
Abstract
Background
Psyllids (Hemiptera: Psylloidea) comprise a group of plant sap-sucking insects that includes important agricultural pests. They have close associations not only with plant pathogens, but also with various microbes, including obligate mutualists and facultative symbionts. Recent studies are revealing that interactions among such bacterial populations are important for psyllid biology and host plant pathology. In the present study, to obtain further insight into the ecological and evolutionary behaviors of bacteria in Psylloidea, we analyzed the microbiomes of 12 psyllid species belonging to the family Psyllidae (11 from Psyllinae and one from Macrocorsinae), using high-throughput amplicon sequencing of the 16S rRNA gene.
Results
The analysis showed that all 12 psyllids have the primary symbiont, Candidatus Carsonella ruddii (Gammaproteobacteria: Oceanospirillales), and at least one secondary symbiont. The majority of the secondary symbionts were gammaproteobacteria, especially those of the family Enterobacteriaceae (order: Enterobacteriales). Among them, symbionts belonging to “endosymbionts3”, which is a genus-level monophyletic group assigned by the SILVA rRNA database, were the most prevalent and were found in 9 of 11 Psyllinae species. Ca. Fukatsuia symbiotica and Serratia symbiotica, which were recognized only as secondary symbionts of aphids, were also identified. In addition to other Enterobacteriaceae bacteria, including Arsenophonus, Sodalis, and “endosymbionts2”, which is another genus-level clade, Pseudomonas (Pseudomonadales: Pseudomonadaceae) and Diplorickettsia (Diplorickettsiales: Diplorickettsiaceae) were identified. Regarding Alphaproteobacteria, the potential plant pathogen Ca. Liberibacter europaeus (Rhizobiales: Rhizobiaceae) was detected for the first time in Anomoneura mori (Psyllinae), a mulberry pest. Wolbachia (Rickettsiales: Anaplasmataceae) and Rickettsia (Rickettsiales: Rickettsiaceae), plausible host reproduction manipulators that are potential tools to control pest insects, were also detected.
Conclusions
The present study identified various bacterial symbionts including previously unexpected lineages in psyllids, suggesting considerable interspecific transfer of arthropod symbionts. The findings provide deeper insights into the evolution of interactions among insects, bacteria, and plants, which may be exploited to facilitate the control of pest psyllids in the future.
Similar content being viewed by others

Background
Jumping plant lice or psyllids (Hemiptera: Sternorrhyncha: Psylloidea) are plant sap-sucking insects comprising about 4000 described species in the world [1, 2]. As with other sternorrhynchan insects [3,4,5], psyllids feed exclusively on phloem sap throughout their life [1, 2, 6]. Due to this feeding habit, some species transmit plant pathogens, including Candidatus Liberibacter spp. (Alphaproteobacteria: Rhizobiales) and Ca. Phytoplasma spp. (Bacilli: Acholeplasmatales), which makes them notorious agricultural or horticultural pests [7,8,9]. Also, as the phloem sap diet is deficient in nutrients including essential amino acids and some vitamins [10,11,12], psyllids depend on vertically transmitted bacterial mutualists to compensate for the nutritional deficiency. They typically harbor two distinct symbionts [13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31] within a specialized abdominal organ called the bacteriome [32]. The ‘primary symbiont’, assumed to be present in all psyllid species [13,14,15,16, 18,19,20,21,22,23,24,25,26,27,28,29,30,31, 33,34,35], is Ca. Carsonella ruddii (Gammaproteobacteria: Oceanospirillales) [33], which provides the host with essential amino acids [21, 22, 31, 35]. Molecular phylogenetic analyses demonstrated cospeciation between psyllids and Carsonella, resulting from a single acquisition of a Carsonella ancestor by a psyllid common ancestor and its stable vertical transmission since then [16, 19, 25, 33, 34]. Another bacterial lineage harbored in the bacteriome is categorized as a ‘secondary symbiont’. The secondary symbionts are phylogenetically diverse among psyllid species and genera, suggesting repeated infections and replacements during the evolution of Psylloidea [14, 16, 17, 19, 21, 24, 25, 27, 28]. Although secondary symbionts in various insect hosts range from parasites to mutualists [36,37,38,39,40,41,42,43,44], those in the psyllid bacteriome appear consistently to have obligate mutualistic features like the primary symbionts [21, 22, 24,25,26, 28, 31]. Because such features are characteristic of nutritional symbionts [36, 45,46,47,48,49,50,51,52,53], secondary symbionts in the psyllid bacteriome are generally believed to have a nutritional basis [19, 26, 28, 29], as confirmed in Ctenarytaina eucalypti (Aphalaridae: Spondyliaspidinae) and Heteropsylla cubana (Psyllidae: Ciriacreminae) [21]. A unique exception is Ca. Profftella armatura (Gammaproteobacteria: Burkholderiales) found in psyllids of the genus Diaphorina (Psyllidae: Diaphorininae) [22, 30, 31, 54], whose main role appears to be protection of the holobiont (host + symbionts) from natural enemies [22, 31, 55,56,57]. In addition to these bacteriome-associated obligate mutualists, psyllids may harbor various secondary symbionts of a facultative nature, including Wolbachia (Alphaproteobacteria: Rickettsiales), Rickettsia (Alphaproteobacteria: Rickettsiales), Rickettsiella (Gammaproteobacteria: Diplorickettsiales), and Diplorickettsia (Gammaproteobacteria: Diplorickettsiales), which can cause systemic infection in the host insects [19,20,21, 23, 24, 26, 28, 29]. Moreover, recent studies are revealing that interactions among psyllid bacterial populations, including those associated with the bacteriome, facultative symbionts, and plant pathogens, are important for psyllid biology and host plant pathology [58,59,60,61,62]. Thus, elucidating microbiomes of various psyllid lineages, which reflect the ecological and evolutionary behaviors of bacterial populations in Psylloidea, would guide strategies to better control pest species.
According to the definition recently revised by Burckhardt et al. [2], psyllids are classified into seven extant families: Aphalaridae, Calophyidae, Carsidaridae, Liviidae, Mastigimatidae, Psyllidae, and Triozidae. Among them, Psyllidae is the most species-rich family (1381 species), whose largest constituent is the subfamily Psyllinae (795 species) [63]. Whereas several high-throughput amplicon-sequencing analyses have been performed on psyllid microbiomes, the target psyllids were biased toward the two most devastating pests Diaphorina citri (Psyllidae: Diaphorininae) and Bactericera cockerelli (Triozidae) as well as Aphalaridae species [23, 24, 27,28,29,30, 64,65,66]. Although recently published study analyzed psyllids from five families, the analysis was based on clustering sequences with a similarity threshold of 97%, resulting in a lower resolution [67]. In the present study, Illumina sequencing of 16S rRNA genes followed by resolving sequence variants down to the level of single-nucleotide differences was performed to assess the microbiomes of 12 Psyllidae species collected in Japan, focusing especially on Psyllinae (Table 1). Whereas these psyllids include agricultural pests (Anomoneura mori for the mulberry; Cacopsylla biwa for the loquat; Cacopsylla burckhardti and Cacopsylla jukyungi for the pear; and Cacopsylla coccinea for the akebi), none are known to vector plant pathogens. However, some other Cacopsylla spp. transmit phytoplasmas [7], and recent analyses have detected potentially pathogenic microbes from unexpected psyllid species [29, 30, 67]. Thus, the present study was performed to elucidate the ecological and evolutionary behaviors of various bacteria in psyllids, aiming to facilitate better pest management in the future.
Results and discussion
All 12 Psyllidae species have Carsonella and at least one other symbiont
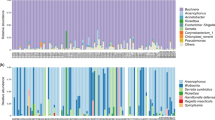
MiSeq sequencing of the amplicon libraries yielded 46,568–73,470 pairs of forward and reverse reads for the 12 psyllid species (Supplementary Table 1). Denoising and joining of the paired-end reads along with removal of low-quality or chimeric reads resulted in 37,901–63,866 non-chimeric high-quality reads (Supplementary Table 1). Dereplication of these reads resulted in 207 independent sequence variants (SVs), among which only 43 SVs accounted for > 1% of the total reads (Supplementary Table 2). We focused on these 43 SVs, because the targets of the present study were relatively abundant symbionts with close association with the host psyllids, and filtering with the threshold of 1% was shown to be among the most effective and accurate methods to remove potential contaminants derived from environments and experimental reagents [68]. SVs with a relative abundance of less than 1% are collectively categorized as ‘others’ in Fig. 1, which correspond to 0.16 – 3.56% reads in total in each psyllid species (Supplementary Table 2). Notably simple bacterial communities like these have been reported for sternorrhynchan insects with bacteriomes, including aphids, whiteflies, and other psyllid species [24, 28, 30, 37, 64,65,66, 69]. All the SVs with a relative abundance of greater than 1% were highly similar to the sequences that were reported to be of insect symbionts (see below). Taxonomic classification by QIIME2 (Supplementary Table 2) followed by independent BLAST searches and phylogenetic analyses showed that all the 12 psyllid species possess distinct lineages of Carsonella (Fig. 1). Because Carsonella has been repeatedly shown to be cospecified with host psyllids [16, 19, 25, 33, 34], the phylogenetic relationship of Carsonella is assumed to be useful to infer that of the host psyllids. In the maximum likelihood (ML) tree, the Carsonella sequences from Psyllinae species formed a clade with those of psyllids belonging to the subfamily Ciriacreminae, and the sequence from Epiacizzia kuwayamai formed an independent clade with those of Aphalaroidinae species (Fig. 2). The exclusion of E. kuwayamai from Psyllinae is consistent with the current classification of this species to the subfamily Macrocorsinae. However, these clades were only poorly supported by bootstrap values (47% for the E. kuwayamai-Aphalaroidinae clade and 39% for the Psyllinae-Ciriacreminae clade), requiring further studies to clarify the phylogenetic position of E. kuwayamai. Besides, the SVs from eight Cacopsylla species did not form a clade (Fig. 2), implying their polyphyly as presumed by Burckhardt et al. [2]. However, this branching pattern also lacked robust statistical support (< 50%). Two types each of Carsonella sequences were detected in Cacopsylla peninsularis and Psylla morimotoi (Fig. 1, Supplementary Table 2). In C. peninsularis, SV19 and SV29 were 99.8% identical (Supplementary Table 2) and formed a clade supported by a bootstrap value of 71% (Fig. 2). SV32 and SV42 from P. morimotoi were also 99.8% identical (Supplementary Table 2), forming a clade supported by a bootstrap value of 97% (Fig. 2). These may reflect sequence variations in each lineage of Carsonella. Although we cannot exclude the possibility that they are artifacts due to polymerase chain reaction (PCR)/sequencing errors, the latter seems less likely because the dada2 plugin corrects sequencing errors during the denoising process [70, 71]. Some previous studies that analyzed psyllid microbiomes using ‘universal primers’ detected only a trace amount of Carsonella reads [27,28,29, 64, 66, 67]; however, the present study, which used primers appropriately modified to improve sensitivity to highly AT-biased symbiont genes [21, 22, 31, 35], detected a large percentage of Carsonella reads (Fig. 1, Supplementary Table 2), which reflects actual populations more precisely [30].

Composition of bacterial populations in psyllids of the family Psyllidae. Relative abundances of Illumina reads belonging to assigned bacterial taxa are shown

Maximum likelihood phylogram of Carsonella. A total of 427 aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. Designations other than those for outgroups refer to psyllid hosts. Families and subfamilies (if applicable) of the host psyllids are shown in brackets. Sequences from this study are shown in bold. DDBJ/EMBL/GenBank accession numbers for sequences are provided in parentheses. The sequence from E. kuwayamai is highlighted in red. The bar represents nucleotide substitutions per position. The outgroups were Ca. Portiera aleyrodidarum; the primary symbiont of the whitefly Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea), and a gammaproteobacterium symbiont of the weevil Metapocyrtus yonagunianus (Coleoptera: Curculionidae)
Besides Carsonella, all 12 psyllids analyzed in the present study possessed at least one other symbiont (Fig. 1).
Various Enterobacteriaceae bacteria reside in Psyllidae
Of the 43 SVs obtained in the present study, 39 corresponded to gammaproteobacteria, among which 22 belonged to the family Enterobacteriaceae (order Enterobacteriales) (Supplementary Table 2). Enterobacteriaceae is a group of bacteria that encompasses an especially large fraction of intimate insect symbionts, including those associated with the bacteriome [36, 46]. Enterobacteriaceae bacteria identified in the present study include Arsenophonus, Fukatsuia, Serratia, Sodalis, endosymbionts2, and endosymbionts3. Among them, the most prevalent was endosymbionts3, which is a genus-level monophyletic group of endosymbionts assigned by the SILVA rRNA database project [72].
Prevalent endosymbionts3
Ten SVs corresponding to distinct lineages of endosymbionts3 were detected in 9 of 11 Psyllinae species (Fig. 1, Supplementary Table 2). Namely, one SV each for endosymbionts3 was observed in Anomoneura mori (SV9: 42.9% of the total denoised reads in A. mori), Cacopsylla biwa (SV1: 70.5% of the C. biwa reads), Cacopsylla coccinea (SV4: 56.1% of the C. coccinea reads), Cacopsylla jukyungi (SV16: 32.8% of the C. jukyungi reads), Cacopsylla kiushuensis (SV25: 22.5% of the C. kiushuensis reads), Cacopsylla peninsularis (SV26: 22.4% of the C. peninsularis reads), Cacopsylla satsumensis (SV8: 61.5% of the C. satsumensis reads), and Cyamophila hexastigma (SV2: 65.9% of the Cy. hexastigma reads). Two SVs corresponding to endosymbionts3, which may reflect sequence variations, were detected in Cacopsylla fatsiae (SV11 and SV22: 42.4 and 24.9%, respectively, of the C. fatsia reads). These SVs were 93.4% (SV2 vs SV11) − 99.8% (SV11 vs SV22) identical to one another. SV9 was 100% identical to the ‘Y-symbiont’ sequence of A. mori (AB013087), which was previously detected via cloning methods [15]. The other nine SVs were 96.2% − 97.7% identical to the sequences of the “Arsenophonus” symbionts of Cacopsylla pyricola (Psyllidae: Psyllinae) (KX077196) and the bat fly Trichobius caecus (Diptera: Streblidae) (DQ314768) [73]. These sequences formed a moderately supported clade (bootstrap: 57%) in the ML tree (Fig. 3). Although these references were named “Arsenophonus”, they were only 84.3% − 87.7% identical to the sequence of the type species Arsenophonus nasoniae (CP038613) [74], and were excluded from the robustly supported clade (bootstrap: 100%) formed by Arsenophonus nasoniae, Ca. Arsenophonus triatominarum, and SV27, assigned as Arsenophonus by QIIME2 in the present study (see below) (Fig. 3). Moreover, this Arsenophonus nasoniae clade formed a strongly supported clade (bootstrap: 96%) with other well-known insect symbionts, including Fukatsuia, Hamiltonella, Regiella, and Serratia, excluding the clade of endosymbionts3 (Fig. 3). These findings may suggest reconsideration of the naming of “Arsenophonus” symbionts that clustered with the endosymbionts3 bacteria. Although little is known about the functions of endosymbionts3-type symbionts [72], the prevalence among analyzed psyllids, high abundance of corresponding reads within each psyllid, and relatively low G + C% (< 50%) of the reads (Fig. 1, Supplementary Table 2) suggest that endosymbionts3 are ancient bacteriome-associated secondary symbionts in these psyllid lineages, which potentially complement the partially deficient functions of Carsonella.

Maximum likelihood phylogram of bacteria belonging to Enterobacteriaceae. A total of 428 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. The scale bar indicates substitutions per site. For symbiotic bacteria, host organisms are shown in brackets. Symbionts of animals other than psyllids are shown in blue. Symbionts of psyllids are shown in red. Sequences from this study are shown in bold. DDBJ/EMBL/GenBank accession numbers are provided in parentheses. Carsonella was used as an outgroup
Arsenophonus symbiont
In addition to Carsonella and an endosymbionts3 bacterium, Arsenophonus was observed in C. jukyungi (Fig. 1). Specifically, QIIME2 assigned SV27, which was derived from 20.0% of denoised C. jukyungi reads (Supplementary Table 2), to Arsenophonus. SV27 was 99.5% identical to the sequence of Arsenophonus symbionts of a wide variety of insects, including other psyllid species, Cardiaspina tenuitela (Aphalaridae: Spondyliaspidinae) (KY428657) and Glycaspis brimblecombei (Aphalaridae: Spondyliaspidinae) (EU043378) [25, 28]. It was 99.3% identical to Arsenophonus nasoniae (CP038613), the type species of Arsenophonus found in the parasitoid wasp Nasonia vitripennis (Hymenoptera: Pteromalidae), and 98.4% identical to Ca. Arsenophonus triatominarum (DQ508185) found in the assassin bug Triatoma rubrofasciata (Hemiptera: Reduviidae). As mentioned above, SV27 formed a robustly supported clade (bootstrap: 100%) with these Arsenophonus symbionts (Fig. 3). Whereas Arsenophonus shows a wide range of associations from parasitic to obligately mutualistic to the host insects [74, 75], its ecological role in psyllids is currently unknown.
Detection of Ca. Fukatsuia symbiotica in psyllids
SV31, which was derived from 11.0% of denoised P. morimotoi reads, was 100% identical to Ca. Fukatsuia symbiotica (CP021659) of the pea aphid Acyrthosiphon pisum (Hemiptera: Sternorrhyncha: Aphidoidea: Aphididae) (Fig. 1, Supplementary Table 2). It was 99.3 and 98.6% identical to Fukatsuia from other aphid species, Cinara confinis (Aphididae) (LT600381) and Maculolachnus submacula (Aphididae) (FJ655539), respectively. SV31 formed a robustly supported clade (bootstrap: 100%) with these sequences in the ML tree (Fig. 3). To our knowledge, this is the first formal report of Fukatsuia detected in psyllids. Fukatsuia has only been recognized as a secondary symbiont of aphids, with a wide variety of reported roles, including pathogen, parasite, defensive symbiont, and obligate nutritional symbiont [76]. The recently revealed culturability of Fukatsuia [76] indicates its ability to survive outside aphids, which would facilitate horizontal transfer to other insects, including psyllids. It would be interesting to assess the prevalence and functional role of Fukatsuia in Psylloidea.
Detection of Serratia symbiotica in psyllids
Six SVs found in C. coccinea corresponded to the sequence of Serratia symbiotica, known as a prevalent secondary symbiont of aphids. Namely, SV35, SV37, SV38, SV39, SV40, and SV41, which accounted for 3.0, 2.0, 2.0, 2.0, 1.8, and 1.8% of the denoised C. coccinea reads, respectively, were 98.8 – 99.8% identical to a single sequence of S. symbiotica (AB522706) (Fig. 1, Supplementary Table 2). This reference sequence was derived from various aphid lineages, including Acyrthosiphon pisum, Aphis fabae, Aphis gossypii, Cinara pinikoraiensis, Cinara ponderosae, and Trama caudata (all Aphididae). The SVs and S. symbiotica sequence from aphids formed a robustly supported clade (bootstrap: 97%) in the ML tree (Fig. 3). To our knowledge, this is the first formal report of S. symbiotica or its close relative detected in psyllids, although there was a previous mention with no concrete data [77]. These SVs were 98.4% (SV35 vs SV41) – 99.8% (SV38 vs SV40) identical to one another. The similarities both in nucleotide sequences and read frequencies imply that the SVs correspond to multiple copies of the 16S rRNA gene in a single S. symbiotica genome. This is consistent with the fact that genomes of several S. symbiotica strains encode more than a single copy of the 16S rRNA gene [42, 43], which contrasts the case of primary symbionts with an extremely streamlined genome encoding only a single copy. Similar to Fukatsuia, the ecological role of S. symbiotica is reported to be widely varied depending on aphid lineages [43, 44]; however, its role in psyllids is currently unknown. Further studies are required to assess this aspect. As Pons et al. showed that S. symbiotica can enter plants and cause new infection in aphids, host plants are likely media for intra-and interspecific horizontal transmission of this bacterium [77].
Sodalis symbionts and its relative
Sodalis endosymbionts were detected in C. burckhardti and C. kiushuensis (Fig. 1, Supplementary Table 2). SV14, which was derived from 30.5% of the C. burckhardti reads, was 96.3% identical to the sequence of the type species Sodalis glossinidius (AP008232), a secondary symbiont of the tsetse fly Glossina morsitans (Diptera: Glossinidae). The sequence was 96.7% identical to that of Ca. Sodalis pierantonius (AF548137), the primary symbiont of the rice weevil Sitophilus oryzae (Coleoptera: Curculionidae). The sequence was 95.6 – 97.9% identical to those of Sodalis endosymbionts from various insects. SV28, which was derived from 13.3% of the C. kiushuensis reads, was 96.7% identical to the Sodalis glossinidius sequence. It was 96.0% identical to the sequence of Ca. Sodalis pierantonius, and 94.6 – 97.2% identical to those of the above-mentioned Sodalis endosymbionts from various insects. These sequences were clustered with that of a Sodalis endosymbiont from another psyllid Cardiaspina maniformis (Aphalaridae: Spondyliaspidinae) (KY428659) [28] and SV34 (see below), whose branching pattern was moderately supported (bootstrap:75%) (Fig. 3). Sodalis symbionts have been detected in a wide variety of insects and are known to have replaced more ancient predecessor symbionts in weevils (Coleoptera: Curculionoidea) [78] and spittlebugs (Hemiptera: Cercopoidea) [79]. In this context, the distribution of the Sodalis symbiont in Cacopsylla spp. may be of interest. Whereas endosymbionts3 appear dominant (presumably bacteriome-associated) secondary symbionts in Cacopsylla spp., C. kiushuensis additionally has a Sodalis symbiont, and C. burckhardti has only Carsonella and Sodalis. This might imply, though speculative, that replacement of endosymbionts3 by Sodalis is at initial stage in C. kiushuensis, and is completed in C. burckhardti.
Regarding SV34, which was derived from 6.1% of denoised P. morimotoi reads, QIIME2 failed to assign a genus-level taxonomy (Fig. 1, Supplementary Table 2). The BLAST best hit of SV34 was Sodalis endosymbiont of the psyllid Cardiaspina maniformis (Aphalaridae: Spondyliaspidinae) (KY428659) [28]. These sequences formed a cluster in the ML tree (Fig. 3). However, this branching pattern was only poorly supported (bootstrap: 31%), and their sequence identity was 93.0%, which was below the generally used arbitrary genus threshold of 94.5 – 95% [80, 81]. Thus, we refrained from assigning this symbiont to a particular genus.
Putative endosymbionts2 symbiont
QIIME2 assigned SV21, which was derived from 31.1% of denoised E. kuwayamai reads, to endosymbionts2 (Fig. 1, Supplementary Table 2), another monophyletic group of endosymbionts assigned by SILVA [72]. The BLAST best hit of SV21 was a secondary endosymbiont of Cacopsylla myrthi (AF263559) [17], but the sequence identity was only 90.9%. SV21 branched basally to other Enterobacteriaceae bacteria in the ML tree (Fig. 3). It would be interesting to assess the prevalence of endosymbionts2 in the subfamily Macrocorsinae, in the context of the apparent prevalence of endosymbionts3 among Psyllinae species analyzed in the present study.
Psylla morimotoi has Pseudomonas and Diplorickettsia
Non-Enterobacteriales gammaproteobacteria found in the present study were Carsonella (Oceanospirillales: Halomonadaceae) mentioned above, Pseudomonas (Pseudomonadales: Pseudomonadaceae), and Diplorickettsia (Diplorickettsiales: Diplorickettsiaceae); of these, the latter two were detected from P. morimotoi. QIIME2 assigned SV12 and SV30, which were derived from 51.5 and 13.3% of denoised P. morimotoi reads, respectively (Fig. 1, Supplementary Table 2), to Pseudomonas. SV12 and SV30 shared 98.4% identity. SV12 was 100% identical to the sequences of various Pseudomonas strains, including type strains for Pse. graminis (Y11150) and Pse. rhizosphaerae (CP009533). SV30 was 99.5% identical to the sequence of the type strain of Pse. viridiflava (NR_114482). Although Pseudomonas species have been detected in various insects including psyllids, they are largely believed to be transient associates [28]. In contrast to Enterobacteriaceae bacteria, many of which have intimate and stable mutualistic relationships with insect hosts, known examples of Pseudomonas with such associations (vertically-transmitted endosymbionts present in the host hemocoel or cells) are limited in rove beetles (Coleoptera: Staphylinidae) [82] and the adelgid Adelges tsugae (Hemiptera: Sternorrhyncha: Phylloxeroidea: Adelgidae) [83]. Although SV12 and SV30 were not closely related to these symbionts (Fig. 4), the fact that the majority (64.8%) of reads in P. morimotoi corresponded to Pseudomonas (Fig. 1, Supplementary Table 2) implies that the Pseudomonas symbionts potentially play important roles in this psyllid.

Maximum likelihood phylogram of Pseudomonas. A total of 427 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. The scale bar indicates substitutions per site. For symbiotic bacteria, host organisms are shown in brackets. Symbionts of animals other than psyllids are shown in blue. Sequences detected in the present study are shown in bold red. DDBJ/EMBL/GenBank accession numbers are provided in parentheses. Carsonella was used as an outgroup
QIIME2 assigned SV36, which was derived from 3.7% of denoised P. morimotoi reads, to Diplorickettsia (Fig. 1, Supplementary Table 2). SV36 was 99.3% identical to the sequence of Diplorickettsia massiliensis 20B (NR_117407) detected in the European sheep tick Ixodes ricinus (Arachnida: Acari: Ixodidae), 98.8% identical to the sequence of Diplorickettsia sp. (TAAA01000010) recently found in another psyllid species Diaphorina cf. continua (Psyllidae: Diaphorininae) [30], 98.4% identical to that of Diplorickettsia sp. MSebKT1 (AB795342) detected in a leafhopper Macrosteles sexnotatus (Hemiptera: Auchenorrhyncha: Cicadellidae), and 98.1% identical to Diplorickettsia sp. NS15 (JN606082) found in human nasal samples. Molecular phylogenetic analysis showed that SV36 forms a well-supported clade (bootstrap: 85%) with these Diplorickettsia spp. (Fig. 5). Diplorickettsia massiliensis was observed in Ixodes ricinus and serum samples of human patients with suspected tick-borne disease, suggesting that this bacterium is a human pathogen [84, 85]. Subsequently, Diplorickettsia lineages were unexpectedly found in two plant sap-sucking hemipteran insects, M. sexnotatus collected in Japan [86], and D. cf. continua collected in Corsica [30]. The present study adds another example of Diplorickettsia. These findings imply that Diplorickettsia is actually prevalent in various sap-sucking insects. Although their host plants are not shared among M. sexnotatus (Poaceae and Fabaceae), D. cf. continua (Thymelaeaceae), and P. morimotoi (Rosaceae), it would be worth assessing the possibility that the plants are also infected with Diplorickettsia. Diplorickettsia is closely related to the genus Rickettsiella (Diplorickettsiales: Diplorickettsiaceae) (Fig. 5) comprising intracellular bacteria associated with various arthropods, including insects, arachnids, and isopods [19, 28]. Whereas many Rickettsiella spp. are simply pathogenic to arthropods, Ca. Rickettsiella viridis [87] modifies the body color of aphids, potentially affecting the attractiveness of insects to natural enemies [88]. As little is known about the functions of Diplorickettsia in host arthropods, it would be interesting to assess the physiological and ecological effects of Diplorickettsia on psyllids.

Phylogenetic position of Diplorickettsia lineages inferred by the maximum likelihood method. A total of 427 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. The scale bar indicates substitutions per site. For symbiotic bacteria, host organisms are shown in brackets. Symbionts of animals other than psyllids are shown in blue. Symbionts of psyllids are shown in red. The sequence from this study is shown in bold. DDBJ/EMBL/GenBank accession numbers are provided in parentheses. Carsonella was used as an outgroup
First detection of Liberibacter in Anomoneura mori
The analysis detected Ca. Liberibacter europaeus (Alphaproteobacteria: Rhizobiales) for the first time in A. mori, a sericultural pest that feeds on the mulberry plants Morus spp. (Moraceae) (Fig. 1, Supplementary Table 2). Namely, SV33, which was derived from 4.6% of denoised A. mori reads, was 99.8% identical to the sequence of Ca. Liberibacter europaeus previously detected in Cacopsylla pyri (Psyllidae: Psyllinae) (FN678792) and Diaphorina cf. continua (Psyllidae: Diaphorininae) (TAAA01000007) [30]. Molecular phylogenetic analysis showed that these sequences form a robustly supported clade (bootstrap: 96%) within Ca. Liberibacter spp. (Fig. 6).

Maximum likelihood phylogram of Liberibacter spp. A total of 402 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. Symbionts of psyllids are shown in red. The sequence from this study is shown in bold. The host psyllids are shown in brackets. DDBJ/EMBL/GenBank accession numbers for sequences are provided in parentheses. The scale bar represents nucleotide substitutions per position. Wolbachia was used as an outgroup
The genus Liberibacter currently includes nine species: Ca. L. asiaticus, Ca. L. americanus, and Ca. L. africanus, which cause greening disease in citrus (Rutaceae) [8, 89]; Ca. L. capsica, a potential pathogen of solanaceous plants [67]; Ca. L. caribbeanus found in citrus, but with uncertain pathogenicity [90]; Ca. L. solanacearum, which causes diseases in solanaceous and apiaceous plants [91,92,93,94]; Ca. L. brunswickensis, a probable endophyte of solanaceous plants [95]; L. crescens, which is non-pathogenic and the only culturable species in the genus [96]; and Ca. L. europaeus (CLeu) [97,98,99]. CLeu was detected from various psyllids in various locations: Cacopsylla spp. (Psyllidae: Psyllinae) in Italy and Hungary [97, 98], Arytainilla spartiophila (Psyllidae: Psyllinae) in New Zealand and the U.K. [99, 100], and D. cf. continua (Psyllidae: Diaphorininae) in Corsica island [30]. CLeu was also detected from rosaceous plants and the Scotch broom Cytisus scoparius (Fabaceae), which are host plants of Cacopsylla spp. and Ar. spartiophila, respectively [97,98,99,100]. Whereas the presence of CLeu is associated with pathological symptoms in the Scotch broom [99], no symptoms are known in rosaceous plants and Thymelaea tartonraira (Thymelaeaceae), the probable host plant of D. cf. continua [30]. The present study adds another example of CLeu from another psyllid species, A. mori (Psyllidae: Psyllinae) in Japan. Because A. mori is a pest species feeding on mulberry plants, it would be interesting to assess if the host plants, which are distantly related to previously known infected plants, are also infected with CLeu and whether infection causes symptoms of disease.
It appears that Ca. Liberibacter lineages have evolved in close association with Psylloidea, and all known vectors for all Ca. Liberibacter spp. are psyllids [8, 30, 67, 89–91, 94, 95, 97–99]. In this context, the finding that the fecundity and population growth rates of D. citri harboring Ca. L. asiaticus are increased as compared with uninfected insects [101] is particularly interesting. This observed benefit may be an ecological driver for the close association between Ca. Liberibacter spp. and psyllids. Future studies should focus on assessing the general applicability of this hypothesis to other Ca. Liberibacter-psyllid combinations.
Two Wolbachia strains reside in four psyllid species
The analysis identified two SVs corresponding to distinct lineages of Wolbachia (Alphaproteobacteria: Rickettsiales), which were previously detected in D. citri and Diaphorina lycii [30]. SV7, which was derived from 11.1% of denoised C. biwa reads, 9.9% of denoised Cy. hexastigma reads, and 31.1% of denoised E. kuwayamai reads (Supplementary Table 2), was 100% identical to the sequence of Wolbachia_iv previously identified in D. citri (TAAA01000013) collected in Japan. The sequence is also identical to those of Wolbachia detected in D. citri from the U.S.A. and the whitefly Bemisia tabaci (Hemiptera: Sternorrhyncha: Aleyrodoidea: Aleyrodidae) from various locations in the Asia-Pacific region [30]. SV43, derived from 1.1% of denoised P. morimotoi reads (Supplementary Table 2), was 100% identical to the sequence of Wolbachia_i previously identified in D. citri and D. lycii (TAAA01000005) [30]. The sequence is also identical to those of Wolbachia reported from the aphid Cinara cedri collected in Israel [102], and various insects in China, including the planthopper Nilaparvata lugens (Hemiptera: Auchenorrhyncha: Delphacidae), and aphids Phloeomyzus passerinii (Phloeomyzidae) and Cervaphis quercus (Aphididae) [103].
Wolbachia are rickettsial bacteria distributed in a wide variety of arthropods and nematodes [104,105,106], whose strains are currently classified into supergroups A–Q [107]. Whereas supergroups A and B are monophyletic and are the most common supergroups infecting arthropods, supergroups C and D infect nematodes. Supergroups E–Q are found in various hosts, including nematodes, springtails, termites, fleas, aphids, and mites [106]. The molecular phylogenetic analysis placed SV7 and SV43 detected in the present study in the robustly supported clade of Wolbachia supergroup B (bootstrap: 97%) (Fig. 7). The majority of Wolbachia strains manipulate the reproduction of arthropod hosts through cytoplasmic incompatibility, feminization, male killing, and parthenogenesis to increase the prevalence of infected females in host populations [104,105,106]. Due to this ability to boost dissemination, Wolbachia are recognized to be promising agents to control insect pests by affecting their traits or microbiomes, including pathogens therein [108, 109]. Because of the high infection rates of Wolbachia in pest psyllids worldwide [18, 62, 66, 110,111,112,113,114,115], and the suggested interactions between Wolbachia and other symbionts [59,60,61,62, 116], the application of Wolbachia to control pest psyllids and/or plant pathogens is anticipated [59, 62, 111, 113, 115]. The present study suggests rampant horizontal transmissions of Wolbachia among various insect lineages, including pest psyllids, implying the feasibility of artificial infection and/or removal of Wolbachia in psyllids. Such techniques would facilitate the exploitation of Wolbachia as a tool to control pest psyllids and/or the plant pathogens they transmit.

Maximum likelihood phylogram of Wolbachia. A total of 402 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. Host organisms are shown in brackets. Symbionts of animals other than psyllids are shown in blue. Symbionts of psyllids are shown in red. The sequence from this study is shown in bold. DDBJ/EMBL/GenBank accession numbers for sequences are provided in parentheses. Supergroups of Wolbachia are shown in angle brackets. The scale bar represents nucleotide substitutions per position. Liberibacter was used as an outgroup
Cacopsylla peninsularis has Rickettsia
The analysis detected Rickettsia sp. (Alphaproteobacteria: Rickettsiales) in C. peninsularis. SV20, which was derived from 31.4% of denoised C. peninsularis reads, was 99.5% identical to the sequence of ‘Rickettsia endosymbiont’ found in various arthropods including the planthopper Nephotettix cincticeps (Delphacidae) (KU586121). The sequence was 99.3 and 99.0% identical to ‘Rickettsia endosymbionts’ of the psyllids, Cacopsylla melanoneura (LR800105) and Chamaepsylla hartigii (LR800074) (Psyllidae: Psyllinae), respectively. Similar sequences were also detected from the drugstore beetle Stegobium paniceum (Coleoptera: Ptinidae) (JQ805029), the booklouse Cerobasis guestfalica (Psocoptera: Trogiidae) (DQ652596), the lacewing Chrysotropia ciliata (Neuroptera: Chrysopidae) (MF156626), and the whitefly B. tabaci (MG063879). These sequences formed a well-supported clade (bootstrap: 87%) in the ML tree (Fig. 8).

Maximum likelihood phylogram of Rickettsia. A total of 402 unambiguously aligned nucleotide sites of 16S rRNA genes were subjected to the analysis. On each branch, bootstrap support values of > 50% are shown. Host organisms are shown in brackets. Symbionts of animals other than psyllids are shown in blue. Symbionts of psyllids are shown in red. The sequence from this study is shown in bold. DDBJ/EMBL/GenBank accession numbers for sequences are provided in parentheses. The scale bar represents nucleotide substitutions per position. Wolbachia was used as an outgroup
Rickettsia is closely related to Wolbachia, both of which belong to the order Rickettsiales. Similar to Wolbachia, some Rickettsia lineages cause reproductive disorders in host insects, including male killing and parthenogenesis [117, 118]. It would be worthwhile to assess whether Rickettsia endosymbionts manipulate the reproduction of psyllids, which will be potentially useful to exploit Rickettsia as a tool to control pest psyllids and/or plant pathogens.
Conclusions
The present study found Ca. Fukatsuia symbiotica and Serratia symbiotica, which were recognized as aphid secondary symbionts, formally for the first time in Psylloidea. The analysis also found the potential plant pathogen, Ca. Liberibacter europaeus (Rhizobiales: Rhizobiaceae), for the first time in a pest psyllid feeding on the mulberry. Furthermore, Wolbachia and Rickettsia, plausible host reproduction manipulators, were detected among analyzed psyllids. The study also identified Arsenophonus, Sodalis, endosymbionts2, endosymbionts3, Pseudomonas, and Diplorickettsia, a plausible human pathogen. These findings suggest considerable interspecific transfer of arthropod symbionts, providing deeper insights into the evolution of interactions among insects, bacteria, and plants. This may be exploited to facilitate the control of pest psyllids with the aid of future studies to determine the localization and genomic structure of the identified bacteria.
Methods
Insects and DNA extraction
Adults of 12 psyllid species belonging to the family Psyllidae were collected from several trees of each host plant species in Japan (Table 1). Although most species were selected from the subfamily Psyllinae, Epiacizzia kuwayamai belongs to the subfamily Macrocorsinae [63]. We included this species because it originally belonged to Psyllinae with the name Psylla kuwayamai and its generic transfer from Psylla to Epiacizzia appeared to be inconsistent with the morphological features [119]. Also, whereas Burckhardt et al. recently proposed to transfer Psylla morimotoi (Psyllinae) to the genus Spanioneura (Psyllinae) based on its ecological features [2], the present study refers to this species as is because its morphological features are more consistent with the definition of Psylla (sensu stricto) [120, 121].
After surface sterilization with ethanol, DNA was extracted from the whole bodies of pooled individuals of five adult males and five adult females of each psyllid species using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). The quality of the extracted DNA was assessed using a NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts, U.S.A.) and the quantity was assessed using a Qubit 2.0 Fluorometer with the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific).
Construction and sequencing of amplicon libraries
Bacterial populations in psyllids were analyzed using the MiSeq system (Illumina, San Diego, California, U.S.A.), as described previously [30]. Briefly, amplicon PCR was performed using the genomic DNA extracted from psyllids, the KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, Massachusetts, U.S.A.), and the primer set 16S_341Fmod (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGYYTAMGGRNGGCWGCAG-3′) and 16S_805R (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′) targeting the V3 and V4 regions of the 16S rRNA gene. Whereas both primers were based on the instructions by Illumina [122], 16S_341F was modified (underlined), where original CC, C, and G were replaced with mixed bases, YY (C or T), M (A or C), and R (A or G). Our preliminary analyses demonstrated that this modification improves sensitivity to detect symbionts with AT-rich genomes including Carsonella, without reducing sensitivity for those with GC-rich genomes. Dual indices and Illumina sequencing adapters were attached to the amplicons with index PCR using the Nextera XT Index Kit v2 (Illumina). The libraries were combined with the PhiX Control v3 (Illumina), and 250 bp of both ends were sequenced on the MiSeq platform (Illumina) with the MiSeq Reagent Kit v2 (500 cycles; Illumina).
Computational analysis of bacterial populations
After the amplicon sequence reads were demultiplexed, the output sequences were imported into the QIIME2 platform (version 2020.2) [123] and processed as described previously [30]. In brief, after primer sequences were removed using the cutadapt plugin [124], paired-end sequences were trimmed, denoised, joined, and dereplicated using the dada2 plugin [70]. During this step, chimeric sequences were detected and removed. The q2-feature-classifier plugin [125] was trained using the V3 and V4 regions of the 16S rRNA gene sequences retrieved from the SILVA database ver. 132 (SILVA_132_QIIME_release/taxonomy/16S_only/99/taxonomy_7_levels.txt) that were clustered at 99% sequence similarity [72]. Subsequently, the denoised and dereplicated amplicon reads were classified and taxonomic information was assigned using the trained q2-feature-classifier. Obtained sequence variants (SVs) were manually checked by performing BLASTN searches against the National Center for Biotechnology Information non-redundant database [126].
Phylogenetic analysis of detected bacteria
Phylogenetic analysis of SVs was performed as described previously [30]. Briefly, after the SVs were aligned to related sequences using SINA (v1.2.11) [127], phylogenetic trees were inferred by the maximum likelihood (ML) method using RAxML (version 8.2.12) [128]. The GTR + Γ model was used with no partitioning of the data matrix, with 1000 bootstrap iterations (options -f a -m GTRGAMMA -# 1000).
Availability of data and materials
The nucleotide sequence data are available in the DDBJ/EMBL/GenBank databases under the accession numbers DRR300288–DRR300299 (MiSeq output) and TAAB01000001–TAAB01000045 (dereplicated sequence variants).
Abbreviations
- Ca :
-
Candidatus
- SV:
-
Sequence variant
- ML:
-
Maximum likelihood
- E.:
-
Epiacizzia
- C.:
-
Cacopsylla
- P.:
-
Psylla
- PCR:
-
Polymerase chain reaction
- A :
-
Anomoneura
- Cy.:
-
Cyamophila
- S.:
-
Serratia
- Pse.:
-
Pseudomonas
- M.:
-
Macrosteles
- D.:
-
Diaphorina
- L :
-
Liberibacter
- CLeu:
-
Candidatus Liberibacter europaeus
- Ar.:
-
Arytainilla
- B.:
-
Bemisia
References
Percy DM, Crampton-Platt A, Sveinsson S, Lemmon AR, Lemmon EM, Ouvrard D, et al. Resolving the psyllid tree of life: phylogenomic analyses of the superfamily Psylloidea (Hemiptera). Syst Entomol. 2018;43:762–76.
Burckhardt D, Ouvrard D, Percy DM. An updated classification of the jumping plant-lice (Hemiptera: Psylloidea) integrating molecular and morphological evidence. Eur J Taxon. 2021;736:137–82.
International Aphid Genomics Consortium. Genome sequence of the pea aphid Acyrthosiphon pisum. PLoS Biol. 2010;8:e1000313. https://doi.org/10.1371/journal.pbio.1000313.
Tamborindeguy C, Monsion B, Brault V, Hunnicutt L, Ju HJ, Nakabachi A, et al. A genomic analysis of transcytosis in the pea aphid, Acyrthosiphon pisum, a mechanism involved in virus transmission. Insect Mol Biol. 2010;19:259–72.
Nakabachi A, Miyagishima S. Expansion of genes encoding a novel type of dynamin in the genome of the pea aphid, Acyrthosiphon pisum. Insect Mol Biol. 2010;19:165–73. https://doi.org/10.1111/j.1365-2583.2009.00941.x.
Hodkinson ID. The biology of the Psylloidea (Homoptera): a review. Bull Entomol Res. 1974;64:325–38.
Jarausch B, Jarausch W. Psyllid vectors and their control. In: Weintraub PG, Jones P, editors. Phytoplasmas: genomes, plant hosts and vectors. Wallingford, Oxfordshire: CAB International; 2010. p. 250–71.
Grafton-Cardwell EE, Stelinski LL, Stansly PA. Biology and management of Asian citrus psyllid, vector of the huanglongbing pathogens. Annu Rev Entomol. 2013;58:413–32. https://doi.org/10.1146/annurev-ento-120811-153542.
Wang N, Stelinski LL, Pelz-stelinski KS, Graham JH, Zhang Y. Tale of the Huanglongbing disease pyramid in the context of the citrus microbiome. Phytopathology. 2017;107:380–7.
Sandstrom J, Moran N, Sandström J, Moran N. How nutritionally imbalanced is phloem sap for aphids? Entomol Exp Appl. 1999;91:203–10.
Ziegler H, Pirson A, Zimmermann MH. Nature of transported substances. In: Zimmermann MH, Milburn JA, editors. Transport in plants I. New York: Springer-Verlag; 1975. p. 59–100.
Nakabachi A, Ishikawa H. Provision of riboflavin to the host aphid, Acyrthosiphon pisum, by endosymbiotic bacteria, Buchnera. J Insect Physiol. 1999;45:1–6. https://doi.org/10.1016/s0022-1910(98)00104-8.
Profft J. Beiträge zur Symbiose der Aphiden und Psylliden. Z Morphol Ökol Tiere. 1937;32:289–326.
Buchner P. Endosymbiosis of animals with plant microorganisms. New York: Interscience; 1965.
Fukatsu T, Nikoh N. Two intracellular symbiotic bacteria from the mulberry psyllid Anomoneura mori (insecta Homoptera). Appl Environ Microbiol. 1998;64:3599–606.
Spaulding AW, von Dohlen CD. Phylogenetic characterization and molecular evolution of bacterial endosymbionts in psyllids (Hemiptera: Sternorrhyncha). Mol Biol Evol. 1998;15:1506–13. https://doi.org/10.1093/oxfordjournals.molbev.a025878.
Thao ML, Clark MA, Baumann L, Brennan EB, Moran NA, Baumann P. Secondary endosymbionts of psyllids have been acquired multiple times. Curr Microbiol. 2000;41:300–4. https://doi.org/10.1007/s002840010138.
Subandiyah S, Nikoh N, Tsuyumu S, Somowiyarjo S, Fukatsu T. Complex endosymbiotic microbiota of the citrus psyllid Diaphorina citri (Homoptera : Psylloidea). Zool Sci. 2000;17:983–9.
Spaulding AW, von Dohlen CD. Psyllid endosymbionts exhibit patterns of co-speciation with hosts and destabilizing substitutions in ribosomal RNA. Insect Mol Biol. 2001;10:57–67. https://doi.org/10.1046/j.1365-2583.2001.00231.x.
Hansen AK, Jeong G, Paine TD, Stouthamer R. Frequency of secondary symbiont infection in an invasive psyllid relates to parasitism pressure on a geographic scale in California. Appl Environ Microbiol. 2007;73:7531–5. https://doi.org/10.1128/AEM.01672-07.
Sloan DB, Moran NA. Genome reduction and co-evolution between the primary and secondary bacterial symbionts of psyllids. Mol Biol Evol. 2012;29:3781–92. https://doi.org/10.1093/molbev/mss180.
Nakabachi A, Ueoka R, Oshima K, Teta R, Mangoni A, Gurgui M, et al. Defensive bacteriome symbiont with a drastically reduced genome. Curr Biol. 2013;23:1478–84. https://doi.org/10.1016/j.cub.2013.06.027.
Arp A, Munyaneza JE, Crosslin JM, Trumble J, Bextine B. A global comparison of Bactericera cockerelli (Hemiptera: Triozidae) microbial communities. Environ Entomol. 2014;43:344–52.
Overholt WA, Diaz R, Rosskopf E, Green SJ, Overholt WA. Deep characterization of the microbiomes of Calophya spp. (Hemiptera: Calophyidae) gall-inducing psyllids reveals the absence of plant pathogenic bacteria and three dominant endosymbionts. PLoS One. 2015;10:1–16.
Hall AAG, Morrow JL, Fromont C, Steinbauer MJ, Taylor GS, Johnson SN, et al. Codivergence of the primary bacterial endosymbiont of psyllids versus host switches and replacement of their secondary bacterial endosymbionts. Environ Microbiol. 2016;18:2591–603.
Fromont C, Riegler M, Cook JM. Phylogeographic analyses of bacterial endosymbionts in fig homotomids (Hemiptera: Psylloidea) reveal codiversification of both primary and secondary endosymbionts. FEMS Microbiol Ecol. 2016;92:fiw205.
Fromont C, Riegler M, Cook JM. Relative abundance and strain diversity in the bacterial endosymbiont community of a sap-feeding insect across its native and introduced geographic range. Microb Ecol. 2017;74:722–34.
Morrow JL, Hall AAG, Riegler M. Symbionts in waiting : the dynamics of incipient endosymbiont complementation and replacement in minimal bacterial communities of psyllids. Microbiome. 2017;5:58.
Morrow JL, Om N, Beattie GAC, Chambers GA, Donovan NJ, Liefting LW, et al. Characterization of the bacterial communities of psyllids associated with Rutaceae in Bhutan by high throughput sequencing. BMC Microbiol. 2020;20:215.
Nakabachi A, Malenovský I, Gjonov I, Hirose Y. 16S rRNA sequencing detected Profftella, Liberibacter, Wolbachia, and Diplorickettsia from relatives of the Asian citrus psyllid. Microb Ecol. 2020;80(2):410-22. https://doi.org/10.1007/s00248-020-01491-z.
Nakabachi A, Malenovský I, Hirose Y. Comparative genomics underlines multiple roles of Profftella, an obligate symbiont of psyllids: providing toxins, vitamins, and carotenoids. Genome Biol Evol. 2020;12:1975–87. https://doi.org/10.1093/gbe/evaa175.
Nakabachi A, Koshikawa S, Miura T, Miyagishima S. Genome size of Pachypsylla venusta (Hemiptera: Psyllidae) and the ploidy of its bacteriocyte, the symbiotic host cell that harbors intracellular mutualistic bacteria with the smallest cellular genome. Bull Entomol Res. 2010;100:27–33. https://doi.org/10.1017/S0007485309006737.
Thao ML, Moran NA, Abbot P, Brennan EB, Burckhardt DH, Baumann P. Cospeciation of psyllids and their primary prokaryotic endosymbionts. Appl Environ Microbiol. 2000;66:2898–905.
Thao ML, Clark MA, Burckhardt DH, Moran NA, Baumann P. Phylogenetic analysis of vertically transmitted psyllid endosymbionts (Candidatus Carsonella ruddii) based on atpAGD and rpoC: comparisons with 16S-23S rDNA-derived phylogeny. Curr Microbiol. 2001;42:419–21. https://doi.org/10.1007/s002840010240.
Nakabachi A, Yamashita A, Toh H, Ishikawa H, Dunbar HE, Moran NA, et al. The 160-kilobase genome of the bacterial endosymbiont Carsonella. Science. 2006;314:267. https://doi.org/10.1126/science.1134196.
Moran NA, McCutcheon JP, Nakabachi A. Genomics and evolution of heritable bacterial symbionts. Annu Rev Genet. 2008;42:165–90. https://doi.org/10.1146/annurev.genet.41.110306.130119.
Nakabachi A, Ishikawa H, Kudo T. Extraordinary proliferation of microorganisms in aposymbiotic pea aphids, Acyrthosiphon pisum. J Invertebr Pathol. 2003;82:152–61. https://doi.org/10.1016/S0022-2011(03)00020-X.
Wu M, Sun LV, Vamathevan J, Riegler M, Deboy R, Brownlie JC, et al. Phylogenomics of the reproductive parasite Wolbachia pipientis wMel: a streamlined genome overrun by mobile genetic elements. PLoS Biol. 2004;2:E69. https://doi.org/10.1371/journal.pbio.0020069.
Oliver KM, Degnan PH, Burke GR, Moran NA. Facultative symbionts in aphids and the horizontal transfer of ecologically important traits. Annu Rev Entomol. 2010;55:247–66. https://doi.org/10.1146/annurev-ento-112408-085305.
Johnson KN. The impact of Wolbachia on virus infection in mosquitoes. Viruses. 2015;7:5705–17.
Ballinger MJ, Perlman SJ. The defensive Spiroplasma. Curr Opin Insect Sci. 2019;32:36–41. https://doi.org/10.1016/j.cois.2018.10.004.
Burke GR, Moran NA. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol Evol. 2011;3:195–208.
Manzano-Marín A, Latorre A. Snapshots of a shrinking partner: genome reduction in Serratia symbiotica. Sci Rep. 2016;6:32590. https://doi.org/10.1038/srep32590.
Perreau J, Patel DJ, Anderson H, Maeda GP, Elston KM, Barrick JE, et al. Vertical transmission at the pathogen-symbiont interface: Serratia symbiotica and aphids. mBio. 2021;12:e00359–21.
McCutcheon JP, Moran NA. Extreme genome reduction in symbiotic bacteria. Nat Rev Microbiol. 2012;10:13–26. https://doi.org/10.1038/nrmicro2670.
Moran NA, Bennett GM. The tiniest tiny genomes. Annu Rev Microbiol. 2014:195–215. https://doi.org/10.1146/annurev-micro-091213-112901.
Nakabachi A, Shigenobu S, Sakazume N, Shiraki T, Hayashizaki Y, Carninci P, et al. Transcriptome analysis of the aphid bacteriocyte, the symbiotic host cell that harbors an endocellular mutualistic bacterium, Buchnera. Proc Natl Acad Sci U S A. 2005;102:5477–82.
Nikoh N, McCutcheon JP, Kudo T, Miyagishima S, Moran NA, Nakabachi A. Bacterial genes in the aphid genome: absence of functional gene transfer from Buchnera to its host. PLoS Genet. 2010;6:e1000827.
Nikoh N, Nakabachi A. Aphids acquired symbiotic genes via lateral gene transfer. BMC Biol. 2009;7:12. https://doi.org/10.1186/1741-7007-7-12.
Shigenobu S, Richards S, Cree AGG, Morioka M, Fukatsu T, Kudo T, et al. A full-length cDNA resource for the pea aphid, Acyrthosiphon pisum. Insect Mol Biol. 2010;19(SUPPL. 2):23–31.
Sloan DB, Nakabachi A, Richards S, Qu J, Murali SC, Gibbs RA, et al. Parallel histories of horizontal gene transfer facilitated extreme reduction of endosymbiont genomes in sap-feeding insects. Mol Biol Evol. 2014;31:857–71. https://doi.org/10.1093/molbev/msu004.
Nakabachi A, Ishida K, Hongoh Y, Ohkuma M, Miyagishima S. Aphid gene of bacterial origin encodes protein transported to obligate endosymbiont. Curr Biol. 2014;24:R640–1.
Nakabachi A. Horizontal gene transfers in insects. Curr Opin Insect Sci. 2015;7:24–9. https://doi.org/10.1016/j.cois.2015.03.006.
Dan H, Ikeda N, Fujikami M, Nakabachi A. Behavior of bacteriome symbionts during transovarial transmission and development of the Asian citrus psyllid. PLoS One. 2017;12:e0189779.
Yamada T, Hamada M, Floreancig P, Nakabachi A. Diaphorin, a polyketide synthesized by an intracellular symbiont of the Asian citrus psyllid, is potentially harmful for biological control agents. PLoS One. 2019;14:e0216319.
Nakabachi A, Okamura K. Diaphorin, a polyketide produced by a bacterial symbiont of the Asian citrus psyllid, kills various human cancer cells. PLoS One. 2019;14:e0218190.
Nakabachi A, Fujikami M. Concentration and distribution of diaphorin, and expression of diaphorin synthesis genes during Asian citrus psyllid development. J Insect Physiol. 2019;118:103931. https://doi.org/10.1016/j.jinsphys.2019.103931.
Nakabachi A, Nikoh N, Oshima K, Inoue H, Ohkuma M, Hongoh Y, et al. Horizontal gene acquisition of Liberibacter plant pathogens from a bacteriome-confined endosymbiont of their psyllid vector. PLoS One. 2013;8:e82612. https://doi.org/10.1371/journal.pone.0082612.
Kruse A, Fattah-Hosseini S, Saha S, Johnson R, Warwick E, Sturgeon K, et al. Combining ‘omics and microscopy to visualize interactions between the Asian citrus psyllid vector and the Huanglongbing pathogen Candidatus Liberibacter asiaticus in the insect gut. PLoS One. 2017;12:e0179531. https://doi.org/10.1371/journal.pone.0179531.
Jain M, Fleites LA, Gabriel DW. A small Wolbachia protein directly represses phage lytic cycle genes in “Candidatus Liberibacter asiaticus” within psyllids. mSphere. 2017;2:e00171–17.
Mann M, Fattah-Hosseini S, Ammar E-D, Stange R, Warrick E, Sturgeon K, et al. Diaphorina citri nymphs are resistant to morphological changes induced by “Candidatus Liberibacter asiaticus ” in midgut epithelial cells. Infect Immun. 2018;86:e00889–17.
Hosseinzadeh S, Shams-Bakhsh M, Mann M, Fattah-Hosseini S, Bagheri A, Mehrabadi M, et al. Distribution and variation of bacterial endosymbiont and “Candidatus Liberibacter asiaticus” titer in the Huanglongbing insect vector, Diaphorina citri Kuwayama. Microb Ecol. 2019;78:206–22.
Ouvrard D. Psyl’list – the world Psylloidea database. 2021. https://www.hemiptera-databases.org/psyllist. Accessed 10 Jul 2021.
Nachappa P, Levy J, Pierson E, Tamborindeguy C. Diversity of endosymbionts in the potato psyllid, Bactericera cockerelli (Triozidae), vector of zebra chip disease of potato. Curr Microbiol. 2011;62:1510–20. https://doi.org/10.1007/s00284-011-9885-5.
Jing X, Wong ACN, Chaston JM, Colvin J, McKenzie CL, Douglas AE. The bacterial communities in plant phloem-sap-feeding insects. Mol Ecol. 2014;23:1433–44.
Meng L, Li X, Cheng X, Zhang H. 16S rRNA gene sequencing reveals a shift in the microbiota of Diaphorina citri during the psyllid life cycle. Front Microbiol. 2019;10:1948.
Kwak Y, Sun P, Meduri VRS, Percy DM, Mauck KE, Hansen AK. Uncovering symbionts across the psyllid tree of life and the discovery of a new Liberibacter species, “Candidatus” Liberibacter capsica. Front Microbiol. 2021;12:739763.
Karstens L, Asquith M, Davin S, Fair D, Gregory WT, Wolfe AJ, et al. Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments. mSystems. 2019;4:e00290–19.
Russell JA, Weldon S, Smith AH, Kim KL, Hu Y, Łukasik P, et al. Uncovering symbiont-driven genetic diversity across north American pea aphids. Mol Ecol. 2013;22:2045–59.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2 : High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
Prodan A, Tremaroli V, Brolin H, Zwinderman AH, Nieuwdorp M, Levin E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS One. 2020;15:e0227434.
Glöckner FO, Yilmaz P, Quast C, Gerken J, Beccati A, Ciuprina A, et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J Biotechnol. 2017;261:169–76. https://doi.org/10.1016/j.jbiotec.2017.06.1198.
Trowbridge RE, Dittmar K, Whiting MF. Identification and phylogenetic analysis of Arsenophonus- and Photorhabdus-type bacteria from adult Hippoboscidae and Streblidae (Hippoboscoidea). J Invertebr Pathol. 2006;91:64–8.
Gherna RL, Werren JH, Weisburg W, Cote R, Woese CR, Mandelco L, et al. Arsenophonus nasoniae gen. Nov., sp. nov., the causative agent of the son-killer trait in the parasitic wasp Nasonia vitripennis. Int J Syst Bacteriol. 1991;41:563–5.
Novakova E, Hypsa VV, Moran NA, Nováková E, Hypsa VV, Moran NA. Arsenophonus, an emerging clade of intracellular symbionts with a broad host distribution. BMC Microbiol. 2009;9:143.
Patel V, Chevignon G, Manzano-Marın A, Brandt JW, Strand MR, Russell JA, et al. Cultivation-assisted genome of Candidatus Fukatsuia symbiotica; the enigmatic “X-type” Symbiont of aphids. Genome Biol Evol. 2019;11:3510–22.
Pons I, Renoz F, Noël C, Hance T. Circulation of the cultivable symbiont Serratia symbiotica in aphids is mediated by plants. Front Microbiol. 2019;10:764.
Toju H, Tanabe AS, Notsu Y, Sota T, Fukatsu T. Diversification of endosymbiosis: replacements, co-speciation and promiscuity of bacteriocyte symbionts in weevils. ISME J. 2013;7:1378–90. https://doi.org/10.1038/ismej.2013.27.
Koga R, Bennett GM, Cryan JR, Moran NA. Evolutionary replacement of obligate symbionts in an ancient and diverse insect lineage. Environ Microbiol. 2013;15:2073–81. https://doi.org/10.1111/1462-2920.12121.
Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer K-H, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–45. https://doi.org/10.1038/nrmicro3330.
Barco RA, Garrity GM, Scott JJ, Amend JP, Nealson KH, Emerson D. A genus definition for bacteria and archaea based on a standard genome relatedness index. MBio. 2020;11:e02475–19.
Kellner RLL. Molecular identification of an endosymbiotic bacterium associated with pederin biosynthesis in Paederus sabaeus (Coleoptera: Staphylinidae). Insect Biochem Mol Biol. 2002;32:389–95 http://www.ncbi.nlm.nih.gov/pubmed/11886773.
von Dohlen CD, Spaulding U, Patch KB, Weglarz KM, Foottit RG, Havill NP, et al. Dynamic acquisition and loss of dual-obligate symbionts in the plant-sap-feeding Adelgidae (Hemiptera: Sternorrhyncha: Aphidoidea). Front Microbiol. 2017;8:1037.
Mediannikov O, Sekeyova Z, Birg M-L, Raoult D. A novel obligate intracellular gamma-Proteobacterium associated with Ixodid ticks, Diplorickettsia massiliensis, Gen. Nov., Sp. Nov. PLoS One. 2010;5:e11478.
Subramanian G, Mediannikov O, Angelakis E, Socolovschi C, Kaplanski G, Martzolff L, et al. Diplorickettsia massiliensis as a human pathogen. Eur J Clin Microbiol Infect Dis. 2012;31:365–9.
Ishii Y, Matsuura Y, Kakizawa S, Nikoh N, Fukatsu T. Diversity of bacterial endosymbionts associated with macrosteles leafhoppers vectoring phytopathogenic phytoplasmas. Appl Environ Microbiol. 2013;79:5013–22.
Tsuchida T, Koga R, Fujiwara A, Fukatsu T. Phenotypic effect of “Candidatus Rickettsiella viridis,” a facultative symbiont of the pea aphid (Acyrthosiphon pisum), and its interaction with a coexisting symbiont. Appl Environ Microbiol. 2014;80:525–33.
Tsuchida T, Koga R, Horikawa M, Tsunoda T, Maoka T, Matsumoto S, et al. Symbiotic bacterium modifies aphid body color. Science (80- ). 2010;330:1102–4. https://doi.org/10.1126/science.1195463.
Bové JM. Huanglongbing: a destructive, newly-emerging, century-old disease of citrus. J Plant Pathol. 2006;88:7–37.
Keremane ML, Ramadugu C, Castaneda A, Diaz JE, Peñaranda EA, Chen J, et al. Report of Candidatus Liberibacter caribbeanus, a new citrus- and psyllid-associated Liberibacter from Colombia, South America. In: APS Annual Meeting 2015:101-O; 2015.
Lin H, Lou B, Glynn JM, Doddapaneni H, Civerolo EL, Chen C, et al. The complete genome sequence of “Candidatus Liberibacter solanacearum”, the bacterium associated with potato zebra chip disease. PLoS One. 2011;6:e19135. https://doi.org/10.1371/journal.pone.0019135.
Nelson W, Fisher T, Munyaneza JE. Haplotypes of “Candidatus Liberibacter solanacearum” suggest long-standing separation. Eur J Plant Pathol. 2011;130:5–12.
Nelson WR, Sengoda VG, Alfaro-Fernandez AO, Font MI, Crosslin JM. A new haplotype of “Candidatus Liberibacter solanacearum” identified in the Mediterranean region. Eur J Plant Pathol. 2013;135:633–9.
Teresani GR, Bertolini E, Alfaro-fernández A, Martínez C, André F, Tanaka O, et al. Association of ‘Candidatus Liberibacter solanacearum’ with a vegetative disorder of celery in Spain and development of a real-time PCR method for its detection. Phytopathology. 2014;104:804–11.
Morris J, Shiller J, Mann R, Smith G, Yen A, Rodoni B. Novel ‘Candidatus Liberibacter’ species identified in the Australian eggplant psyllid, Acizzia solanicola. Microb Biotechnol. 2017;10:833–44.
Fagen JR, Leonard MT, Coyle JF, McCullough CM, Davis-Richardson AG, Davis MJ, et al. Liberibacter crescens gen. Nov., sp. nov., the first cultured member of the genus Liberibacter. Int J Syst Evol Microbiol. 2014;64:2461–6.
Raddadi N, Gonella E, Camerota C, Pizzinat A, Tedeschi R, Crotti E, et al. “Candidatus Liberibacter europaeus” sp. nov. that is associated with and transmitted by the psyllid Cacopsylla pyri apparently behaves as an endophyte rather than a pathogen. Env Microbiol. 2011;13:414–26. https://doi.org/10.1111/j.1462-2920.2010.02347.x.
Camerota C, Raddadi N, Pizzinat A, Gonella E, Crotti E, Tedeschi R, et al. Incidence of ‘Candidatus Liberibacter europaeus’ and phytoplasmas in Cacopsylla species (Hemiptera: Psyllidae) and their host/shelter plants. Phytoparasitica. 2012;40:213–21.
Thompson S, Fletcher JD, Ziebell H, Beard S, Panda P, Jorgensen N, et al. First report of “Candidatus Liberibacter europaeus” associated with psyllid infested scotch broom. New Dis Rep. 2013;27:6.
Tannières M, Fowler SV, Manaargadoo-Catin L, Lange C, Shaw R. First report of “Candidatus Liberibacter europaeus” in the United Kingdom. New Dis Rep. 2020;41:3.
Pelz-Stelinski KS, Killiny N. Better together: association with ‘Candidatus Liberibacter asiaticus’ increases the reproductive fitness of its insect vector, Diaphorina citri (Hemiptera: Liviidae). Ann Entomol Soc Am. 2016;109:371–6.
Augustinos AA, Santos-Garcia D, Dionyssopoulou E, Moreira M, Papapanagiotou A, Scarvelakis M, et al. Detection and characterization of Wolbachia infections in natural populations of aphids: is the hidden diversity fully unraveled? PLoS One. 2011;6:e28695.
Tang M, Lv L, Jing S, Zhu L, He G. Bacterial symbionts of the brown planthopper, Nilaparvata lugens (Homoptera: Delphacidae). Appl Environ Microbiol. 2010;76:1740–5. https://doi.org/10.1128/AEM.02240-09.
Stouthamer R, Breeuwer JA, Hurst GD. Wolbachia pipientis: microbial manipulator of arthropod reproduction. Annu Rev Microbiol. 1999;53:71–102. https://doi.org/10.1146/annurev.micro.53.1.71.
Werren JH, Baldo L, Clark ME. Wolbachia: master manipulators of invertebrate biology. Nat Rev Microbiol. 2008;6:741–51. https://doi.org/10.1038/nrmicro1969.
Pascar J, Chandler CH. A bioinformatics approach to identifying Wolbachia infections in arthropods. PeerJ. 2018;6:e5486.
Lindsey ARI, Bordenstein SR, Newton ILG, Rasgon JL. Wolbachia pipientis should not be split into multiple species: A response to Ramírez-Puebla et al., “Species in Wolbachia? Proposal for the designation of ‘Candidatus Wolbachia bourtzisii’, ‘Candidatus Wolbachia onchocercicola’. Syst Appl Microbiol. 2016;39:220–222.
Flores HA, O’Neill SL. Controlling vector-borne diseases by releasing modified mosquitoes. Nat Rev Microbiol. 2018;16:508–18.
Brinker P, Fontaine MC, Beukeboom LW, Salles JF. Host, symbionts, and the microbiome: the missing tripartite interaction. Trends Microbiol. 2019;27:480–8.
Dossi FCA, da Silva EP, Cônsoli FL. Population dynamics and growth rates of endosymbionts during Diaphorina citri (Hemiptera, Liviidae) ontogeny. Microb Ecol. 2014;68:881–9.
Chu CC, Gill TA, Hoffmann M, Pelz-Stelinski KS. Inter-population variability of endosymbiont densities in the Asian citrus psyllid (Diaphorina citri Kuwayama). Microb Ecol. 2016;71:999–1007.
Saha S, Hunter WB, Reese J, Morgan JK, Marutani-Hert M, Huang H, et al. Survey of endosymbionts in the Diaphorina citri metagenome and assembly of a Wolbachia wDi draft genome. PLoS One. 2012;7:e50067. https://doi.org/10.1371/journal.pone.0050067.
Guidolin AS, Cônsoli FL. Molecular characterization of Wolbachia strains associated with the invasive Asian citrus psyllid Diaphorina citri in Brazil. Microb Ecol. 2013;65:475–86. https://doi.org/10.1007/s00248-012-0150-7.
Lashkari M, Manzari S, Sahragard A, Malagnini V, Boykin LM, Hosseini R. Global genetic variation in the Asian citrus psyllid, Diaphorina citri (Hemiptera: Liviidae) and the endosymbiont Wolbachia: links between Iran and the USA detected. Pest Manag Sci. 2014;70:1033–40.
Chu C, Hoffmann M, Braswell WE, Pelz-Stelinski KS. Genetic variation and potential coinfection of Wolbachia among widespread Asian citrus psyllid (Diaphorina citri Kuwayama) populations. Insect Sci. 2019;26:671–82.
Hosseinzadeh S, Ramsey J, Mann M, Bennett L, Hunter WB, Shams-bakhsh M, et al. Color morphology of Diaphorina citri influences interactions with its bacterial endosymbionts and ‘Candidatus Liberibacter asiaticus’. PLoS One. 2019;14:e0216599. https://doi.org/10.1371/journal.pone.0216599.
Lawson ET, Mousseau TA, Klaper R, Hunter MD, Werren JH. Rickettsia associated with male-killing in a buprestid beetle. Heredity (Edinb). 2001;86(Pt 4):497–505. https://doi.org/10.1046/j.1365-2540.2001.00848.x.
Hagimori T, Abe Y, Date S, Miura K. The first finding of a Rickettsia bacterium associated with parthenogenesis induction among insects. Curr Microbiol. 2006;52:97–101.
Li F. Psyllidomorpha of China (Insecta: Hemiptera). Beijing: Science Press; 2011. p. 546–7.
Inoue H. The generic affiliation of Japanese species of the subfamily Psyllinae (Hemiptera: Psyllidae) with a revised checklist. J Nat Hist. 2010;44:333–60.
Cho G, Malenovský I, Lee S. Higher-level molecular phylogeny of jumping plant lice (Hemiptera: Sternorrhyncha: Psylloidea). Syst Entomol. 2019;44:638–51.
Illumina. 16S metagenomic sequencing library preparation Part#15044223 Rev.B. 2013;:http://jp.support.illumina.com/content/dam/illumin.
Bolyen E, Rideout JR, Dillon MR, Bokulich N, Abnet C, Al-Ghalith GA, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–7.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–2.
Bokulich NA, Kaehler BD, Rideout J, Dillon M, Bolyen E, Knight R, et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome. 2018;6:90.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421.
Pruesse E, Peplies J, Glöckner FO. SINA : accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Acknowledgements
The DNA sequencing facility was supported by the Department of Applied Chemistry and Life Science and the University-Community Partnership Promotion Center in Toyohashi University of Technology.
Funding
This work was supported by the Japan Society for the Promotion of Science (https://www.jsps.go.jp) KAKENHI [grant numbers 26292174 and 20H02998 to AN, and 25850035 to HI], and research grants from Tatematsu foundation and Nagase Science and Technology Foundation to AN. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
AN designed experiments. HI prepared materials. AN and YH performed microbiome analyses. AN and HI drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Nakabachi, A., Inoue, H. & Hirose, Y. Microbiome analyses of 12 psyllid species of the family Psyllidae identified various bacteria including Fukatsuia and Serratia symbiotica, known as secondary symbionts of aphids. BMC Microbiol 22, 15 (2022). https://doi.org/10.1186/s12866-021-02429-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-021-02429-2