
Abstract
Background
The genomic diversity of different species within the genus Lactococcus and the relationships between genomic differentiation and environmental factors remain unclear. In this study, type isolates of ten Lactococcus species/subspecies were sequenced to assess their genomic characteristics, metabolic diversity, and phylogenetic relationships.
Results
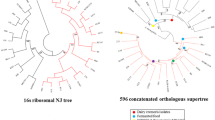
The total genome sizes varied between 1.99 (Lactococcus plantarum) and 2.46 megabases (Mb; L. lactis subsp. lactis), and the G + C content ranged from 34.81 (L. lactis subsp. hordniae) to 39.67% (L. raffinolactis) with an average value of 37.02%. Analysis of genome dynamics indicated that the genus Lactococcus has an open pan-genome, while the core genome size decreased with sequential addition at the genus and species group levels. A phylogenetic dendrogram based on the concatenated amino acid sequences of 643 core genes was largely consistent with the phylogenetic tree obtained by 16S ribosomal RNA (rRNA) genes, but it provided a more robust phylogenetic resolution than the 16S rRNA gene-based analysis.
Conclusions
Comparative genomics indicated that species in the genus Lactococcus had high degrees of diversity in genome size, gene content, and carbohydrate metabolism. This may be important for the specific adaptations that allow different Lactococcus species to survive in different environments. These results provide a quantitative basis for understanding the genomic and metabolic diversity within the genus Lactococcus, laying the foundation for future studies on taxonomy and functional genomics.
Similar content being viewed by others


Background
Lactococcus is a genus of catalase-negative, gram-positive, non-motile, facultative anaerobic bacteria that typically inhabit animals, plants, and their related products, particularly fermented products; they are generally considered nonpathogenic toward humans [1]. Lactococcus species have variable and abundant nutritional requirements. They usually grow within a temperature range of 10–40 °C, although some species are capable of growing at temperatures as low as 7 °C over a prolonged incubation period of 10–14 days [2]. Most Lactococcus species can grow in 4.0% (w/v) NaCl; in media, they grow best at pH 7.0 and cease to grow if the pH drops to 4.5 [3].
Lactococcus lactis was the first species in the genus to be identified (by Joseph Lister in 1873) and then it was later renamed Streptococcus lactis [1]. Schleifer et al. defined the genus Lactococcus as distinct from the genus Streptococcus [4]. To date, there are 12 recognized species with four further subspecies in the genus (http://www.bacterio.net/lactococcus.html, December 2016). Some species, especially L. lactis, are important in the manufacturing of fermented milk products and some species produce antimicrobial compounds, such as bacteriocins, nisin, and lactococcin [5, 6], and recombinant proteins [7]. Some probiotic foods contain these species, and there have been more studies regarding the relationships between these probiotic-containing functional foods and the maintenance of human intestinal health [8, 9]. One species, Lactococcus garvieae, is a major pathogen of fish causing fatal hemorrhagic septicemia [10]. However, L. garvieae has also been reported as a common component of fermented dairy manufactured from raw milk [11,12,13,14], and phenotypically it is closely related to L. lactis.
The number of studies using high-throughput sequencing to study bacterial metabolism, evolution, and speciation within genera has increased markedly with the advances and decreasing costs of sequencing technologies [15,16,17]. Whole genomic sequences allow reconstruction of reliable phylogenies based on much larger sequence data sets than previously available. This makes it possible to gain an understanding of the genomic diversification of entire genera in great detail [18]. The first genome of a lactic acid bacterium (LAB), L. lactis isolate IL1403, was sequenced in 2001 [19]. Subsequently, there have been increasing reports on whole genome sequences of LAB. However, of the 16 currently recognized species/subspecies in the genus Lactococcus, only seven have been fully sequenced (National Center for Biotechnology information [NCBI]): L. lactis subsp. lactis, L. lactis subsp. cremoris, L. garvieae, L. raffinolactis, L. plantarum, L. fujiensis, and L. piscium [20,21,22,23]. With many new species being identified in the genus Lactococcus, a genomic view could provide important information to understand bacterial evolution and adaptation to different environments. Moreover, comparative genomic of species within the same genus can reveal the genomic diversity within a genus and provide insights into how environmental conditions affect the evolution of different species.
In the present study, the genomes of ten type species/subspecies in the genus Lactococcus were sequenced. We then used comparative genomic approaches to define the pan-genome, core genome, and unique genes; assess genetic diversity and species taxonomy; infer phylogenetic relationships; and study the mechanisms of carbohydrate metabolism.
Methods
Bacterial isolates, DNA extraction and carbohydrate utilization
The ten type isolates of Lactococcus species/subspecies, as defined by the LPSN (http://www.bacterio.net/) were collected from the German Collection of Microorganisms and Cell Cultures (DSMZ; Table 1). These isolates were cultured in M17 broth (Oxoid Ltd., Basingstoke, UK) at 37 °C for 24 h.
Bacterial DNA extraction kits (OMEGA D3350–02) were used for DNA extraction from each isolate according to the manufacturer’s instructions. Quantitative and qualitative analysis of genomic DNA was achieved using electrophoresis on 1% agarose gels and a TBS-380 mini fluorometer (Turner BioSystems Inc., Sunnyvale, CA). Only good quality DNA samples (OD260/280 = 1.8~2.0, >6μg) were used to construct fragment libraries (200 to 300 bp).
Carbohydrates fermentation was tested using the API 50 CH systems (Biomerieux, France) based on the manufacturer’s instructions.
Genome sequencing, assembly, prediction and annotation of coding sequences (CDS), and pathway mapping
The ten genomes of the the Lactococcus type species/subspecies were sequenced using an Illumina HiSeq 2000 (Illumina Inc. USA) by generating paired-end libraries. The average length was 300 bp, and 838 Mb of high quality data was generated for each isolate consistent with a sequencing depth of 129- to 388-fold. Assembly of the paired-end reads was performed using SOAPdenovo v2 [24]. The software Gap Closer was used to fill the local inner gaps and correct single base errors (http://sourceforge.net/projects/soapdenovo2/files/GapCloser/). Gene prediction and functional annotation of predicted genes were achieved using RAST 2.0 [25, 26]. The individual genome assemblies were deposited in the NCBI, the accession numbers are shown in Table 2. Carbohydrate metabolism related pathway information was identified from the pathways described in the Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.kegg.jp/) [27].
Core genome, pan-genome and unique gene analysis
For predicting the possible dynamic changes in genome size, the sizes of the core genome (common genes, mutually conserved), pan-genome (gene repertoire) and unique genes (specific genes, only found in one genome) were calculated. Owing to large variation were happened in the homologous genes between species within a genus, the concept of ‘gene family’ was used to take the place of the generally used term ‘gene’. According to previous research [28, 29], the core genome, pan-genome and unique genes [30] of Lactococcus were estimated. A pair of genes was placed in the same gene family, when their amino acid sequences identity was >50%, and when more than 50% of the amino acid sequences of the longer gene could be covered by the shorter one.
For construction of the pan-genome, all of the predicted genes were first grouped into possible gene families for each genome and then the gene families of ten genomes were accumulated. The core genome was built by counting the number of commonly shared gene families within ten genomes, while the unique gene family was defined as the gene families that could only be found in one genome.
The average nucleotide identity (ANI) and phylogenetic analysis
The pair-wise ANI values of ten Lactococcus species/ subspecies were calculated using the methods of Goris et al. [30]. To infer phylogenetic relationships among Lactococcus species, we constructed a maximum likelihood (ML) tree based on the concatenated amino acid sequences of 643 core genes. Streptococcus (S.) thermophilus LMG 18311 was used as an out-group because it is phylogenetically closely related to the genus Lactococcus [31]. Amino acid sequences of the core genes were aligned by MUSCLE v3.8.31 [32], and the software PHYML was used to construct the ML tree with the WAG model and 500 bootstrap iterations [33]. In addition, a Neighbour-Joining (NJ) tree was inferred based on 16S rRNA sequences downloaded from Genbank for all ten type species/subspecies of Lactococcus using Mega 6.0 [34] (http://www.megasoftware.net) with 1000 bootstrap iterations; again S. thermophilus LMG 18311 was used as an out-group.
Results
General features
The estimated genomes of the type isolates of ten Lactococcus species/subspecies were sequenced and assembled into 43–226 scaffolds. Across species, the total genome size varied from 1.99 (L. plantarum) to 2.46 megabases (Mb; L. lactis subsp. lactis), with a mean value of 2.27 Mb (Table 2). In addition, the G + C content ranged from 34.81 (L. lactis subsp. hordniae) to 39.67% (L. raffinolactis) with an average value of 37.02% (Table 2). The numbers of predicted coding genes varied from 1861 (L. plantarum) to 2684 (L. lactis subsp. hordniae).
Pan-genome and core genome analysis
The pan-genome, defined as the full complement of genes from the ten Lactococcus genomes evaluated, contained 8036 gene families and grew continuously as each genome was added; a mean of 662 gene families were added for each genome (Fig. 1a). Compare to the pan-gene families, the amount of core gene families decreased sharply along with the number of genomes increased, reaching a minimum value of 643 for all ten genomes (Fig. 1b). The proportion of core genes varied between 23.9 (L. plantarum) and 34.6% (L. lactis subsp. hordniae) of the total predicted coding genes for the ten Lactococcus species/subspecies. These observations indicated a high degree of genomic diversity among the ten type species/subspecies of the genus Lactococcus. Such genome diversity in the genus Lactococcus is not surprising considering the range of dairy, plant, and animal environments from which the species were isolated.

Pan-genome and core genomes of species/subspecies in the Lactococcus genus. The gene accumulation curves show the number of new genes (pan-genome) and genes in common (core-genome) obtained by adding a new genome to a previous set. The pan-genome curve is the least squares fit of the power law to these values
To examine the functional characteristics of the core genes, the Cluster of Orthologous Group (COG) database was used to investigate the distribution of core proteins mapped to each COG category (Fig. 2). There were eight subcategories in metabolism, seven subcategories in cellular processes and signaling, three subcategories in information storage and processing and two subcategories that were poorly characterized. The information storage and processing category accounted for 32.3% of the clusters, and the cellular processes and signaling category and metabolism categories accounted for 17.4 and 29.7% of clusters, respectively (Fig. 2). Furthermore, 18.66% of core genes were belong to translation, ribosomal structure, and biogenesis (J); 7.93% were involved in replication, recombination, and repair (L); and 6.22% were involved in cell wall/membrane/envelope biogenesis (M). Other than the functional subcategories J, L, and M, the 17 remaining functional subcategories included 6–38 core genes. It is notable that most of the core genes played important roles in maintaining growth and reproduction in the genus Lactococcus; these genes are indispensable and constitute the basic framework of the Lactococcus genome.

Distribution of functional categories of core gene families in the Lactococcus genus. Cluster of Orthologous Group (COG) functional classification description — Information Storage and Processing: [A] RNA processing and modification; [B] chromatin structure and dynamics; [J] translation, ribosomal structure, and biogenesis; [K] transcription; [L] replication, recombination, and repair. Cellular Processes and Signaling: [D] cell cycle control, cell division, chromosome partitioning; [M] cell wall/membrane/envelope biogenesis; [N] cell motility; [O] posttranslational modification, protein turnover, chaperones; [T] signal transduction mechanisms; [U] intracellular trafficking, secretion, and vesicular transport; [V] defense mechanisms; [W] extracellular structures; [Y] nuclear structure; [Z] cytoskeleton. Metabolism: [C] energy production and conversion; [E] amino acid transport and metabolism; [F] nucleotide transport and metabolism; [H] coenzyme transport and metabolism; [G] carbohydrate transport and metabolism; [I] lipid transport and metabolism; [P] inorganic ion transport and metabolism; [Q] secondary metabolites biosynthesis, transport, and catabolism. Poorly Characterized: [R] general function prediction only; [S] function unknown
Phylogenetic relationships of Lactococcus strains
In order to infer phylogenetic position of ten species within the genus Lactococcus, phylogenetic trees were constructed using the 16S ribosomal RNA (rRNA) gene and the concatenated amino acid sequences of 643 core genes from the ten type isolates. The ML tree generated a reliable delineation of phylogenetic relationships across the Lactococcus species, because of more nodes were supported by 100% of the bootstrap iterations (Fig. 3a). According to this highly robust ML tree, the ten Lactococcus species/subspecies were divided into two branches; one branch contained L. raffinolactis ATCC 43920T, L. chungangensis DSM 22330T, L. plantarum DSM 20686T, and L. piscium DSM 6634T, while the other branch included four L. lactis subspecies, L. fujiensis JCM 16395T, and L. garvieae DSM20684T.

Phylogenetic relationships among species in the Lactococcus genus. The trees were constructed with concatenated amino acid sequences of core genes (a) and the 16S rRNA gene (b) using maximum likelihood and neighbor-joining methods, respectively. Streptococcus thermophilus LMG 18311 served as outgroup for both trees
The topologies of the 16S rRNA gene trees were similar to the ML tree (Fig. 3b). However, the resolution and accuracy of the tree of concatenated amino acid sequences was better than that of the 16S rRNA gene tree. For instance, ATCC 19257T and L. lactis subsp. tructae DSM 21502T, L. lactis subsp. hordniae DSM 20450T and L. lactis subsp. lactis ATCC19435T were clustered closely in the 16S rRNA gene tree and shared > 99.90% identity, but were divided in the orthologous proteins tree. Both trees proved that the ten isolates clustered together to form a single genus.
Genetic diversity and species taxonomy
The ANI value is always used to estimate the genetic distance between isolates, which means the sequence identities of the conserved regions between two genomes [29]. The pair-wise ANI values of ten Lactococcus species/ subspecies were calculated to study the interspecies genetic relatedness within the genus Lactococcus. Two clusters formed when the ANI for Lactococcus species was analyzed (Fig. 4). One cluster contained two subgroups: subgroup I (L. lactis subsp. hordniae and L. lactis subsp. lactis) and subgroup II (L. lactis subsp. cremoris and L. lactis subsp. tructae). The 16S rRNA gene identity for the four subspecies was > 99% [35]; however, the ANI value for this cluster was 86.1–97.9%. It is worth noting that the pairwise ANI values between the two subgroups range from 86.1–86.5%, below the recommended 95% threshold value for species circumscription [36]. To further determine the relationships between the four subspecies, we performed additional comparative genomics with 15 genomic sequences of L. lactis subsp. lactis and L. lactis subsp. cremoris from NCBI. The ANI value distribution of Lactococcus is shown in Additional file 1: Table S1, and the heat map was built based on pairwise ANI values (Fig. 4). Based on these results, the four strains were clearly divided into two subgroups, and these values were in line with the DNA–DNA hybridization results. DNA relatedness values between the type strains L. lactis subsp. tructae and L. lactis subsp. hordniae, L. lactis subsp. lactis, L. lactis subsp. cremoriswere 60, 62, and 90%, respectively [35].

Pairwise average nucleotide identity (ANI) values across ten Lactococcus genomes. The colours in the heat map represent pairwise ANI values, with a gradient from yellow (low identity) to blue (high identity)
Therefore, we suggest that L. lactis subsp. cremoris ATCC 19257T and L. lactis subsp. tructae DSM 21502T should be separated from L. lactis subsp. hordniae DSM 20450T and L. lactis subsp. lactis ATCC 19435T as a single species on the basis of the genetic distance and the phylogenetic relationships shown in ML tree (Fig. 3a). The other cluster contained six type isolates with 16S rRNA gene identity of 91–97% and ANI of 67.4–85.0%. These are the classical delimitation criteria for different species within one genus.
Carbohydrate metabolism
Carbohydrate metabolism plays a key role in the growth of Lactococcus species. On average, 12.5% of the genes in each Lactococcus genome are associated with carbohydrate metabolism. Among the core genes of the ten Lactococcus species/subspecies evaluated, a total of 38 genes (5.9%) were associated with carbohydrate metabolism. The majority were involved in glycolysis/gluconeogenesis and the pentose phosphate pathway (PPP). Twenty-eight key enzymes that contribute to glycolysis/gluconeogenesis and PPP were encoded (Table 3, Additional file 2: Figure S1). Our results indicated that glycolysis/gluconeogenesis and the PPP are the central carbohydrate metabolism pathways of Lactococcus species. In addition, all strains have genes encoding fructokinase (EC 2.7.1.4), phosphoenolpyruvate–protein phosphotransferase (EC 2.7.1.199), and glucose-6-phosphate isomerase (EC 5.3.1.9), which are key enzymes in fructose and mannose metabolism. Thus, all strains could ferment d-glucose, d-fructose, and d-mannose (Table 4). Pathways enabling the use of l-arabinose, l-sorbose, l-rhamnose, xylitol, d-lyxose, and d-tagatose as energy were not found in the genome sequences of the ten strains examined.
Some differences in carbohydrate metabolism genes were also found among the ten Lactococcus species/subspecies. The number of carbohydrate metabolism genes in the ten species/subspecies evaluated varied from 194 (L. plantarum DSM 20686T) to 366 (L. lactis subsp. tructae DSM 21502T). Pairwise comparisons among the ten Lactococcus species/subspecies showed that the number of strain-specific carbohydrate metabolism genes varied between 2 and 15, and appeared to reflect the functional diversity observed for each isolate. To detect differences in functional genes related to carbohydrate metabolism, acid production from carbohydrates was tested using the API 50 CH system (bioMérieux, Inc., Marcy l’Etoile, France) (Table 4). Raffinose can be enzymatically hydrolyzed to melibiose by β-fructofuranoside (EC 3.2.1.26), and melibiose is further hydrolyzed to d-glucose (α-galactosidase; EC 3.2.1.22), and enters the glycolysis pathway. Three strains, L. lactis subsp. tructae DSM 21502T, L. piscium DSM 6634T, and L. raffinolactis ATCC 43920T, can utilize raffinose and melibiose, because they possess the key enzymes (EC 3.2.1.26 and EC 3.2.1.22) for raffinose and melibiose metabolism, while L. plantarum DSM 20686T, L. lactis subsp. hordniae DSM 20450T, and L. chungangensis DSM 22330T with β-fructofuranoside and no α-galactosidase, cannot utilize raffinose and melibiose. Furthermore, β-fructofuranoside is a key enzyme for sucrose hydrolysis, and so the four strains lacking this enzyme cannot utilize sucrose. The other six strains can hydrolyze sucrose to d-glucose-6P and d-fructose. The role of L. lactis as an important industrial starter strain is mainly due to the rapid conversion of lactose to lactic acid. Lactose is hydrolyzed by β-galactosidase (EC 3.2.1.23) to give α-d-glucose and d-galactose, and it can also be catalyzed to lactose 6-phosphate (Lac-6P) by lactose phosphotransferase (EC 2.7.1.207). Lac-6P is subsequently hydrolyzed by 6-phospho-β-galactosidase (EC 3.2.1.85) to give galactose 6-phosphate (Gal-6P), and Gal-6P is catabolized by the tagatose 6-phosphate (Tag-6P) pathway [37, 38]. Comparative genomic analysis showed that most strains have the key enzymes for lactose metabolism, with the exception of L. chungangensis DSM 22330T, L. fujiensis JCM16395T, L. plantarum DSM 20686T, and L. lactis subsp. tructae DSM 21502T. These four strains lack lactose phosphotransferase and β-galactosidase, but L. lactis subsp. tructae DSM 21502T and L. fujiensis JCM16395T could utilize lactose in the API 50 test (Table 4). Therefore, we inferred that they have additional pathways for lactose metabolism. Ribokinase (EC 2.7.1.15) is the key enzyme in d-ribose metabolism. L. raffinolactis ATCC 43920T, L. plantarum DSM 20686T, L. lactis subsp. hordniae DSM 20450T, and L. chungangensis DSM 22330T have no genes encoding ribokinase, so they cannot utilize d-ribose.
Discussion
It is considerable realistic significance for applying Lactococcus strains to systematically analyze the evolutionary history and phylogenetic position of the Lactococcus species/subspecies. Therefore, we completed the whole-genome sequencing of ten type strains in Lactococcus genus to assess their genomic characteristics, metabolic diversity, and phylogenetic relationships.
Across ten Lactococcus species, the total genome size varied from 1.99 (L. plantarum) to 2.46 Mb (L. lactis subsp. lactis) (Table 2). The genome size of the Lactococcus species evaluated was relatively small compared with other LAB genera, such as Enterococcus (2.31–5.27 Mb) and Lactobacillus (1.23–4.91 Mb) [32]. A smaller genome size was suggested to indicate adaptations for reproductive efficiency or competitiveness in new environments [39]. The G + C content of 10 strains ranged from 34.81 (L. lactis subsp. hordniae) to 39.67% (L. raffinolactis). The G + C content of genome is influenced by selection and mutation involving multiple factors, including the symbiotic lifestyle, environment, nitrogen fixation ability, aerobiosis, and how the polIIIa subunits are combined [40]. In addition, analysis of the trends in pan-genome size of Lactococcus species/subspecies proved that Lactococcus genus has an open pan-genome. This may be associated with the range of environments colonized by different Lactococcus species and the existence of numerous ways of exchanging genetic material. Previous reports showed that bacterial genomes change when they adapt to variable conditions, and that greater niche diversity requires larger pan-genomes [41].
Currently, the 16S rRNA gene sequencing technique and genomic DNA–DNA hybridization are considered the gold standards for identification of isolates at the species or genus level. However, because of conservative property, 16S rRNA gene could provide sufficient resolution at the species levels [42, 43], and DNA–DNA hybridization is rather time-consuming and labor-intensive. With advances in sequencing and computational technologies, the combination of core genome phylogeny and ANI values could provide accurate taxonomic guidance for LAB based on whole-genome sequences [44]. The lowest value for the Lactococcus genus was 67.4%, while the highest ANI value was 97.9%, revealing that the genomic sequence identities between different pairs of species had marked differences. The ANI values were mainly distributed in the region of 67–68% (Additional file 1: Table S1), while the distribution of Bifidobacterium genus was concentrated in the region of 71–74% [29]. These results suggest that the majority of sequence identities between Lactococcus genomes were low, only slightly higher than those between species from different genera. This could be used as a reference characteristic for identification of Lactococcus species.
Differences in carbohydrate metabolism genes among the ten Lactococcus species/subspecies showed that each could utilize various different carbohydrates. One interesting application of comparative genomics is the identification of associations between strains and their origins. Previous studies using comparative genomics in the genera Nocardiopsis [45] and Bifidobacterium [29] demonstrated specificity to particular ecological niches. In this study, the genotypic and phenotypic carbohydrate metabolism analyses suggested that the lactococcal species may correlate with respect to their origins, but regularities were not found. This is probably due to the unequal and small numbers of isolates from different origins. Three species were isolated from milk and four species from fish, bovine, and leafhopper, whereas only two species were isolated from plants and one species from activated sludge foam (Table 1). As more species are discovered from different countries and origins, further genomic evidence may become available for the genus Lactococcus, which would provide strong indications of the factors that have affected its evolutionary history. The majority of the carbohydrate fermentation results are in accordance with the metabolism pathway predicted by the genome sequence, but some differences were also detected, for which there are at least two explanations: predicted gene function based on sequence data is not completely accurate, and the pathways of carbohydrate metabolism are not yet well understood in KEGG.
Conclusion
In the present study, ten type isolates of species/subspecies from the genus Lactococcus were sequenced and analyzed. Comparative genomic analysis revealed that species in the genus Lactococcus had relatively small and diverse genomes. The high degrees of diversity in genome size, gene content, and carbohydrate metabolism may be because the isolates originated from a wide range of host types and ecological niches. The genus Lactococcus has an open pan-genome, and thus the size of the pan-genome is as yet underestimated and will increase as additional isolates and species are sequenced. Since we had only a limited number of isolates, gene evolution in the genus Lactococcus was not analyzed. Nonetheless, this study provided insights into genomic and metabolic diversity and phylogenetic relationships of the majority of type species/subspecies in the genus Lactococcus, which will lay the foundation for future studies of their taxonomy and functional genomics.
Abbreviations
- ANI:
-
Average nucleotide identity
- CDS:
-
Coding sequences
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- L. :
-
Lactococcus
- LAB:
-
Lactic acid bacterium
- NCBI:
-
National Center for Biotechnology Information
- S. :
-
Streptococcus
References
Sun Z, Yu J, Dan T, Zhang W, Zhang H. Phylogenesis and evolution of lactic acid bacteria. In: Zhang H, Cai Y, editors. Lactic acid bacteria. Netherlands: Springer; 2014. p. 1–101.
Sakala RM, Hayashidani H, Kato Y, Hirata T, Makino Y, Fukushima A, Yamada T, Kaneuchi C, Ogawa M. Change in the composition of the microflora on vacuum-packaged beef during chiller storage. Int J Food Microbiol. 2002;74(1–2):87–99.
Teuber M. Genus II. Lactococcus Schleifer, Kraus, Dvorak, Kilpper-Balz,Collins and Fischer 1986. In: De Vos P, Garrity GM, Jones D, Krieg NR, Ludwig W, Rainey FA, Schleifer KH, Whitman WB, editors. Bergey’s manual of systematic bacteriology, vol. 3. Berlin: Springer; 2009. p. 711–22.
Schleifer KH, Kraus J, Dvorak C, Kilpper-Bälz R, Collins MD, Fischer W. Transfer of Streptococcus lactis and related streptococci to the genus Lactococcus gen. Nov. Syst Appl Microbiol. 1985;6(2):183–95.
Park SH, Itoh K, Kikuchi E, Niwa H, Fujisawa T. Identification and characteristics of nisin Z-producing Lactococcus lactis subsp. lactis isolated from Kimchi. Curr Microbiol. 2003;46(5):385–8.
Rattanachaikunsopon P, Phumkhachorn P. Lactic acid bacteria: their antimicrobial compounds and their uses in food production. Ann Biol Res. 2010;1(4):218–28.
Morello E, Bermúdez-Humarán LG, Llull D, Solé V, Miraglio N, Langella P, Poquet I. Lactococcus lactis, an efficient cell factory for recombinant protein production and secretion. J Mol Microbiol Biotechnol. 2008;14(1–3):48–58.
Fang H, Elina T, Heikki A, Seppo S. Modulation of humoral immune response through probiotic intake. FEMs Immunol Med Mic. 2000;29:47–52.
Yadav K, Bhardwaj A, Kaur G, Iyer R, De S, Kumar N, Malik RK. Potential of Lactococcus lactis as a probiotic and functional lactic acid bacteria in dairy industry. Int J Probiotics Prebiotics. 2009;4:219–28.
Teixeira LM, Merquior VL, Vianni MC, Carvalho MG, Fracalanzza SE, Steigerwalt AG, Brenner DJ, Facklam RR. Phenotypic and genotypic characterization of atypical Lactococcus garvieae strains isolated from water buffalos with subclinical mastitis and confirmation of L. garvieae as a senior subjective synonym of Enterococcus seriolicida. Int J Syst Bacteriol. 1996;46(3):664–8.
Alegría A, Alvarezmartín P, Sacristán N, Fernández E, Delgado S, Mayo B. Diversity and evolution of the microbial populations during manufacture and ripening of Casín, a traditional Spanish, starter-free cheese made from cow's milk. Int J Food Microbiol. 2009;136:44–51.
Fortina MG, Ricci G, Acquati A, Zeppa G, Gandini A, Manachini PL. Genetic characterization of some lactic acid bacteria occurring in an artisanal protected denomination origin (PDO) Italian cheese, the Toma piemontese. Food Microbiol. 2003;20(02):397–404.
Foschino R, Picozzi C, Borghi M, Cerliani MC, Cresci E. Investigation on the microflora of Caprino Lomeardo cheese from raw goat milk.Ital. J Food Sci. 2006;18(1):33–49.
Yu J, Wang HM, Zha MS, Qing YT, Bai N, Ren Y, Xi XX, Liu WJ, Menghe BLG, Zhang HP. Molecular identification and quantification of lactic acid bacteria in traditional fermented dairy foods of Russia. J Dairy Sci. 2015;98(8):5143–454.
Touchon M, Cury J, Yoon EJ, Krizova L, Cerqueira GC, Murphy C, Feldgarden M, Wortman J, Clermont D, Lambert T. The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol Evol. 2014;6(10):2866–82.
Gao XY, Zhi XY, Li HW, Klenk HP, Li WJ. Comparative genomics of the bacterial genus Streptococcus illuminates evolutionary implications of species groups. PLoS One. 2014;9:e101229.
Kant R, Blom J, Palva A, Siezen RJ, de Vos WM. Comparative genomics of Lactobacillus. J Microbial Biotechnol. 2010;4(3):323–32.
Lugli GA, Milani C, Turroni F, Duranti S, Ferrario C, Viappiani A, Mancabelli L, Mangifesta M, Taminiau B, Delcenserie V, van Sinderen D, Ventura M. Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl Environ Microbiol. 2014;80:6383–94.
Bolotin A, Wincker P, Mauger S, Jaillon O, Malarme K, Weissenbach J, Ehrlich SD, Sorokin A. The complete genome sequence of the lactic acid bacterium Lactococcus lactis ssp. lactis IL1403. Genome Res. 2001;11(5):731–53.
Yang CH, Wu CC, Cheng WS, Chung MC, Tsai YC, Chang CH. A17, the first sequenced strain of Lactococcus lactis subsp. cremoris with potential immunomodulatory functions. Genome Announc. 2015;3(1):e01563–14.
Andreevskaya M, Johansson P, Laine P, Smolander OP, Sonck M, Rahkila R, Jääskeläinen E, Paulin L, Auvinen P, Björkroth J. Genome sequence and transcriptome analysis of meat spoilage lactic acid bacterium Lactococcus piscium MKFS47. Appl Environ Microbiol. 2015;81:3800–11.
Zhao F, Ma H, Ying L, Teng K, Kang X, Wang F, Yang X, Jin Z. Complete genome sequence of Lactococcus lactis S0, an efficient producer of nisin. J Biotechnol. 2015;198:15–6.
Meslier V, Loux V, Renault P. Genome sequence of Lactococcus raffinolactis strain 4877, isolated from natural dairy starter culture. J Bacteriol. 2012;194(22):6364.
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Qi P, Liu Y. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience. 2012;1(1):1–6.
Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M. The RAST server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014;42(Database issue):D206–14.
Kanehisa M, Sato Y, Kawashima M, Furumichi M, Mao T. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2015;44(D1):D457–62.
Lukjancenko O, Ussery DW, Wassenaar TM. Comparative genomics of Bifidobacterium, Lactobacillus and related probiotic genera. Microb Ecol. 2012;63(3):651–73.
Sun Z, Zhang W, Guo C, Yang X, Liu WJ, Wu YR, Song YQ, Kwok LY, Cui YJ, Menghe BLG, Yang TF, Hu LP, Zhang HP. Comparative genomic analysis of 45 type strains of the genus Bifidobacterium: a snapshot of its genetic diversity and evolution. PLoS One. 2015;10(2):e0117912.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(1):81–91.
Sun Z, Harris HMB, Mccann A, Guo C, Argimón S, Zhang W, Yang X, Jeffery IB, Cooney JC, Kagawa TF. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat Commun. 2015;6:8322.
Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC bioinformatics. 2004;5:113.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33(7):1870.
Pérez T, Balcázar JL, Peix A, Valverde A, Velázquez E, De BI, Ruizzarzuela I. Lactococcus lactis subsp. tructae subsp. nov. isolated from the intestinal mucus of brown trout (Salmo Trutta) and rainbow trout (Oncorhynchus Mykiss). Int J Syst Evol Microbiol. 2011;61(8):1894–8.
Richter M, Rosselló-Móra R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106(45):19126–31.
Benthin S, Nielsen J, Villadsen J. Galactose expulsion during lactose metabolism in Lactococcus lactis subsp. cremoris FD1 due to dephosphorylation of intracellular galactose 6-phosphate. Appl Environ Microbiol. 1994;60(4):1254–9.
Lorca G, Reddy L, Nguyen A, Sun EI, Tseng J, Yen MR, Saier MH, Milton H, Saier J. Lactic acid bacteria: comparative genomic analyses of transport systems. In: Mozzi F, Raya RR, Vignolo GM (eds) Biotechnology of lactic acid bacteria: novel applications. Oxford: Wiley-Blackwell. 2010. pp. 73–87. doi:10.1002/9780813820866.ch1.
Burke GR, Moran NA. Massive genomic decay in Serratia symbiotica, a recently evolved symbiont of aphids. Genome Biol Evol. 2011;3(5):195–208.
Hao W, Zhang Z, Hu S, Yu J. On the molecular mechanism of GC content variation among eubacterial genomes. Biol Direct. 2012;7(1):2.
Konstantinidis KT, Tiedje JM. Trends between gene content and genome size in prokaryotic species with larger genomes. Proc Natl Acad Sci U S A. 2004;101(9):3160–5.
Weisburg WG, Barns SM, Pelletier DA, Lane DJ. 16S ribosomal DNA amplification for phylogenetic study. J Bacteriol. 1991;173(2):697–703.
Stackebrandt E, Goebel BM. Taxonomic note: a place for DNA-DNA reassociation and 16S rRNA sequence analysis in the present species definition in bacteriology. Int J Syst Bacteriol. 1994;44(4):846–9.
Xu J. Microbial ecology in the age of genomics and metagenomics: concepts, tools, and recent advances. Mol Ecol. 2006;15(7):9–10.
Li HW, Zhi XY, Yao JC, Zhou Y, Tang SK, Klenk HP, Zhao J, Li WJ. Comparative genomic analysis of the genus Nocardiopsis provides new insights into its genetic mechanisms of environmental adaptability. PLoS One. 2013;8(4):e61528.
Cho SL, Nam SW, Yoon JH, Lee JS, Sukhoom A, Kim W. Lactococcus chungangensis sp. nov., a lactic acid bacterium isolated from activated sludge foam. Int J Syst Evol Microbiol. 2008;58(Pt 8):1844–9.
Cai Y, Yang J, Pang H, Kitahara M. Lactococcus fujiensis sp. nov., a lactic acid bacterium isolated from vegetable matter. Int J Syst Evol Microbiol. 2011;61(Pt7):1590–4.
Williams AM, Fryer JL, Collins MD. Lactococcus piscium sp. nov. a new Lactococcus species from salmonid fish. FEMS Microbiol Letters. 1990;68(1–2):109–13.
Acknowledgements
The authors are indebted to the members of the Key Laboratory of Dairy Biotechnology and Engineering, Inner Mongolia Agricultural University, Hohhot, for their technical supports.
Funding
This research was supported by the National Natural Science Foundation of China (Beijing, China; Grant No. 31622043) and National Natural Science Foundation of China (Beijing, China; Grant No.31601454).
Availability of data and materials
The sequence information of ten Lactococcus type species in this paper has been deposited in the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov), the accession numbers JXKA00000000, JXKB00000000, JXKC00000000, JXJW00000000, JXJX00000000, JXJT00000000, JXJU00000000, JXJV00000000, JXJZ00000000 and JXJY00000000.
Author information
Authors and Affiliations
Contributions
JY and ZHS designed the experiments. YTQ and YR performed the experiments. YQS and WJL analyzed the data. JY drafted the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The Ethical Committee of the Inner Mongolia Agricultural University ruled that no formal ethics approval was required for this study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Glycolysis/gluconeogenesis and the pentose phosphate pathway of ten type strains in the Lactococcus genus. The names of the genes that were present in the genomes of ten type strains in Lactococcus genus are shown in red, and those that were absent in all strains are shown in gray. (JPEG 336 kb)
Additional file 2: Figure S1.
Pairwise ANI values across ten Lactococcus genomes. (DOC 42 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Yu, J., Song, Y., Ren, Y. et al. Genome-level comparisons provide insight into the phylogeny and metabolic diversity of species within the genus Lactococcus . BMC Microbiol 17, 213 (2017). https://doi.org/10.1186/s12866-017-1120-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12866-017-1120-5