
Abstract
Background
Growth regulation is a complex process influenced by genetic and environmental factors. We examined differences between growth hormone (GH) transgenic (T) and non-transgenic (NT) coho salmon to elucidate whether the same loci were involved in controlling body size and gene expression phenotypes, and to assess whether physiological transformations occurring from GH transgenesis were under the influence of alternative pathways. The following genomic techniques were used to explore differences between size classes within and between transgenotypes (T vs. NT): RNA-Seq/Differentially Expressed Gene (DEG) analysis, quantitative PCR (qPCR) and OpenArray analysis, Genotyping-by-Sequencing, and Genome-Wide Association Study (GWAS).
Results
DEGs identified in comparisons between the large and small tails of the size distributions of T and NT salmon (NTLarge, NTSmall, TLarge and TSmall) spanned a broad range of biological processes, indicating wide-spread influence of the transgene on gene expression. Overexpression of growth hormone led to differences in regulatory loci between transgenotypes and size classes. Expression levels were significantly greater in T fish at 16 of 31 loci and in NT fish for 10 loci. Eleven genes exhibited different mRNA levels when the interaction of size and transgenotype was considered (IGF1, IGFBP1, GH, C3–4, FAS, FAD6, GLUT1, G6PASE1, GOGAT, MID1IP1). In the GWAS, 649 unique SNPs were significantly associated with at least one study trait, with most SNPs associated with one of the following traits: C3_4, ELA1, GLK, IGF1, IGFBP1, IGFII, or LEPTIN. Only 1 phenotype-associated SNP was found in common between T and NT fish, and there were no SNPs in common between transgenotypes when size was considered.
Conclusions
Multiple regulatory loci affecting gene expression were shared between fast-growing and slow-growing fish within T or NT groups, but no such regulatory loci were found to be shared between NT and T groups. These data reveal how GH overexpression affects the regulatory responses of the genome resulting in differences in growth, physiological pathways, and gene expression in T fish compared with the wild type. Understanding the complexity of regulatory gene interactions to generate phenotypes has importance in multiple fields ranging from applications in selective breeding to quantifying influences on evolutionary processes.
Similar content being viewed by others

Background
Domestication and artificial selection have long been used to increase size and growth rates of fishes and other vertebrates used for food production. More recently, creation of growth enhanced transgenic organisms through introductions of growth hormone (GH) gene constructs has been the subject of research in many fish species [1,2,3,4]. Growth regulation is a complex process influenced by genetic, cellular and environmental factors. In fishes, growth is mediated primarily via the growth hormone (GH)/insulin-like growth factor-I (IGF-I) pathway [5, 6], and introduction of a GH construct in some species has resulted in greater than 30-fold increases in the size-at-age of transgenic fish [1, 7, 8], with more modest gains in other species [9].
In salmon, GH plays a critical role in somatic growth through promotion of protein synthesis, feed intake, and feed-conversion efficiency [5, 10, 11]. In addition, GH and IGF-1 are involved in many other processes in salmon, including reproduction, feeding behaviours, predator avoidance, and osmoregulation [5, 12]. Effects of GH overexpression, relative to wild type, have also been found to be highly influenced by environmental conditions and by genotype by environment interactions [13, 14].
Recent studies comparing transgenic and non-transgenic salmon have examined the role of GH in regulating genes involved in growth. Genes involved in the GH/IGF-I pathway exhibit differential expression between wild-type and transgenic coho salmon (Oncorhynchus kisutch), with greatly increased expression of GH and IGF-I in the latter, and multiple differences between genotypes in other pathways, including transcription, amino acid metabolism, respiration, stress/immune function, lipid metabolism/transport, brain/neuron function, and carbohydrate metabolism [10, 15,16,17]. Levels of myostatin 2, a protein involved in muscle development and growth, was found to vary between transgenic and wild-type salmon, with higher levels in red muscle of transgenic fish and lower levels in white muscle [18]. Genes involved in appetite and feeding response (e.g., AgRP1) are also strongly differentially expressed (approximately 15-fold) in the brain and pituitary gland of transgenic coho salmon compared with wild-type fish [19].
Comparisons of the effects of GH transgenesis among strains with different genetic backgrounds has found variable growth responses. For example, a highly domesticated (fast-growing) strain of rainbow trout (Oncorhynchus mykiss) showed little or no increase in size compared with wild strains following introduction of the GH transgene [7], whereas in a wild-type (slow-growing) trout strain variable responses were detected [17]. In contrast, in coho salmon, additive effects of domestication and GH transgenesis were observed [16]. Similar strain effects have also been observed in GH transgenic mice [20]. Thus, the genetic background into which the GH transgene construct is introduced appears to influence observed changes in phenotype. Recent studies have indicated that phenotypic effects of GH transgenesis and domestication may arise from similar influences on gene expression and physiological pathways. Indeed, previous measurements in domesticated salmonids have found elevated levels of GH and IGF-I relative to wild type [21, 22] as occurs in GH transgenic fish [10], indicating this growth-regulating pathway is affected in similar ways by these two types of genetic change. However, it is not clear if all types of fast-growing strains, or all fast-growing individuals within strains, exhibit similar phenotypes due to parallel changes in gene expression and physiology.
In order to more directly examine if genetic background affects the phenotypic outcomes of GH transgenesis, and whether such influences affect phenotype in non-transgenic (NT) and GH transgenic (T) siblings in the same or distinct ways, we identified differentially expressed genes (DEGs) between fish size classes (large vs. small) within both T and NT salmon. The analysis examined whether the presence of a GH transgene affected expression (mRNA levels) of genes associated with growth (and other pathways of interest) in GH transgenic and non-transgenic fish. We further performed a Single Nucleotide Polymorphism (SNP)-based genome-wide association study (GWAS) to identify loci that affected body size as well as the expression of an array of genes involved in growth and other pathways affected by GH. Specifically, we examined whether the same or different regulatory loci are involved in controlling body size and gene expression variation between T and NT fish, with the objective of assessing whether the physiological transformations occurring from GH transgenesis are under the influence of alternative gene regulation pathways than those affecting size variation in NT salmon. The analysis found multiple regulatory loci affecting gene expression between fast-growing and slow-growing fish within T or NT groups, but few such regulatory loci were found to be shared between NT and T groups. These data have revealed how GH overexpression alters the regulatory responses of the genome to the shift in growth, physiological pathways, and gene expression associated with GH transgenesis.
Results
RNA-Seq, differentially expressed genes, and GO analysis
From the RNA-Seq analysis, an average of 14,529,510; 14,492,284; 14,298,225; and 14,099,226 RNA sequencing reads were detected in the technical replicates for NTLarge, NTSmall, TLarge and TSmall, respectively.
DEGs from RNA-Seq analyses comparing fish from the large and small tails of the size distributions of T and NT salmon (NTLarge, NTSmall, TLarge and TSmall) spanned a broad range of biological processes, indicating a wide-spread influence of the transgene on gene expression. However, the response to the transgene differed between size groups. In a comparison of TLarge and NTLarge fish, 939 genes were found to be differentially expressed with a greater than 3-fold change in expression (Supplemental Material Table S1); of these, 593 genes had higher expression in TLarge fish while 346 had higher expression in NTLarge. In contrast, 1518 genes were differentially expressed between TSmall and NTSmall fish (Table S2); 805 DEGs had higher expression in TSmall and 713 had higher expression in NTSmall. Of the 346 genes that were overexpressed in NTLarge fish, 191 were also overexpressed in NTSmall fish in the comparison with TSmall (Fig. 1a). Similarly, 408 genes were overexpressed in T fish (i.e., in TLarge when compared with NTLarge and in TSmall when compared with NTSmall; Fig. 1a).

Venn diagrams showing unique and shared Differentially Expressed Genes (DEGs) between fish identified in comparisons by size or by transgenotype (large transgenic, TLarge; small transgenic, TSmall; large non-transgenic, NTLarge; and small non-transgenic, NTSmall). a DEGs identified in comparisons within sizes across transgenotypes (TSmall and NTSmall or TLarge and NTLarge) that had higher expression in the indicated group and an indication of whether these DEGS were unique or shared with other groups. For example, 346 DEGS were overexpressed in NTLarge compared with TLarge; 191 of these were also overexpressed in NTSmall in a comparison with TSmall. In other words, 191 DEGS were overexpressed in non-transgenic fish as compared with size matched transgenic fish. b DEGs identified in comparisons within transgenotypes (NTSmall vs. NTLarge and TLarge vs. TSmall) that had higher expression in the indicated group and an indication of whether these DEGS were unique or shared with other groups
DEGs were identified in comparisons between size groups (Large vs. Small) within transgenotypes (T or NT), albeit considerably fewer than between transgenotypes (T vs. NT). In a comparison between TLarge and TSmall, only 37 DEGs were identified, of which 12 genes were more highly expressed in TLarge and 25 genes were more highly expressed in TSmall (Table S3, Fig. 1b). A greater number of DEGs were identified in comparisons of large and small NT fish, with 87 more highly expressed in NTSmall and 71 more highly expressed in NTLarge (Table S4, Fig. 1b). No DEGs were consistently upregulated in large fish across transgenotypes or in small fish across transgenotypes (Fig. 1b).
Gene Ontogeny (GO) analysis was used to compare biological processes affected by the presence of the transgene. In the comparison between NTLarge and TLarge, DEGS were assigned to 547 Biological Process GO terms (Table S5). The numbers of DEGs differed significantly from expectation for 204 GO terms (χ2; p < 0.05); for 194 terms, the number of observed DEGs was significantly greater than expected (Table S5). In the comparison between NTSmall and TSmall, DEGs were assigned to 609 Biological Process GO terms (Table S5). A total of 197 categories differed significantly from expectation (χ2; p < 0.05), with 184 of these having more DEGs observed than expected. The shared biological processes in the comparison of large and small fish across the two transgenotypes reflect the genotype differences that are not due to differences in body size (Figure S1, Table S5). Processes that differed between transgenotypes included regulation of cell cycle progression and cell division, altered DNA replication, increased catabolism of essential macromolecules and changes in endocrine control (Fig. 2a, b, Figure S1).

Gene Ontology (GO) Biological Process categories for the differentially expressed genes (DEGs) identified in comparisons between transgenotypes (transgenic fish, T, and non-transgenic fish, NT) for large and small fish. a GO terms associated with catabolism and metabolism/biosynthesis of fatty acids; b Endocrine control; c Sacromerogenesis; and d Immune response (see Supplemental Materials Figure S1 for complete set of DEGs)
Overexpression of growth hormone also led to changes in distinct regulatory pathways between transgenotypes at both ends of the body size spectrum. When TLarge fish were compared to NTLarge fish, we observed enrichment of genes in pathways that regulated DNA repair; DNA damage sensing mechanisms; demethylation; and responses to hyperosmotic salinity, fungus, light and UV exposure (Fig. 2, Table S5). In contrast, the comparison of TSmall and NTSmall yielded enriched GO terms associated with metabolism and biosynthesis of various fatty acids; development of skeletal muscles (sarcomerogenesis); response to immune stresses (interferon-gamma) and toxic stresses (unfolded protein, cadmium ion); regulation of macromitophagy; and sensory perception of pain (Fig. 2a, c, d, Table S5).
DEGs assigned to Biological Process GO terms were assessed for size classes within transgenotypes. DEGs were assigned to 152 and 24 Biological Process GO terms from comparisons between NT size groups and T size groups, respectively (Table S6). Differences between size classes were unique to each transgenotype (Fig. 3). In NT fish, DEG enrichment was most notable in pathways that affect carboxylic acid catabolism and biosynthesis; endocytosis (phagocytosis); generation of superoxide anion (activates glycolysis); regulation of immunoglobulin secretion; salt tolerance (related to the GH affecting carbonic anhydrase II and copper tolerance); regulation of key hormone and peptide secretions; and gluconeogenesis pathways. In the T fish, differences in GO terms between body size classes were less frequent and depended on genes that negatively regulate proteolysis; hydrolysis; transcription; and RNA and cellular macromolecular biosynthetic process (Fig. 3, Table S6).

Gene Ontology (GO) Biological Process categories for the differentially expressed genes identified in comparisons between large vs. small fish within transgenotypes (transgenic fish, T, and non-transgenic fish, NT)
Quantitative PCR
Transgenotype (T vs. NT) had a strong influence on gene expression of genes assayed individually. Twenty-six of the assessed genes exhibited significant differences in mRNA expression level between T and NT individuals (Table 1; gene name abbreviations are as for Table S9). Gene expression was greater in transgenic fish for 16 of those genes, while non-transgenic fish had higher gene expression for 10 genes. GH/IGF pathway genes exhibited significant differences between T and NT, and as expected, hepatic GH was not expressed at detectable levels in NT fish. GHR, IGF-I and IGF-II mRNA levels were elevated in T vs. NT, whereas IGFBPI, IGFBP2B2 and IGFIR were downregulated; these results were consistent with the overall stimulation of growth via the GH/IGF pathway. Differences between T and NT were particularly large for three genes: LEPTIN, GLK and G6PASE1. LEPTIN, was found at levels 12.1-fold and 8.4-fold greater in small and large NT fish as compared with their small and large T counterparts, respectively. Similarly, G6PASE1 was found at levels 5.8-fold and 4.8-fold higher in NTSmall and NTLarge compared with size matched T fish. In contrast, GLK expression levels were 23.1-fold and 10.2-fold higher in TSmall and TLarge than in NT fish.
Eleven genes exhibited different mRNA levels when the interaction of size and transgenotype was considered (IGF1, IGFBP1, GH, C3–4, FAS, FAD6, GLUT1, G6PASE1, GOGAT, MID1IP1; ANOVA p < 0.05; Table 1, Figure S2). There was no clear pattern for which groups differed significantly. For example, for G6PASE1 and IGFBP1 there was no difference within T fish, but T and NT fish differed and size classes differed within NT (Tukey, p < 0.05). In contrast, with IGF1 and C3–4 there was no difference in expression within NT fish, but T differed from NT and size classes differed within T fish (Tukey, p < 0.05). For FAS and GLUT1 there were significant differences between NTLarge and TLarge but not within transgenotypes or between small fish, while with GOGAT and MID1PI1, the TSmall group differed from all others but there were no significant differences between large fish. For ALB, AST, and GLDH, both size and transgenotype were significant factors in determining gene expression when assessed individually, although the intersection of transgenotype and size was not significant (ANOVA, p > 0.05).
SNP discovery and GWAS
One transgenic fish was removed from SNP discovery due to missing sequence data. A total of 619,839 barcoded reads were considered for discovery; of these 80.2% were successfully mapped to the coho salmon reference genome. After merging multiple-aligned tags and filtering low quality reads, 62,058 unique SNPs were identified. Average read depth was 14.7x. After the additional filtering steps described above, 13,588 SNPs were considered for subsequent association analysis. SNPs were distributed fairly evenly across all 30 linkage groups with an average of 312 ± 98 SNPs per group (Figure S3). An additional 4237 SNPs were found on unassigned genome contigs and scaffolds.
For body size traits, a total of 17 SNPs were significantly associated with weight in T fish, while only 4 SNPs were associated with weight in NT fish (FDR q < 0.05; Table 2, Table S7). Similarly, 15 and 8 SNPs were associated with length in T and NT fish, respectively. In T fish, 11 SNPs were significantly associated with both weight and length, while in NT fish only 3 SNPs were significantly associated with both traits. Interestingly, condition factor was associated with 299 SNPs in NT fish but with only 7 SNPs in T fish. When fish were examined by size group (Small vs. Large), most of the significant SNPs were identified in NTSmall fish (Table 2). Further, 374 SNPs were associated with condition factor for NTSmall fish. Of these, 249 were also significantly associated with condition factor when all NT fish were examined together. A large number of SNPs were associated with length in NTSmall fish (311 SNPs) and there was considerable overlap in SNPs associated with CF and length (but not weight) in NTSmall fish (95.7% similarity).
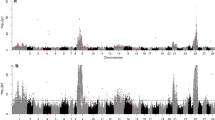
For gene expression traits, SNPs were identified that were significantly associated with 29 of the 31 assessed genes, although SNPs were distributed unevenly across size/transgenotype groups (Table 2, Table S7). A total of 649 unique SNPs were associated with at least one trait in one of the groups, with most SNPs associated with one of the following 7 traits: C3_4, ELA1, GLK, IGF1, IGFBP1, IGFII, or LEPTIN (Table 2). Interestingly, only 2 SNPs were associated with the transgene (GH) and only in TLarge, although more SNPs were associated with expression levels of other components of the growth hormone axis in the different transgenotype and size groups (Table 2). None of the SNPs identified as being associated with trait variation were located near (i.e., less than 1 Mb) the target gene (as determined by the current annotation in GenBank). For some traits, SNPs were broadly distributed across multiple linkage groups, while for others SNPs tended to be clustered on one linkage group (Figures S5, S6). For example, in T, SNPs for C3_4 and LEPTIN were found on 20 or more linkage groups, with 30 and 34 SNPs respectively (Fig. 4a, b), while the 10 SNPs for GHR were located primarily on linkage group 4 (Fig. 4c). For the phenotypic traits weight and length, SNPs were clustered on linkage groups 6 and 10, or on scaffold fragments (Fig. 5a, b).

Manhattan plots of SNP number and linkage group (1–30, and unassigned (U)) for select expression traits in transgenic (T) fish. For all traits, please see Supplemental Figure S5. Significant SNPs (FDR = 0.05) are indicated by red triangles; dotted line indicates significant q value. a C3_4; b Leptin; c GHR (abbreviations are as for Table S1)

Manhattan plots of SNP number and linkage group (1–30, and unassigned (U)) for length and weight in transgenic (T) fish. For all traits, please see Supplemental Figure S6. Significant SNPs (FDR = 0.05) are indicated by red triangles; dotted line indicates significant q value. a Length; b Weight
Despite both T and NT groups possessing the same genetic variation on average, only 1 SNP significantly associated with at least one phenotype was found to be in common between T and NT fish when large and small fish were analyzed together. Further, no SNPs were found in common between transgenotypes when associations were analyzed within size categories (Fig. 6). Within transgenotypes, TSmall and TLarge fish shared only 2 SNPs in common while NT size groups had only 1 SNP in common (Fig. 6, Table S9). A total of 440 SNP markers were associated with at least two traits within the same analysis, i.e., fish grouped by size or comparisons between transgenotypes only (e.g., SNP S1_495516192 was associated with both ACTB and AST in TLarge and also with C3–4 in NTSmall fish; Table S9). Of these markers, 93% were significantly associated with more than one trait in NTSmall. The majority of the markers associated with multiple traits were explained by the correlation between CF and Length, with 280 SNPs associated with both of those traits but not with others. One SNP, S1_1372865070, located on chromosome 23, was associated with 5 traits (ALB, GLDH, GLK, IGFBP2B2, and PEPCK) in TLarge fish and with 4 traits (ALB, GLDH, Length and Weight) in T fish when size was not considered.

Venn Diagram of the number of significant SNPs associated with all phenotypes in each size and transgenotype category and shared between groups. T: transgenic, NT: non-transgenic
Discussion
Here we used multiple methods (RNA-Seq/DEG analysis; quantitative PCR; GBS with SNP discovery; and GWAS) to examine the effects of a GH transgene on body size phenotypes and on expression of key growth-related genes in coho salmon. We further examined how the GH transgene influenced trans-acting regulatory loci affecting variation at these morphological and gene expression traits. It is well documented that the presence of the GH transgenesis results in overexpression of growth hormone in transgenic fish with correspondingly faster growth rates and larger average size (e.g. 10). Here too, we found that transgenic fish had elevated growth rate as well as a higher average condition factor.
Differentially expressed genes and GO analysis
Although multiple studies have reported the physiological difference between non-transgenic and GH transgenic coho salmon (e.g., 4), it is not well understood which pathways are involved in causing size variation within and between transgenotypes (as opposed to being a secondary consequence of growth modification). Using DEG analysis, we found changes to a diversity of pathways that were unique to each transgenotype. Considerably more DEGs were identified in comparisons between transgenotypes (i.e., between NTSmall and TSmall, or between NTLarge and TLarge), than between size classes within transgenotypes. Shared differences in biological processes observed in the comparison of large fish between transgenotypes, and of small fish between transgenotypes, reflect the influence of the transgene rather than effects of body size. As expected, the present study found that overexpression of GH in transgenic fish led to negative regulation of cell cycle progression and cell division, altered DNA replication, increase catabolism of essential macromolecules and changes in endocrine control resulting in differentially expressed genes associated with accelerated developmental changes in neurogenesis, skeletal muscle, hemopoietic stem cell differentiation and cardiac muscle development.
Interestingly, overexpression of growth hormone also led to changes in gene expression between T vs. NT fish that differed between ends of the body size distribution. When TLarge fish were compared to NTLarge fish, there was enrichment of genes in pathways that regulate DNA repair, DNA damage sensing mechanism, demethylation, response to hyperosmotic salinity, fungus, light and UV. In contrast, the comparison of TSmall and NTSmall yielded enriched GO terms for metabolism of arachidonic acid and prostanoid, biosynthesis of terpenoid and icosanoids, development of skeletal muscles (sarcomerogenesis), response to immune stresses (interferon-gamma), and toxic stresses (unfolded protein, cadmium ion). There was also enrichment of GO terms for regulation of macromitophagy and sensory perception of pain in the comparison of TSmall and NTSmall. We note that several of these pathways specifically respond to hyperosmotic stress and immune responses, effects observed between transgenic and wild-type fishes. For example, GH transgenic zebrafish had increased mortality when exposed to salinity stress, and all osmoregulatory genes were down regulated under hyperosmotic stress [23]. Presence of a GH transgene has also been found to influence the development of saltwater tolerance in coho salmon undergoing smoltification [24]. In the present study, we found that expression levels of C3_4, a component of the complement system involved in immune responses, differed between large and small fish; however, in non-transgenic fish, expression was elevated in small fish while in transgenic fish, expression was elevated in large fish. Transgenic coho salmon also have a dampened immune response in comparison with wild-type fish [25], including changes in baseline expression of immune related genes in the IGF system [26].
We also compared size classes within transgenotypes. The pathways that exhibit differential gene expression in large vs. small fish differ between transgenotypes (i.e., comparing NTLarge vs. NTSmall and TLarge vs. TSmall) further suggest that different genomic mechanisms may be leading to the large (or small) size phenotype in transgenic vs. non-transgenic coho salmon. For example, in the comparison of NT fish, differential gene expressions were most notable in pathways that affect carboxylic acid catabolism and biosynthesis, endocytosis (phagocytosis), generation of superoxide anion (activates glycolysis), regulation of immunoglobulin secretion, salt tolerance (related to the GH affecting carbonic anhydrase II and copper tolerance), regulation of key hormone and peptide secretions, and gluconeogenesis pathways. In the comparison within T fish, DEGs between size groups was involved in negative regulation of proteolysis, hydrolysis, transcription, RNA and cellular macromolecular biosynthetic processes.
Gene expression and morphological phenotypes
Within each of the T and NT progeny groups, there was also significant variation in the weight and length between large and small T fish, comparable to the variation seen between large and small NT salmon with the same median size. To investigate the basis of this variation, we examined GH expression among size and transgenotypes groups and found GH levels to be related both to transgenotype and body size within transgenotypes (GH was not expressed in the livers of non-transgenic fish as expected). Higher GH expression was seen in TLarge than in TSmall groups, suggesting variation in transgene expression may exist within the strain. Other genes in the growth hormone axis demonstrated differences in expression across transgenotypes, with GHR, IGF1, and IGFII being overexpressed in transgenic fish, while IGFBP1, IGF1R and IGFBP2B2 were underexpressed. For IGF1 and IGFBP1, there was also a significant interaction between transgenotype and size, with the highest expression levels in TLarge and NTSmall respectively. These results are generally consistent with findings from previous studies of transgenic coho salmon [10] and with other studies of GH and IGF binding proteins in fishes [6, 27]. GH-transgenic zebrafish exhibited an increase in expression of IGF1 and IGFII, and a decrease in IGF1R [28]. In GH-transgenic Nile tilapia, IGFBP1 was lower than in wild-type fish [29]. GH and other genes in the growth hormone axis are involved in several important traits beyond growth, including hypo-osmoregulatory changes during smoltification and upon initial entry to saltwater, regulation of sexual maturity, and in feeding behaviour and aggression (reviewed in 5). Thus, large differences in gene expression patterns are expected between GH transgenic and wild-type salmon.
Of the expression traits surveyed, 84% differed significantly between transgenic and non-transgenic fish, with one-third also differing between size categories. In addition to the genes in the growth axis discussed above, two other genes stand out as being highly differentially expressed between transgenotypes: LEPTIN and G6PASE1. Expression of G6PASE1 also differed significantly between size groups within and between transgenotypes, with the highest levels of expression occurring in NTSmall followed by NTLarge. G6PASE1 is involved in regulation of carbohydrate metabolism and appears to not be under dietary control in salmonids [30]. Reduced expression of G6PASE1 in transgenic fish would suggests that these fish have limited ability to metabolize glucose in the liver or utilize this energy source. However, direct experimental assessment [31] suggests that transgenic fish may have elevated ability to use carbohydrates. Previous measures of G6PASE1/2 have found differing results depending on whether fish were in a fed or food-deprived state. In the present study and that of Abernathy et al. [32], fish were sampled while in a fed and growing state, and in both cases G6PASE mRNA levels were seen to be reduced in T relative to NT. In contrast, Panserat et al. [33] analyzed fish in a food deprived state and did not detect a difference in mRNA level for G6PASE. This discordance in results is intriguing and fosters speculation that the G6PASE gene expression changes observed may be strongly sensitive to experimental conditions and the nutritional status of the fish. Glucokinase was found to be elevated in the present study as well as in previous assessments that found enhanced potential for carbohydrate utilization in GH transgenic salmon [32, 33]. LEPTIN also had large changes in expression between transgenotypes, but not between size classes. Here we observed a decrease in LEPTIN levels in GH transgenic fish which is consistent with the known role of GH in suppressing leptin [34], as well as the findings of other studies of transgenic fish [29, 35, 36]. Leptin is considered a ‘pleiotropic hormone’ in fish with roles in regulating food intake and weight gain, development and maturation, and stress response and acclimation [37, 38].
Genome-wide association study
The GWAS found that, in general, more SNPs were associated with traits within non-transgenic fish than within transgenic fish, and when fish were analyzed by size group within transgenotypes, most SNPs were detected in NTSmall. However, there were almost no SNPs shared among groups and very few SNPs shared between traits, with the exception of SNPs shared between CF and length in NTSmall. This is in accord with another recent study examining body size variation throughout the life history of transgenic coho salmon. In this case, none of 243 quantitative trait loci (QTL) for growth were found to be shared between GH transgenic and non-transgenic fish [39]. Surprisingly, a similar relationship was not observed between CF and weight, and in fact this overlap in SNPs was not observed in the comparison of T and NT where a large number of SNPs associated with CF were detected but only 8 SNPs were associated with length. However, we note that as fish were matched for length at sampling, the reduced variation between T and NT may have led to reduced power to detect SNPs associated with length.
In the current study, most gene expression traits were associated with fewer than 10 SNPs. The power of GWAS to identify a true association between SNP and trait depends on phenotypic variance, and thus rare variants, or variants of small effect size, are likely to be missed. However, some traits were associated with many more SNPs (including up to 157 for C3–4), although there was considerable variation in the number of SNPs associated with a given trait between size categories across transgenotypes. Surprisingly, only 2 SNPs associated with G6PASE1 variation were identified in transgenic fish, and only 1 SNP in non-transgenic salmon. When fish were analyzed between sizes, only 1 SNP was found in TSmall and TLarge, and none were identified in NTSmall or NTLarge. Unlike G6PASE1, LEPTIN expression was associated with a large number of SNPs in transgenic fish (34 in TAll; and 81 in TLarge when fish were grouped by size), indicating many loci play a role in regulating this hormone in coho salmon.
To examine the influence of regulatory loci on gene expression and body-size phenotypes, we undertook a GWA analysis to identify SNPs associated with variation in these traits and further assessed these influences in the presence and absence of the GH transgene. For many of the traits associated with larger numbers of influential SNPs (N > 30), those SNPs tend to be broadly distributed across the genome. For example, SNPs associated with LEPTIN expression in transgenic fish were identified on 14 linkage groups (and several unassigned scaffolds). It is likely such a broad association between expression levels and the various regulatory elements rises from the diverse pathways in which LEPTIN plays a role. SNPs associated with C3–4, another gene associated with a diversity of functions [40, 41] and controlled by numerous regulatory elements [42], were also widely dispersed throughout the genome. In comparison, expression of GHR, which has a limited function in the growth hormone axis [43], was associated with just 10 SNPs in transgenic fish of which 8 were located on linkage group 4.
None of the SNPs identified here were located within the genes of the associated trait, and most were on different chromosomes from the gene being assessed, indicating the regulatory effects are for the most part acting in trans. Indeed, many studies employing a GWAS approach have not been able to identify causal sites despite extensive sequencing of areas around SNPs or other markers, indicating that in many cases the GWAS association is likely not a direct causal variant acting in a cis fashion at the gene being assessed [44]. We note that SNPs that are significantly associated with traits, regardless of the physical proximity of the SNP to the gene of interest, can still be valuable predictors of a phenotype [45].
Conclusions
Here, we examined the impact of GH transgenesis in coho salmon relative to wild type, and have assessed genomic influences between large vs. small fish in NT and T genotypes. The results indicate that there are wide-spread regulatory influences acting to influence body size and gene expression traits, in addition to effects of GH transgenesis. The results reported here arise from one family, thus allowing us to sensitively examine differences caused by the presence of the GH transgene within the same genetic background, on average. Reducing genetic background effects is particularly useful for reducing the number of differentially expressed genes or SNPs identified using RNA-Seq approaches [46]; by decreasing genetic heterogeneity, we increase the power to detect correlations between phenotypes and specific genetic variants [45]. Thus, while the current data are family specific, the breadth of analysis supports the hypothesis that specific genetic loci influence body size and gene expression phenotypes, and that different loci are acting in transgenic salmon than act in wild type.
Trans-acting regulatory loci and/or epistatic or pleiotropic interactions are expected given that GH transgenes affect expression of proteins in complex physiological and cellular pathways, and interact with other loci and their pathways to modulate phenotypic effects. Studies with model organisms found many genetic modifiers that were capable of altering in trans the penetrance and expressivity of other loci. Indeed, evidence for trans-acting loci that act inversely to their dose to affect gene expression have been found to be wide-spread in Drosophila and plants [47,48,49]. In transgenic mammals, modifier loci affecting physiological processes and oncogenesis have been mapped. For example, a study in mice mapping modifier loci affecting the consequences of a mammary tumour-inducing transgene under different feeding conditions found at least 13 QTL affecting the onset, severity and metastasis, as well as QTL by diet interactions [50]. Similarly, Saito and Suzuki [51] found three modifiers affected tumour induction caused by a transgene expressing the K-rasG12V oncogene. In rats, Kantachuvesiri et al. [52] found that the effects of a transgene causing malignant hypertension were highly dependent on strain genetic background. In plants, modifiers affecting insect resistance were found to act additively with a Bacillus thuringiensis (Bt transgene to reduce impacts of corn earworm on soybean [53]. Thus, regulatory loci play significant roles in causing variation in traits in a variety of organisms, including those whose phenotype has been modified by transgenesis. Genetic modifiers of transgenic phenotypes likely act through interacting gene expression pathways, and via influences on the pathways their expressed proteins modulate. The precise mechanism of how regulatory effects act, and how GH transgenesis can modify these effects, is not known. We know that overexpression of GH strongly affects many gene expression and physiological pathway phenotypes, in some cases to the point of saturation [54]. As such, the influences of regulatory loci (e.g., transcription factors, proteins acting in complexes to cause epistatic effects, etc.) may be expected to have different capacities to affect pathways between GH transgenic and wild-type strains.
Transgenes are not native members of a genome that have evolved within that genomic environment. In some cases, transgenes have been found to be subject to silencing via epigenetic processes [55]. In addition, modifier loci that affect the expression of variegating transgenes have been identified in Drosophila where methylation of DNA does not occur [56]. We do know that some transgenes (e.g., GFP-expressing) in salmonids can show strong variegation that differs in extent among strains (unpublished), and we note that some transgenic individuals in the present strain (M77) do not show full growth stimulation suggesting some gene silencing mechanisms may be operating [54]. However, formally, we do not know whether the GH transgene in coho salmon used in the current study is subject to variegation affecting its expression and effects on growth, although we note that its chromosomal position has been determined to be centromeric [57] and its molecular environment is highly enriched in repetitive DNA [58], which is known to cause varied effects on gene expression. Thus, it is possible that some of the regulatory loci identified in the present study are acting to influence transgene silencing mechanisms and thus cause suppression of growth stimulation in some individuals. Further study to examine the mechanisms of transgene silencing, how this may influence interactions with other loci, and how or if these affects vary across a population, would be valuable.
The present data extend our understanding of background genetic effects beyond strain-level studies conducted previously. The findings are valuable to understand the role of background genetics in controlling phenotype specifically in GH transgenic organisms, but are also generally informative where analysis of pleiotropic effects and variable expressivity of traits are being examined. Understanding the complexity of regulatory gene interactions to generate phenotype has importance in multiple fields ranging from selective breeding to quantifying influences on evolutionary processes. The significant trans regulation of traits observed, and the finding that different loci affect phenotypic variability in GH transgenic and non-transgenic individuals, could allow selection to result in retention of different regulatory alleles between NT and T transgenotypes. Further understanding the degree to which regulatory controls differ between T and NT individuals is important for ecological risk assessments examining the potential consequences of transgenic organisms in nature where selection of variation in T organisms may not be optimal for maximum fitness of NT individuals [4, 59].
Methods
Experimental design and sample collection
Animals used in this study were from an outcrossed coho salmon family generated on February 28, 2012 by crossing a transgenic (T) female hemizygous for a growth hormone gene construct (strain M77) [13] and a non-transgenic (NT) male that was derived from a hatchery-supported natural population from the Chehalis River in British Columbia. This cross produced approximately a 1:1 ratio of the transgenotypes (T vs. NT) as expected for Mendelian segregation of the M77 transgene at a single insertion locus in the coho salmon genome. A total of 427 fish from this cross were reared in 200 L tanks in aerated fresh well water (10 ± 0.5 °C) under a simulated natural photoperiod, with fish densities kept below 5 kg/m3. Fish were fed to satiation 3 times daily with commercial salmon feed (Skretting Canada Ltd., Vancouver BC, Canada). Fish were reared at Fisheries and Oceans Canada’s West Vancouver Laboratory which is specially designed to prevent the escape of transgenic fish into the environment.
Sampling protocols have been described elsewhere [60]. In brief, fast- and slow-growing morphs were grown under the same rearing conditions, with both groups fed to satiety thrice daily using commercial salmon diets. The fish were reared together until August 14–16, 2012 when the fast-growing T fish reached a mean fork length of 9.7 cm, at which time they were sampled. NT siblings continued to be reared until November 13–15, 2012 when their mean length (9.5 cm) was approximately equal to that of the T salmon (Table S8, Figure S4). For tissue collection, coho salmon were anaesthetized using a procedure approved for use in salmon by the Canadian Council on Animal Care DFO Pacific Region Animal Care Committee (Management Procedure 3.7). Fish were netted from their rearing tank into an aerated bucket containing 200 mg/L tricaine methane sulphonate (MS222, Syndel Laboratories, Nanaimo, BC, Canada) buffered with a 2-fold weight of sodium bicarbonate in fresh well water (10 °C) to initiate aqueous absorption of the anaesthetic. Progress of anaesthetization was monitored by observing the fish’s ability to maintain equilibrium, and by noting opercular (gill cover) movement. Fish were removed from the anaesthetic bath immediately upon cessation of ventilation, at which point the unconscious animals were decapitated and tissues recovered by dissection. Lengths and weights were measured, fin clips were collected and stored in 95% ethanol for subsequent use, and liver samples were taken from the same fish and stored in RNAlater (Qiagen, Germany) at − 20 °C. Sex was determined by internal gonadal morphology and by use of a tightly linked Y-chromosomal marker (growth hormone pseudogene, GHΨ) that is genetically inseparable from the sex-determining locus [61]. Transgenotype was confirmed using a transgene-specific quantitative PCR (qPCR) test [62].
We employed an extreme phenotype sampling strategy to select fish for further genetic analysis; previous studies suggest that sampling from the tails of a phenotypic distribution has similar or increased power to detect genotype/trait associations as sampling from the full population which allows us to avoid performing costly genomic analysis on a prohibitively large number of individuals [63,64,65]. The roughly 20% (numbers were adjusted slightly to accommodate a 96-well format) largest and smallest fish by weight from each experimental group were selected in descending and ascending rank order, respectively, for subsequent analyses: NTLarge (n = 47; weight range 9.8–15.2 g), NTSmall (n = 46; 4.5–7.0 g), TLarge (n = 46; 15.0–22.0 g), and TSmall (n = 47; 3.0–9.3 g). We note that this strain of GH transgenic coho salmon does show a skewed distribution of size (Figure S4) such that some individuals show only partial growth enhancement, potentially arising from gene silencing mechanisms (e.g., methylation or other epigenetic influences) [54]. We included these individuals in our analyses as we wished to capture all sources of genetic influences on phenotype, but acknowledge that this added variance may have reduced our power to detect some weaker regulatory loci. However, we note that an unbalanced sampling design has been shown to be as effective as a balanced design when sampling extreme phenotypes for association mapping in at least one other study [63].
Differential gene expression and gene ontology analysis
Differentially expressed genes (DEGs) in T and NT coho salmon were analyzed by RNA-Seq to assess the number and type of genes that were commonly affected by body size in NT and T groups. Total RNA was extracted from liver tissue using an RNeasy mini kit (Qiagen, Valencia, CA, USA) following the manufacturer’s recommendations. RNA concentration and purity were measured using a Nanodrop (Thermo Scientific, DE, USA). RNA quality was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, CA, USA) with RIN values > 9.0.
RNA from the 12 largest and 12 smallest fish from each transgenotype were pooled for a total of 4 groups (NTLarge, NTSmall, TLarge and TSmall). These samples were replicated for each group, and were sent for RNA sequencing and bioinformatic analysis at BGI Genomics (Cambridge, MA, USA). Polyadenylated mRNA was enriched with oligo (dT) magnetic beads, fragmented to 200 bp lengths and reverse transcribed with first-strand cDNA synthesis. The double-stranded cDNA library was then size-selected, amplified by PCR, and 2x50bp paired-end sequencing was performed using the Illumina HiSeq2000 platform (Illumina, CA, USA). Raw reads were filtered to remove adaptor sequences, ambiguous reads (reads with > 10% of bases given as N), and low-quality reads (> 50% of the read had base quality values < 5). Retained reads were then mapped to the reference transcriptome and genome of coho salmon (Assembly GCF_002021735.1; 62) with Soap2.21 [66] allowing for 2 mismatches and 3 mismatches, respectively. RNAseq data has been submitted to the NCBI database (accession: SUB6704126, PRJNA597081). Gene expression levels were calculated using the RPKM method [67]: RPKM = 109 C/NL, where C is the total number of reads mapped onto a gene, N is the total number of mapped reads, and L is the sum of genes in base pairs. DEGs were then screened between technical replicates from the same group using a Poisson distribution model and between different treatment groups using the NOISeq method [68]. Groups were compared to each other by calculating the log2 ratio of normalized expression levels as follows: NTLarge vs. TLarge, NTSmall vs. TSmall, NTLarge vs. NTSmall, and TLarge vs. TSmall.
An enrichment analysis was performed to determine which Gene Ontology (GO) terms were over- or under-represented in DEG sets for groups NTLarge, NTSmall, TLarge and TSmall. GO annotations were comprised of functional annotations from the published coho salmon transcriptome (GenBank Accession GDQG00000000.1) and BLAST searches performed against the NCBI non-redundant protein database (as of Oct. 23, 2016), as well as the SwissProt, InterProScan, and EggNOG databases, using Blast2Go v. 4.0.7 [69]. While GO-terms were not assigned to all DEGs due to the limitations of the available databases, a total of 31,151 GO annotations (mean GO-level 7.12) were considered for distribution analysis. GO distribution analysis was performed separately for each ontology (i.e., Biological Process, Molecular Function, and Cellular Component) using 6th level GO terms for DEGs with a ≥ 3-fold change in expression. The Chi-Square test was used to identify significant ontologies (p < 0.05) in comparison groups as follows: (χ2 = ∑(O − E)2/E), where the observed frequency (O) is the sample frequency for a particular GO term, and the expected frequency (E) is the frequency of each GO term in the transcriptome multiplied by the total number of differentially expressed genes with a greater than 3-fold change. Of the significant ontologies, those with > 1 observation were explored further. Only results for the Biological Process Category are provided here as this category is a reflection of the cellular component and molecular function categories.
The R package gplots [70] was used to identify the number of DEGs that were unique to each DEG set, as well as those shared among groups; results were visualized using Venn diagrams generated using an online tool available at http://bioinformatics.psb.ugent.be/webtools/Venn/. Comparisons were made between large and small fish within a transgenotype (e.g., TLarge vs. TSmall) and between transgenotypes (e.g., TLarge vs. NTLarge).
Quantitative PCR
For each of the 186 fish selected for analysis (47 NTLarge; 46 NTSmall; 46 TLarge; and 47 TSmall), total RNA was extracted from liver tissue as described above; samples with an RNA integrity number (RIN) ≥7.0 were selected for quantitative PCR (qPCR). First-strand cDNA was synthesized from total RNA (0.5 μg) using the High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor (Applied Biosystems, California USA). qPCR reactions were performed with TaqMan® Fast Advance Master Mix (Applied Biosystems) on a 7500 Fast Real-Time PCR System (Applied Biosystems) under Fast conditions. A reference gene, ubiquitin, was used to normalize mRNA levels. Ubiquitin exhibited stable mRNA expression levels among experimental groups (combined sd = 0.90). Relative mRNA expression levels were calculated using the 2-△△Ct method [71]. Eight genes were analyzed in individual assays: growth hormone (GH), growth hormone receptor (GHR), insulin-like growth factor 1 (IGF1), insulin-like growth factor 1 receptor (IGF1R), insulin-like growth factor II (IGFII), insulin-like growth factor binding protein 1(IGFBP1), T-cell receptor beta (TRB) and ubiquitin (UBIQ; used as a house-keeping gene).
Gene expression was also assessed for an additional 24 genes that were selected based on known or inferred function or their previously observed responses to GH treatment. TaqMan® probes and primers were designed using Primer Express Software® v3.0.1 (Thermo Fisher Scientific, MA, USA) and had a short amplicon length (50-100 bp; Table S9). The primer/probe combinations were used to print TaqMan® OpenArray® chips (Applied Biosystems, Burlington, ON, Canada) for use on a QuantStudio 12 K Flex Real-Time PCR System (Thermo Fisher Scientific, MA, USA). Each chip contained 48 subarrays of 56 through-holes, resulting in a total of 2688 through-holes per chip. Thus for each chip, 48 cDNA samples were run in duplicate for each of 24 genes on each chip. A solution comprised of cDNA (30 ng per sample), 2.5 μL of TaqMan® OpenArray® Real-Time PCR Master Mix (Applied Biosystems, Burlington, ON, Canada), and ddH2O to a total volume of 5 μL was prepared, distributed across a 384-well plate and then loaded onto the TaqMan® OpenArray® chips using the OpenArray® AccuFill System to reduce inter-assay variation. The through-holes on the chips were pre-loaded with the primer and probe sequences for each of the 24 original genes by the manufacturer. A total of 5 chips were used for 186 cDNA samples; one chip was used to run duplicate samples to determine whether there were any chip effects. ExpressionSuite Software (Thermo Fisher Scientific, MA, USA) was used to calculate raw critical threshold (CT) values. The transcription data was normalized to the average expression of the non-transgenic samples.
Significant differences in mRNA levels among NTLarge, NTSmall, TLarge and TSmall were determined by type II ANOVA, with transgenotype, size group and sex as factors, using the car package in R v. 3.1.1, with a Tukey HSD posthoc test [72, 73]. Outliers were removed prior to other analyses using the generalized extreme studentized deviate (GESD) test for outliers [74] with a = 0.01; GESD analyses were implement in R v 3.1.1.
GBS sequencing and SNP discovery
Genotype by Sequencing (GBS) and Single Nucleotide Polymorphism (SNP) discovery were undertaken as described in McClelland et al. (2016). Briefly, genomic DNA was extracted from fin samples for each of the T and NT fish selected for analysis (as described above) using a DNeasy kit (Qiagen, Germany) following the manufacturer’s instructions. DNA sequencing and SNP discovery were performed at the Institute for Genomic Diversity at Cornell University using their discovery pipeline implemented in TASSEL v. 3 [75, 76]. DNA was digested with the restriction enzyme EcoT221 and strands were identified using individual-specific barcodes. Tags with fewer than 10 reads per tag were removed. Barcoded sequences were combined into a list of unique sequence tags. These tags were then aligned to the coho salmon reference genome (NCBI Assembly GCF_002021735.1 Okis_V1), and the genomic position of the tags was recorded. SNP discovery was performed using the aligned tags with genotypes determined by a binomial likelihood ratio method [76].
Genome-wide association analysis
A genome-wide association (GWAS) analysis was performed using the GBS SNPs and the following phenotypes: weight, length, condition factor, and qPCR expression data for the genes described above (Table 1). Condition factor was calculated as: \( CF=100\ast \frac{W}{L^3} \), where W is the weight (g) and L is the fork length (cm) [77]. A filtering approach was applied to remove spurious SNPs that had call rates < 80% and minor allele frequency (MAF) < 0.1. SNPs that did not have bi-allelic loci were also removed as were individuals with more than 50% missing data. To minimize the presence of duplicated loci and error prone loci, loci with Mendelian errors greater than 5% were excluded using PLINK [78]. Association analyses were performed using TASSEL v. 5 [79] with sex as a covariate. Associations between SNP markers and phenotypes were tested using a general linear model (GLM) in TASSEL with the default settings. TASSEL analyses were performed using the complete data set (results not shown since the transgenotype dominates effects on phenotype) and for subsets comprised of transgenic-only (T) and non-transgenic-only (NT) fish. Significant associations were determined using a false discovery rate (FDR) of 0.05 [80]. Venn diagrams summarizing significant findings were generated using the online tool available at http://bioinformatics.psb.ugent.be/webtools/Venn/.
Availability of data and materials
The datasets generated and/or analysed during the current study are available from the following locations: RNAseq and GBS data have been submitted to the NCBI BioProject database (https://www.ncbi.nlm.nih.gov/bioproject; accession: PRJNA597081); coho salmon whole genome sequence data used for GO annotation is also available in the NCBI GenBank database (accession: GDQG00000000.1); differentially expressed genes are given in Tables S1, S2, S3, S4; GO terms are given in Tables S5, S6.
Abbreviations
- ACTB:
-
β-actin
- AGBL5:
-
Cytosolic carboxypeptidase-like protein 5
- ALB:
-
Albumin
- AST:
-
Aspartate aminotransferase
- C3–4:
-
Complement component C3–4
- CF:
-
Condition factor
- CPT1:
-
Carnitine palmitoyltransferase 1b
- DEGs:
-
Differentially expressed genes
- ELA1:
-
Elastase 1
- FAD6:
-
Fatty acid desaturase 6
- FAS:
-
Fatty acid synthase medium chain
- FBP:
-
Fructose 1,6 biphosphatase
- FDR:
-
False discovery rate
- G6PASE1:
-
Glucose-6-phosphatase
- G6PDH:
-
Glucose 6-phosphate dehydrogenase
- GBS:
-
Genotype by sequencing
- GH:
-
Growth hormone
- GHR:
-
Growth hormone receptor
- GLDH:
-
Glutamate dehydrogenase
- GLK:
-
Glucokinase
- GLM:
-
General linear model
- GLUT1:
-
Glucose transporter 1
- GOGAT:
-
Glutamine synthetase
- GWAS:
-
Genome-wide association study
- HK1:
-
Hexokinase 1
- HOAD:
-
Hydroxyacyl dehydrogenase
- IGF1:
-
Insulin-like growth factor I
- IGF1R:
-
Insulin-like growth factor I receptor
- IGFBP1:
-
Insulin-like growth factor binding protein I
- IGFBP2B2:
-
Insulin-like growth factor binding protein 2B2
- IGF-I:
-
Insulin-like growth factor
- IGFII:
-
Insulin-like growth factor II
- MAF:
-
Minor allele frequency
- MID1IP1:
-
Mid1-interacting protein
- NT:
-
Non-transgenic
- PEPCK:
-
Phosphoenolpyruvate carboxy kinase
- PLCE1:
-
1-phosphatidylinositol-4,5-bisphosphate phosphodiesterase ε-1
- qPCR:
-
Quantitative polymerase chain reaction
- QTL:
-
Quantitative trait loci
- SNP:
-
Single nucleotide polymorphism
- T:
-
Transgenic
- TRB:
-
Thyroid hormone receptor b
- UBQ:
-
Ubiquitin
- X6PFK:
-
6-phosphofructokinase
References
Devlin RH, Yesaki TY, Donaldson EM, Du SJ, Hew C-L. Production of germline transgenic Pacific salmonids with dramatically increased growth performance. Can J Fish Aquat Sci. 1994;52:1376–84.
Martínez R, Estrada M, Berlanga J, Guillén I, Hernández O, Cabrera E, et al. Growth enhancement in transgenic tilapia by ectopic expression of tilapia growth hormone. Mol Mar Biol Biotechnol. 1996;5(1):62–70.
Du SJ, Gong Z, Fletcher GL, Shears MA, King MJ, Idler DR, et al. Growth enhancement in transgenic Atlantic salmon by the use of an “all fish” chimeric growth hormone gene construct. Nat Biotechnol. 1992;10:176–81.
Devlin RH, Sundström LF, Leggatt RA. Assessing ecological and evolutionary consequences of growth-accelerated genetically engineered fishes. Bioscience. 2015;65(7):685–700.
Björnsson BT. The biology of salmon growth hormone: from daylight to dominance. Fish Physiol Biochem. 1997;17(1–6):9–24.
Beckman BR. Perspectives on concordant and discordant relations between insulin-like growth factor 1 (IGF1) and growth in fishes. Gen Comp Endocrinol. 2011;170:233–52.
Devlin RH, Biagi CA, Yesaki TY, Smailus DE, Byatt JC. Growth of domesticated transgenic fish. Nature. 2001;409:781–2.
Nam YK, Noh JK, Cho YS, Cho HJ, Cho KN, Kim CG, et al. Dramatically accelerated growth and extraordinary gigantism of transgenic mud loach, Misgurnus mizolepis. Transgenic Res. 2001;10:353–62.
Figueiredo MA, Lanes CFC, Almeida DV, Proietti MC, Marins LF. The effect of GH overexpression on GHR and IGF-I gene regulation in different genotypes of GH-transgenic zebrafish. Comp Biochem Physiol Part D Genomics Proteomics. 2007;2(3):228–33.
Raven PA, Uh M, Sakhrani D, Beckman BR, Cooper K, Pinter J, et al. Endocrine effects of growth hormone overexpression in transgenic coho salmon. Gen Comp Endocrinol. 2008;159(1):26–37.
Oakes JD, Higgs DA, Eales JG, Devlin RH. Influence of ration level on the growth performance and body composition of non-transgenic and growth-hormone-transgenic coho salmon (Oncorhynchus kisutch). Aquaculture. 2007;265(1–4):309–24.
Campbell B, Dickey JT, Beckman BR, Young G, Pierce A, Fukada H, et al. Previtellogenic oocyte growth in salmon: relationships among body growth, plasma insulin-like growth Factor-1, estradiol-17beta, follicle-stimulating hormone and expression of ovarian genes for insulin-like growth factors, Steroidogenic-Acute Regulatory P. Biol Reprod. 2006;75(1):34–44.
Devlin RH, Andrade MD, Uh M, Biagi CA. Population effects of growth hormone transgenic coho salmon depend on food availability and genotype by environment interactions. Proc Natl Acad Sci U S A. 2004;101(25):9303–8.
Sundström LF, Lõhmus M, Tymchuk WE, Devlin RH. Gene-environment interactions influence ecological consequences of transgenic animals. Proc Natl Acad Sci U S A. 2007;104(10):3889–94.
Rise ML, Douglas SE, Sakhrani D, Williams J, Ewart KV, Rise M, et al. Multiple microarray platforms utilized for hepatic gene expression profiling of GH transgenic coho salmon with and without ration restriction. J Mol Endocrinol. 2006;37:259–82.
Devlin RH, Sakhrani D, Tymchuk WE, Rise ML, Goh B. Domestication and growth hormone transgenesis cause similar changes in gene expression in coho salmon (Oncorhynchus kisutch). Proc Natl Acad Sci U S A. 2009;106(9):3047–52.
Devlin RH, Sakhrani D, White S, Overturf KE. Effects of domestication and growth hormone transgenesis on mRNA profiles in rainbow trout (Oncorhynchus mykiss). J Anim Sci. 2013;11:5247–58.
Roberts SB, McCauley LAR, Devlin RH, Goetz FW. Transgenic salmon overexpressing growth hormone exhibit decreased myostatin transcript and protein expression. J Exp Biol. 2004;207(21):3741–8.
Kim JH, Leggatt RA, Chan M, Volkoff H, Devlin RH. Effects of chronic growth hormone overexpression on appetite-regulating brain gene expression in coho salmon. Mol Cell Endocrinol. 2015;413:178–88.
Eisen EJ, Fortman M, Chen WY, Kopchick JJ. Effect of genetic background on growth of mice hemizygous for wild-type or dwarf mutated bovine growth hormone transgenes. Theor Appl Genet. 1993;87:161–9.
Fleming IA, Agustsson T, Finstad B, Johnsson JI, Björnsson BT. Effects of domestication on growth physiology and endocrinology of Atlantic salmon (Salmo salar). Can J Fish Aquat Sci. 2002;59:1323–30.
Tymchuk WE, Beckman BR, Devlin RH. Altered expression of growth hormone/insulin-like growth factor I axis hormones in domesticated fish. Endocrinology. 2009;150:1809–16.
Almeida DV, de MG MC, Figueiredo MA, Lanes CFC, Bianchini A, Fernando ML. Growth hormone transgenesis affects osmoregulation and energy metabolism in zebrafish (Danio rerio). Transgenic Res. 2013;22(1):75–88.
Bystriansky JS, Clarke WC, Alonge MM, Judd SM, Schulte PM, Devlin RH. Salinity acclimation and advanced parr-smolt transformation in growth-hormone transgenic coho salmon (Oncorhynchus kisutch). Can J Zool. 2017;95(9):633–43.
Kim JH, Balfry S, Devlin RH. Disease resistance and health parameters of growth-hormone transgenci and wild-type coho salmon, Oncorhynchus kisutch. Fish Shellfish Immunol. 2013;34:1553–9.
Alzaid A, Kim JH, Devlin RH, Martin SAM, Macqueen DJ. Growth hormone transgenesis in coho salmon disrupts muscle immune function impacting cross-talk with growth systems. J Exp Biol. 2018. https://doi.org/10.1242/jeb.173146.
Shimizu M, Dickhoff WW. Circulating insulin-like growth factor binding proteins in fish: their identities and physiological regulation. Gen Comp Endocrinol. 2017;252:150–61.
Nornberg BF, Figueiredo MA, Marins LF. Expression profile of IGF paralog genes in liver and muscle of a GH-transgenic zebrafish. Gen Comp Endocrinol. 2016;226:36–41.
Chen J, Cao M, Zhang A, Shi M, Tao B, Li Y, et al. Growth hormone overexpression disrupts reproductive status through actions on Leptin. Front Endocrinol (Lausanne). 2018;9(March):1–10.
Panserat S, Médale F, Brèque J, Kaushik S. Lack of significant long-term effect of dietary carbohydrates on hepatic glucose-6-phosphatase expression in rainbow trout (Oncorhynchus mykiss ). J Nutr Biochem. 2000;11:22–9.
Higgs DA, Sutton JN, Kim H, Oakes JD, Smith J, Biagi C, et al. Influence of dietary concentrations of protein, lipid and carbohydrate on growth, protein and energy utilization, body composition, and plasma titres of growth hormone and insulin-like growth factor-1 in non-transgenic and growth hormone transgenic coho s. Aquaculture. 2009;286(1–2):127–37.
Abernathy J, Panserat S, Welker T, Plagne-Juan E, Sakhrani D, Higgs DA, et al. Food shortage causes differential effects on body composition and tissue-specific gene expression in salmon modified for increased growth hormone production. Mar Biotechnol. 2015;17(6):753–67.
Panserat S, Kamalam BS, Fournier J, Plagnes-Juan E, Woodward K, Devlin RH. Glucose metabolic gene expression in growth hormone transgenic coho salmon. Comp Biochem Physiol A Mol Integr Physiol. 2014;170:38–45.
Miyakawa M, Tsushima T, Murakami H, Isozaki O, Demura H, Tanaka T. Effect of growth hormone (GH) on serum concentrations of leptin: study in patients with acromegaly and GH deficiency. J Clin Endocrinol Metab. 1998;83(10):3476–9.
Kim JH, Chatchaiphan S, Crown MT, White SL, Devlin RH. Effect of growth hormone overexpression on gastric evacuation rate in coho salmon. Fish Physiol Biochem. 2018;44(1):119–35.
Fuentes EN, Kling P, Einarsdottir IE, Alvarez M, Valdés JA, Molina A, et al. Plasma leptin and growth hormone levels in the fine flounder (Paralichthys adspersus) increase gradually during fasting and decline rapidly after refeeding. Gen Comp Endocrinol. 2012;177(1):120–7.
Gorissen M, Flik G. Leptin in teleostean fish, towards the origins of leptin physiology. J Chem Neuroanat. 2014;61–62:200–6.
Copeland DL, Duff RJ, Liu Q, Prokop J, Londraville RL. Leptin in teleost fishes: an argument for comparative study. Front Physiol. 2011;2:1–11.
Kodama M, Naish KA, Devlin RH. Influence of a growth hormone transgene on the genetic architecture of growth-related traits: a comparative analysis between transgenic and wild-type coho salmon. Evol Appl. 2018;11:1886–900 accepted.
Causey DR, Pohl MAN, Stead DA, Martin SAM, Secombes CJ, Macqueen DJ. High-throughput proteomic profiling of the fish liver following bacterial infection. BMC Genomics. 2018;19:1–17.
Houde ALS, Schulze AD, Kaukinen KH, Strohm J, Patterson DA, Beacham TD, et al. Transcriptional shifts during juvenile Coho salmon (Oncorhynchus kisutch) life stage changes in freshwater and early marine environments. Comp Biochem Physiol Part D Genomics Proteomics. 2019;29:32–42.
Rebl A, Goldammer T. Under control: the innate immunity of fish from the inhibitors’ perspective. Fish Shellfish Immunol. 2018;77(April):328–49.
Reinecke M. Influences of the environment on the endocrine and paracrine fish growth hormone-insulin-like growth factor-I system. J Fish Biol. 2010;76(6):1233–54.
Dickson SP, Wang K, Krantz I, Hakonarson H, Goldstein DB. Rare Variants Create Synthetic Genome-Wide Associations. PLoS Biol. 2010;8(1):e1000294.
Korte A, Farlow A. The advantages and limitations of trait analysis with GWAS : a review. Plant Methods. 2013;9(29):1–9.
Habib A, Powell JJ, Stiller J, Liu M, Shabala S, Zhou M, et al. A multiple near isogenic line (multi-NIL) RNA-seq approach to identify candidate genes underpinning QTL. Theor Appl Genet. 2018;131(3):613–324.
Birchler JA. Aneuploidy in plants and flies: the origin of studies of genomic imbalance. Semin Cell Dev Biol. 2013;24:315–9.
Devlin RH, Holm DG, Grigliatti TA. The influence of whole-arm trisomy on gene expression in drosophila. Genetics. 1988;118:87–101.
Maroni G, Plaut W. Dosage compensation in Drosophila melanogaster triploids. II. Glucose-6-phosphate dehydrogenase activity. Genetics. 1973;74:331–42.
Gordon RR, Hunter KW, La Merrill M, Sorensen P, Threadgill DW, Pomp D. Genotype x diet interactions in mice predisposed to mammary cancer: II. Tumors and metastasis. Mamm Genome. 2008;19(3):179–89.
Saito H, Suzuki N. K-rasG12V mediated lung tumor models identified three new quantitative trait loci modifying events post-K-ras mutation. Biochem Biophys Res Commun. 2014;452(4):1067–70.
Kantachuvesiri S, Haley CS, Fleming S, Kurian K, Whitworth CE, Wenham P, et al. Genetic mapping of modifier loci affecting malignant hypertension in TGRmRen2 rats. Kidney Int. 1999;56(2):414–20.
Zhu S, Walker R, Boerma HR, All JN, Parrott WA. Effects of defoliating insect resistance QTLs and a cry1Ac transgene in soybean near-isogenic lines. Theor Appl Genet. 2008;116(4):455–63.
Devlin RH, Sakhrani D, Biagi CA, Smith JL, Fujimoto T, Beckman BR. Growth and endocrine effect of growth hormone transgene dosage in diploid and triploid coho salmon. Gen Comp Endocrinol. 2014;196:112–22.
Martin C, McGowan R. Genotype specific modifiers of transgene methylation and expression in the zebrafish, Danio rario. Genet Res (Camb). 1995;65:21–8.
Sun F-L, Cuaycong MH, Craig CA, Wallrath LL, Locke J, Elgin SCR. The fourth chromosome of Drosophila melanogaster: interspersed euchromatic and heterochromatic domains. Proc Natl Acad Sci U S A. 2000;97(10):5340–5.
Phillips R, Devlin RH. Integration of growth hormone gene constructs in transgenic strains of coho salmon (Oncorhynchus kisutch) at centromeric or telomeric sites. Genome. 2010;53(1):79–82.
Uh M, Khattra J, Devlin RH. Transgene constructs in coho salmon (Oncorhynchus kisutch) are repeated in a head-to-tail fashion and can be integrated adjacent to horizontally-transmitted parasite DNA. Transgenic Res. 2006;15(6):711–27.
Ahrens RNM, Devlin RH. Standing genetic variation and compensatory evolution in transgenic organisms: a growth-enhanced salmon simulation. Transgenic Res. 2011;20(3):583–97.
McClelland EK, Chan M, Sakhrani D, Devlin RH. Identification of SNPs associated with transgenic and sex phenotypes in coho salmon (Oncorhynchus kisutch). Conserv Genet Resour. 2016;8(4):569–80.
Du SJ, Devlin RH, Hew C-L. Genomic structure of growth hormone genes in Chinook salmon (Oncorhynchus tshawytscha): presence of two functional genes, GH-I and GH-II, and a male-specific psuedogene, GH-PSI. DNA Cell Biol. 1993;12:739–51.
Fitzpatrick JL, Akbarashandiz H, Sakhrani D, Biagi CA, Pitcher TE, Devlin RH. Cultured growth hormone transgenic salmon are reproductively out-competed by wild-reared salmon in semi-natural mating arenas. Aquaculture. 2011;312(1–4):185–91.
Li Y, Levran O, Kim J, Zhang T, Chen X, Suo C. Extreme sampling design in genetic association mapping of quantitative trait loci using balanced and unbalanced case- control samples. Sci Rep. 2019;9:15504.
Bjornland T, Bye A, Ryeng E, Wisloff U, Langaas M. Powerful extreme phenotype sampling designs and score tests for genetic association studies. Stat Med. 2018;37(28):4234–51.
Berndt SI, Gustafsson S, Magi R, Ganna A, et al. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013;45(5):501–12.
Li R, Yu C, Li Y, Lam T-W, Yiu S-M, Kristiansen K, et al. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 2009;25(15):1966–7.
Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–8.
Tarazona S, Furió-Tarí P, Turrà D, Di Pietro A, Nueda MJ, Ferrer A, et al. Data quality aware analysis of differential expression in RNA-seq with NOISeq R/Bioc package. Nucleic Acids Res. 2015;43(21):e140.
Conesa A, Götz S, García-Gómez JM, Terol J, Talón M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–6.
Warnes G, Bolker B, Bonebakker L, Gentlemen R, Huber W, Liaw A, et al. gplots: Various R programming tools for plotting data. R package version 2.11.0. http://CRAN.R-project.org/package=gplots. 2012.
Livak KJ, Schmittgen TD. Analysis of relatvie gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8.
R Core Team. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2012. ISBN 3–900051–07-0, URL http://www.R-project.org/.
Fox J, Weisberg S. An {R} companion to applied regression. 2nd ed. Thousand Oaks: Sage; 2011.
Rosner B. Percentage points for a generalized ESD many-outlier procedure. Technometrics. 1983;25(2):165–72.
Lu F, Lipka AE, Glaubitz JC, Elshire RJ, Chemey JH, Casler MD, et al. Switchgrass genomic diversity, ploidy and evolution: novel insights from a network-based SNP discovery protocol. PLoS ONE Genet. 2013;9(1):e1003215.
Glaubitz JC, Casstevens TM, Lu F, Harriman J, Elshire RJ, Sun Q, et al. TASSEL-GBS: a high capacity genotyping by sequencing analysis pipeline. PLoS One. 2015;9(2):e90346.
Froese R. Cube law, condition factor and weight-length relationships: history, meta-analysis and recommendations. J Appl Ichthyol. 2006;22:241–53.
Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience. 2015;4(1):1–16.
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdas Y, Buckler ES. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics. 2007;23:2633–5.
Benjamini Y, Hochberg Y. Controlling the false discovery rate - a practical and powerful approach to multiple testing. J R Stat Soc Ser B Methodol. 1995;57(1):289–300.
Acknowledgements
no additional acknowledgements at this time.
Funding
Funding for this research was provided by Fisheries and Oceans Canada (DFO-61740).
Author information
Authors and Affiliations
Contributions
EKM analyzed and interpreted data, generated figures, and wrote the first draft of the manuscript; MTC and XL analyzed and interpreted data and generated figures; DS, MTC, FV, and JK generated data for the study; RHD conceived of the experiment, acquired funding, interpreted data, and assisted with drafting of the manuscript. DDH and all other authors provided editorial comments on the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Animal husbandry was conducted under Animal Use Protocol 11–020 issued from the DFO Pacific Region Animal Care Committee in compliance with the Canadian Council on Animal Care guidelines.
Consent for publication
Not Applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Table S1.
Genes differentially expressed between large transgenic (TLarge) and non-transgenic (NTLarge) fish with a ≥ 3 fold change in expression level. Negative log2Ratios indicate genes that had higher expression levels in NTLarge while positive values are genes with higher expression levels in TLarge. Table S2. Genes differentially expressed between small transgenic (TSmall) and non-transgenic (NTSmall) fish with a ≥ 3 fold change in expression level. Negative log2Ratios indicate genes that had higher expression levels in NTSmall while positive values are genes with higher expression levels in TSmall. Table S3. Genes differentially expressed between large (TLarge) and small (TSmall) transgenic (T) fish with a ≥ 3 fold change in expression level. Negative log2Ratios indicate genes that had higher expression levels in TLarge while positive values are genes with higher expression levels in TSmall. Table S4. Genes differentially expressed between large (NTLarge) and small non-transgenic (NTSmall) fish with a ≥ 3 fold change in expression level. Negative log2Ratios indicate genes that had higher expression levels in NTLarge while positive values are genes with higher expression levels in NTSmall. Table S5. Gene Ontology (GO) Biological Process categories for differentially expressed genes (DEGs) identified in comparisons between transgenotypes within size groups (large non-transgenic, NTLarge; large transgenic, TLarge; small non-transgenic, NTSmall; and small transgenic, TSmall). The total number of DEGs found in GO categories in the whole genome are given as well as observed (obs) and expected (exp) GO terms represented in the study dataset. The c2 value for GO terms where the observed number of DEGs differed significantly from expected (p < 0.05) is given (c2), along with deviations (dev) from expected as observed - expected, and the relative percent (rel %) of the genome represented by that GO-term. Table S6. Gene Ontology (GO) Biological Process categories for the differentially expressed genes identified in comparisons between size groups within transgenotypes (large non-transgenic, NTLarge; large transgenic, TLarge; small non-transgenic, NTSmall; and small transgenic, TSmall). The total number of DEGs found in GO categories in the whole genome are given as well as observed (obs) and expected (exp) GO terms represented in the study dataset. The c2 value for GO terms where the observed number of DEGs differed significantly from expected (p < 0.05) is given (c2), along with deviations (dev) from expected as observed - expected, and the relative percent (rel %) of the genome represented by that GO-term. Table S7. SNP markers that were significantly associated with expression levels of assessed genes or phenotypic traits in different size groups of either Transgenic (T) or Non-Transgenic (NT) fish. P-values from the GLM analysis are given along with the adjusted q value (FDR = 0.05); significant q values are shown in bold. Traits are as for Table 1; linkage groups (LG) with NCBI Reference numbers (REF), and position (POS) on the linkage group are given, along with the Major and Minor alleles for each SNP. a: SNP marker associated with multiple traits; b: the same marker associated with different traits in T and NT; c: the same marker associated with different traits between size classes. Table S8. The number of transgenic and non-transgenic coho salmon with their mean (standard deviation) weight, length and condition factor (CF) at the time of sampling. Table S9. Gene names and products for traits analysed by qPCR and Open Array. Oligo sequences of forward (F) and reverse (R) primers and probes are given along with gene locations and NCBI transcriptome IDs (for Open Array genes). General functions are indicated.
Additional file 2:
Figure S1. Gene Ontology (GO) Biological Process categories for the differentially expressed genes (DEGs) identified in comparisons between transgenotypes (transgenic fish, T, and non-transgenic fish, NT) for large and small fish. Figure S2. Box plots represent the median and 25% quantiles for relative gene expression for large and small transgenic (TLarge; TSmall) and nontransgenic (NT Large and NTSmall) fish. Groups with the different letters are significantly different (Tukey HSD, p < 0.05). Gene abbreviations are as for Table S2. Figure S3. Distribution of SNPs across Coho Salmon linkage groups. Figure S4. Histogram of lengths (cm) for transgenic (T) and non-transgenic (NT) fish at the time of sampling. A subset of these fish were used for further analysis as described in the text. Figure S5. Manhattan plots of SNP number and linkage group (1–30, and unassigned (U)) for expression traits in transgenic (T) and non-transgenic (NT) fish. Significant SNPs (FDR = 0.05) are indicated by red triangles; dotted line indicates significant q value. Name abbreviations are as for Table S2. Figure S6. Manhattan plots of SNP number and linkage group (1–30, and unassigned (U)) for weight, length and condition factor in transgenic (T) and non-transgenic (NT) fish. Significant SNPs (FDR = 0.05) are indicated by red triangles; the dotted line indicates the significant q value.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
McClelland, E.K., Chan, M.T.T., Lin, X. et al. Loci associated with variation in gene expression and growth in juvenile salmon are influenced by the presence of a growth hormone transgene. BMC Genomics 21, 185 (2020). https://doi.org/10.1186/s12864-020-6586-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-020-6586-0