in the leafhopper Orosius albicinctus (Hemiptera: Cicadellidae). Appl Environ Microbiol. 2013;79:4246–52.
Ishii Y, Matsuura Y, Kakizawa S, Nikoh N, Fukatsu T. Diversity of bacterial endosymbionts associated with Macrosteles leafhoppers vectoring phytopathogenic phytoplasmas. Appl Environ Microbiol. 2013;79:5013–22.
Araújo WL, Marcon J, Maccheroni W, van Elsas JD, van Vuurde JW, Azevedo JL. Diversity of endophytic bacterial populations and their interaction with Xylella fastidiosa in citrus plants. Appl Environ Microbiol. 2002;68:4906–14.
Lacava PT, Araújo WL, Marcon J, Maccheroni W Jr, Azevedo JL. Interaction between endophytic bacteria from citrus plants and the phytopathogenic bacterium Xylella fastidiosa, causal agent of citrus variegated chlorosis. Lett Appl Microbiol. 2004;39:55–9.
Douglas AE. Experimental studies on the mycetome symbiosis in the leafhopper Euscelis incisus. J Insect Physiol. 1988;34:1043–53.
Cheung WW, Purcell AH. Ultrastructure of the digestive system of the leafhopper Euscelidius variegatus Kirshbaum (Homoptera: Cicadellidae), with and without congenital bacterial infections. Int J Insect Morphol Embryol. 1993;22:49–61.
Sreerag RS, Jayaprakas CA, Ragesh L, Kumar SN. Endosymbiotic bacteria associated with the mealy bug, Rhizoecus amorphophalli (Hemiptera: Pseudococcidae). Int Sch Res Not. 2014;2014:268491.
Visôtto LE, Oliveira MG, Ribon AO, Mares-Guia TR, Guedes RN. Characterization and identification of proteolytic bacteria from the gut of the velvetbean caterpillar (Lepidoptera: Noctuidae). Environ Entomol. 2009;38:1078–85.
Leroy PD, Sabri A, Heuskin S, Thonart P, Lognay G, Verheggen FJ, Francis F, Brostaux Y, Felton GW, Haubruge E. Microorganisms from aphid honeydew attract and enhance the efficacy of natural enemies. Nat Commun. 2011;2:348.
Chilton MD, Drummond MH, Merio DJ, Sciaky D, Montoya A, Gordon MP, Nester EW. Stable incorporation of plasmid DNA into higher plant cells: the molecular basis of crown gall tumorigenesis. Cell. 1977;11:263–71.
Crook MB, Mitra S, Ané J-M, Sadowsky MJ, Gyaneshwar P. Complete genome sequence of the Sesbania symbiont and rice growth-promoting endophyte Rhizobium sp. strain IRBG74. Genome Announc. 2013;1(6):e00934–13.
Mathur RN. Notes on the biology of the Psyllidae (Homopt.). Indian Forest Records I. 1935;2:35–71.
Mathur RN. Psyllidae of the Indian subcontinent. New Delhi: Indian Council of Agricultural Research; 1975.
Li FS. Psyllid host plants. In: Li FS, editor. Psyliidomorpha in China (Insecta: Hemiptera) volume I. Beijing: Science Press; 2011. p. 63–78.
Marchesi JR, Sato T, Weightman AJ, Martin TA, Fry JC, Hiom SJ, Wade WG. Design and evaluation of useful bacterium-specific PCR primers that amplify genes coding for bacterial 16S rRNA. Appl Environ Microbiol. 1998;64:795–9.
O'Neill SL, Giordano R, Colbert AM, Karr TL, Robertson HM. 16S rRNA phylogenetic analysis of the bacterial endosymbionts associated with cytoplasmic incompatibility in insects. Proc Natl Acad Sci U S A. 1992;89:2699–702.
Schirmer M, Ijaz UZ, D’Amore R, Hall N, Sloan WT, Quince C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015;43:e37.
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Altschul SF, Gish W, Miller W, Myers EW. Lipman DJ basic local alignment search tool. J Mol Biol. 1990;215:403–10.
R Core Team. R: A Language and Environment for Statistical Computing. http://www.R-project.org/. 2014.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173:697–703.
Acknowledgements
We thank members of the National Plant Protection Centre, Department of Agriculture, Ministry of Agriculture and Forests, Bhutan for their help with this study. We would also like to thank Jocelyn King, Caroline Janitz, and Emma Hackett from the HIE Next Generation Sequencing Facility for the amplicon library preparation and sequencing. We would also like to thank Anna Englezou for her help with the molecular studies related to the new liberibacter species.
Funding
NO was supported by a John Allwright Fellowship awarded by the Australian Centre for International Agricultural Research. JLM was supported by a fellowship from Australian Research Council Industrial Transformation Training Centre for Fruit Fly Biosecurity Innovation. MR was supported by a Hermon Slade Foundation grant.
Author information
Authors and Affiliations
Contributions
JLM, PH and GACB were responsible for experimental design and data analysis. NO and GACB were responsible for the collection of psyllids. LWL was responsible for the design of the 484F/1124R primer pair. JLM, PH, GACB, NO, NJD, LWL, GAC and MR were responsible for data interpretation and the drafting and approval of the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Rarefaction curve of 16S rRNA gene sequences from male and female psyllids. Curves were calculated based on operational taxonomic units (OTUs) at 97% similarity.
Additional file 2: Figure S2.
Phylogenetic analysis of 16S rRNA gene sequences of Rickettsiella-like bacteria found within two of the species of Bhutanese psyllids (emboldened). The evolutionary history was inferred using maximum likelihood based on the Kimura 2-parameter model [54]. The tree with the highest log likelihood (− 1263.03) is shown. A discrete Gamma distribution was used to model evolutionary rate differences among sites (5 categories (+G, parameter = 0.3116)). The rate variation model allowed for some sites to be evolutionarily invariable ([+I], 55.45% sites). The tree is drawn to scale, with branch lengths measured in the number of substitutions per site. (NB OTU numbers are reference sequences in the Greengenes database; see Supplementary Table 3 and [30]).
Additional file 3: Supplementary Table 1.
Primer sequences and PCR conditions.
Additional file 4: Supplementary Table 2.
Sample metadata and alpha diversity metrics.
Additional file 5: Supplementary Table 3.
OTU name and abundance based on the 16S rRNA amplicon dataset rarefied to 5612 sequences, for each of the 48 psyllid samples.
Additional file 6: Supplementary Table 4.
Representative set of 16S rRNA gene sequences of OTUs from five psyllid species from Bhutan.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

{kind=link}
{kind=link}
Cite this article
Morrow, J.L., Om, N., Beattie, G.A.C. et al. Characterization of the bacterial communities of psyllids associated with Rutaceae in Bhutan by high throughput sequencing. BMC Microbiol 20, 215 (2020). https://doi.org/10.1186/s12866-020-01895-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-020-01895-4