
Abstract
Background
The power of genome-wide association studies (GWAS) is often limited by the sample size available for the analysis. Milk fatty acid (FA) traits are scarcely recorded due to expensive and time-consuming analytical techniques. Combining multi-population datasets can enhance the power of GWAS enabling detection of genomic region explaining medium to low proportions of the genetic variation. GWAS often detect broader genomic regions containing several positional candidate genes making it difficult to untangle the causative candidates. Post-GWAS analyses with data on pathways, ontology and tissue-specific gene expression status might allow prioritization among positional candidate genes.
Results
Multi-population GWAS for 16 FA traits quantified using gas chromatography (GC) in sample populations of the Chinese, Danish and Dutch Holstein with high-density (HD) genotypes detects 56 genomic regions significantly associated to at least one of the studied FAs; some of which have not been previously reported. Pathways and gene ontology (GO) analyses suggest promising candidate genes on the novel regions including OSBPL6 and AGPS on Bos taurus autosome (BTA) 2, PRLH on BTA 3, SLC51B on BTA 10, ABCG5/8 on BTA 11 and ALG5 on BTA 12. Novel genes in previously known regions, such as FABP4 on BTA 14, APOA1/5/7 on BTA 15 and MGST2 on BTA 17, are also linked to important FA metabolic processes.
Conclusion
Integration of multi-population GWAS and enrichment analyses enabled detection of several novel genomic regions, explaining relatively smaller fractions of the genetic variation, and revealed highly likely candidate genes underlying the effects. Detection of such regions and candidate genes will be crucial in understanding the complex genetic control of FA metabolism. The findings can also be used to augment genomic prediction models with regions collectively capturing most of the genetic variation in the milk FA traits.
Similar content being viewed by others

Background
Several fatty acids (FAs) of varying carbon chain length (C4-C22) and degree of saturation are present in milk. FAs in milk can originate either through direct transport from the rumen to the mammary gland via the blood, or from de novo synthesis in the mammary gland from acetate, beta-hydroxybutyrate [1] and propionate [2, 3]. Additionally, FAs in the mammary gland can originate from mobilization of body fat reserves. The short and intermediate chain FAs are mostly synthesized de novo in the mammary gland with the exception of C16:0, of which approximately 50% is assumed to be synthesized de novo. The long chain FAs, and approximately 50% of C16:0, are suggested to be derived from blood lipids originating from the diet [4] and mobilization of body fat reserves [1]. Considerable genetic variation has been reported for the fat composition of milk [5, 6]. Part of this genetic variation is attributed to polymorphisms in genes with major effects such as DGAT1 and SCD1 [7]. In addition, several regions on the bovine genome with suggestive effects on milk fat composition have been reported from GWAS [8,9,10]. Identified genes and genomic regions explain a fraction of 3.6 to 53% of the total genetic variation in different milk FA traits [8, 11]. Detection of additional genomic regions requires availability of larger sample size and high-density markers. GC analysis, the current method of choice to quantify milk FA, requires expensive equipment and is time-consuming, thus limiting measurement of the traits to experimental scale.
GWAS for the milk FA traits so far relied on such smaller datasets within different dairy cattle breeds/populations. An option to deal with the limitation in sample size could be to combine the available smaller datasets across populations for joint GWAS. Such analyses can increase detection power depending on the genetic distance between the populations and the marker density [12]. In this study, we undertake multi-population GWAS for milk FA traits by combining samples from Chinese, Danish and Dutch Holstein Friesians with HD genotypes available. Previous studies show high consistency in the linkage disequilibrium (LD) and minor allele frequencies between the populations [13, 14]. Thus, combining samples from these populations for joint GWAS might allow identification of genomic regions explaining even small proportions of the genetic variation in milk FA traits.
A hurdle is that due to the long range of LD in livestock breeds, GWAS often result in detection of large genomic regions [15] containing several positional candidate genes. Identifying the actual causative variants, therefore, requires additional evidence on top of the GWAS. Enrichment analysis is commonly undertaken in GWAS to prioritize positional candidate genes linked to significantly enriched pathways and gene ontology (GO) terms that are believed to be relevant to traits of interest. However, FA synthesis can take place in various mammalian tissues and thus further evidence is needed to determine whether such prioritized genes are relevant particularly to milk FA related mechanisms. Studies have been profiling differential expression of genes in the mammary tissues in various species [16, 17]. Information on expression status of genes in the mammary tissues can been used to further prioritize candidate genes linked to FA related pathways. Furthermore, the mammalian phenotype ontology [18], which provides annotation of mammalian phenotypes in the context of mutations, is increasingly becoming useful in fine-tuning the link between detected genes and phenotypes associated [19].
In this study, we implement GWAS for milk FA composition using multi-population dataset. Furthermore, we undertake post-GWAS analyses to identify, prioritize and functionally annotate genes within detected genomic regions using multiple information sources including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways, mammary gland gene expression status and information in the mammalian phenotype ontology database [18].
Results
Descriptive statistics and genetic parameters
Table 1 presents phenotypic means, additive genetic variances and heritability estimates of the FAs expressed as weight percentage of total fat and the desaturation indexes in the combined multi-population dataset. The 13 FAs studied together amounted to 87.6% of total fat. Of the studied FAs, C18:3n3 and CLA occurred at concentrations less than 1% of total fat in the milk samples. Other FAs including C15:0, C8:0, C14:1 and C16:1 also occurred at low concentrations of total fat (means = 1.09–1.49). Coefficients of variation (not shown) of the FA traits ranged between 0.06% (C18 index) and 0.43% (CLA). Heritability estimates in the studied FA traits ranged from low (0.18) for C18:2n6 to high (0.53) for C14 index. The dataset used in the current study comprises samples from the Chinese, Danish and Dutch Holstein population and details regarding descriptive statistics and genetic parameters within each population can be found in our previous study (Submitted).
Detected genomic regions
Our multi-population GWAS resulted in the detection of 56 genomic regions containing single nucleotide polymorphisms (SNPs) significantly associated with at least one of the studied FA traits (Table 2). Significant associations were detected on all chromosomes except BTA 18. Most of the FA traits showed significant associations with multiple genomic regions on several chromosomes; particularly for C10:0 (14 regions), C16:0 (12 regions), C16:1 (13 regions), C18:1c9 (11 regions) and C16 index (13 regions). Proportions of genetic variance explained by the lead SNPs in the detected regions ranged between 1.4 and 45.3% for the different FA traits studied.
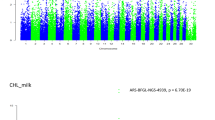
Peak sizes (highest –log 10 p-value) across FA traits ranged from a –log 10 p-value of 6.9 for C18:0 to a –log 10 p-value of 126 for C14 index. Figs. 1, 2, 3 and 4 present Manhattan plots for all FAs according to the different FA groups i.e., de novo FAs (Fig. 1), intermediate to long-chain saturated FAs (Fig. 2), the unsaturated FAs (Fig. 3), and desaturation indexes (Fig. 4). The strongest association for C8:0 (−log10 p-value = 11.39), C15:0 (−log10 p-value = 21), C16:0 (−log10 p-value = 58), C16:1 (−log10 p-value = 55), C18:1c9 (−log10 p-value = 46), C18:2n6 (−log10 p-value = 29), C18:3n3 (−log10 p-value = 24.8), CLA (−log10 p-value = 18.1) and C18 index (−log10 p-value = 19.3) was observed at two variants on BTA 14 (ARS-BFGL-NGS-4939 and BovineHD1400000216). This region (14a) was significantly associated with all studied FA traits except C12:0. The lead SNP in this region explained up to 34% of the genetic variation in C18:1c9 and C18:2n6. Two other regions on BTA 14 remained significantly associated with multiple FA traits after accounting for the fixed effect of the lead SNP from region 14a (ARS-BFGL-NGS-4939). The second region (14b) was also significantly associated with most FA traits except C12:0. The third region on BTA 14 (14c), was significantly associated with C14:1, C16:1, C14 index and C18 index. The lead SNP in this region explained 2.7% of the genetic variation in C18 index and 1.6% in C14 index.

Manhattan plots showing BTAs on the x-axis and -log 10-p values on the y-axis for the de novo synthesized FAs of C8:0 (a), C10:0 (b), C12:0 (c), C14:0(d). Red line indicates the significance threshold (log 10 p-value =5.0)

Manhattan plots with BTAs on the x-axis and -log 10-p values on the y-axis for the medium to long-chain FAs of C15:0 (a), C16:0 (b), C18:0 (c). y-axis for (b) has breaks at –log 10-p-value =15 to show only the highest values of those –log 10-p-value > 25 while keeping the visibility of smaller peaks. Red line indicates the significance threshold (log 10 p-value =5.0)

Manhattan plots showing BTAs on the x-axis and -log 10-p values on the y-axis for the unsaturated FAs of C14:1 (a), C16:1 (b), C18:1c9 (c), C18_2n6 (d), C18_3n3 (e), CLA (f). Y-axis breaks for (b) at –log 10-p-value =20 and for (a), (c) and (d) at –log 10-p-value = 15. Red line indicates the significance threshold (log 10 p-value =5.0)

Manhattan plots showing BTAs on the x-axis and -log 10-p values on the y-axis for the desaturation indexes: C14 index (a), C16 index (b), C18 index (c) with y-axis breaks at –log 10 p-values = 15 for (a) and (b). Red line indicates the significance threshold (log 10 p-value =5.0)
Strongest association for C10:0 (−log10 p-value = 24.3), C12:0 (−log10 p-value = 22) and C14:0 (24.2) was detected with two variants on BTA 19 (BovineHD1900014372 and BovineHD1900014348). Significant associations were also detected for C8:0, C16:0, C18:1c9, C14 index and C18 index with SNPs located between 37.3 to 61.3 Mbp on chromosome 19. Particularly for C14:0, 22.3% of the genetic variation was explained by the lead SNP in this region.
The strongest association for C14:1 (−log10 p-value = 98.8), C14 index (−log10 p-value = 126) and C16 index (−log10 p-value = 39.8) was found with SNPs on chromosome 26 (BovineHD2600005461). Significant associations were also detected for C8:0, C10:0, C12:0, C14:0, C16:0, C16:1, C18:0 and C18 index. The lead SNP in this region explained 39.0% of the genetic variation in C14:1.
Effects of lead SNPs in all the detected genomic regions are presented in Additional file 1. In general for most of the regions, directions of effects were opposite for the de novo synthesized FAs versus the long chain FAs.
Gene assignment and functional annotations
Several genes positioned within the detected genomic regions were retrieved from the ensemble database. These positional candidate genes were further prioritized using enrichment analyses implemented in the DAVID web platform (https://david.ncifcrf.gov), which resulted in different significantly enriched GO terms and KEGG pathways relevant to FA related mechanisms (Table 3).
Among the enriched GO terms and pathways were biosynthesis related, such as ‘GO:0006633~FA biosynthetic process’, binding and transport related, such as ‘GO:0008289~lipid binding’ and ‘GO:0010876~lipid localization’, and metabolism, such as ‘GO:0006631~FA metabolic process’ and ‘bta00564:Glycerophospholipid metabolism’.
Some among the set of genes in all significantly enriched pathways and GO terms (Additional file 2) were also found to be expressed in mammary tissues and epithelial cells across different species. Furthermore, some of the prioritized candidate genes were linked to abnormalities related to FA metabolism in the mammalian phenotype database including ‘increased circulating triglyceride levels’ (MP:0001552), ‘abnormal lipid homeostasis’ (MP:0002118) and ‘abnormal phospholipid level’ (MP:0004777).
Apart from genes, also non-coding genomic features such as micro RNAs were located within the detected genomic regions as presented on Additional file 3.
Discussion
Agreement between detected regions and previous reports
Our multi-population GWAS resulted in detection of large numbers of genomic regions significantly associated with at least one of the 16 milk FA traits studied, indicating the complexity of the milk FA synthesis pathways. Most of the detected genomic regions have been previously reported in connection to milk FA traits, e.g. genomic regions on BTA 14, BTA 19 and BTA 26 [8, 10, 20].
On BTA 14, our analysis indicates three distinct regions significantly associated with several FA traits. The first region is known to contain the DGAT1 gene, of which the effects are well established for multiple FA traits [21, 22]. The second region was previously reported to show significant associations with milk fat percentage [23]. The boundaries of these two regions (14a and 14b) are in close proximity of each other (1.5–5 Mbp and 5.2–20 Mbp) and the regions appear to be highly correlated in terms of associated FA traits and proportions of genetic variance explained for these traits. While our analysis indicates two distinctive regions, Bouwman et al. [8], based on part of the dataset used in our study, reported a single, broader region (0.0–26.3 Mbp) with significant associations to several FA traits. Our hypothesis is that different quantitative trait loci (QTL) underlie these two regions (14a and 14b) but that estimated effects of the QTLs could be confounded, because the high LD at the start of BTA 14 [24] makes it difficult to disentangle the effects of multiple QTL.
The third region on BTA 14 (44.7–49.9 Mbp) was exclusively associated with C14:1 and C16:1 as well as C14 index and C18 index. This region was previously reported for significant associations with C16:1 [8] and milk fat percentage [25]. The region contains the fatty acid binding proteins FABP4, FABP9 and FABP12 as well as the peripheral myelin protein (PMP2), enriching the GO terms of FA metabolic process (GO:0006631) and lipid binding activities (GO:0008289). A study by Nafikov et al. [26] reported a FABP4 haplotype negatively associated with saturated milk FAs and the ratio between saturated and unsaturated FAs while having positive effects on the unsaturated FAs. Marchitelli et al. [27] also reported that the FABP4 affected the ratio of monounsaturated/saturated FA in milk. Additionally, variation in FABP4 is reported to affect other milk production traits such as milk yield [28]. Therefore, results of our analysis and previous studies suggest a role of this region in desaturation of C14:0, C16:0 and C18:0 with the FABP4 as the most likely candidate gene.
Broader regions were detected on BTA 19 (37.3–61.3 Mbp) and BTA 26 (2.9–43.0 Mbp). The genes FASN on BTA 19 [29] and SCD1 on BTA 26 [30] have previously been suggested as the likely candidate genes for FA traits. However, our enrichment analysis indicate additional genes in these regions connected to important FA metabolism processes including the ACLY, STAT5a, PRKAA1, GH on BTA 19 and ELOVL3, ACLS5 on BTA 26. Significant associations were previously reported between variants within some of these genes and some milk FA traits [11, 31].
In our study, more FA traits have been found to have significant associations with the DGAT1 and SCD1 regions than previous GWAS using different parts of the multi-population dataset used in the current analysis [8,9,10,11, 14]. These previous studies might not be considered as independent of the current analysis; however, more associations in the current analysis can be an indication of improved detection power from combining the populations. This was also demonstrated in our previous study (Submitted) in which results of population-specific analyses versus multi-population joint GWAS were compared. Effects of the DGAT1 (ARS-BFGL-NGS-4939) and SCD1 (BovineHD2600005461) loci were similar in direction and highly correlated between the three populations but estimated effects in the Chinese sample were consistently lower across the FAs compared to the Dutch and Danish Holstein samples.
The three regions detected on BTA 5 overlap with previously reported regions for milk FA traits [8, 9, 32]. For region 5c, MGST1 was suggested as the most likely candidate gene [32]. In our analysis, the lead SNP in the region was located within the MGST1 gene. However, our enrichment analysis did not establish any connection to MGST1 with significantly enriched FA related GO terms and pathways. Additionally, PLBD1 and LRP6 genes were connected to several pathways including lipid localization (GO:0010876) and transport (UP_KEYWORDS) suggesting that the significant association observed in the region with 10 FA traits might not be limited to the MGST1 effect.
The region on BTA 13 was previously detected in the Dutch Holstein population [8, 11] and in Danish Jersey [9] with both studies suggesting the ACSS2 as the highly likely candidate gene. Meanwhile, using infrared (IR) predicted phenotypes for the de novo FAs, Olsen et al. [33] suggested that the NCOA6, not the ACSS2, is responsible for significant associations in the region. Our enrichment analysis however links ACSS2 with several significantly enriched pathways while no such links were established for the NCOA6 gene.
Similarly, the first region on BTA 15 (27.2–31.2 Mbp) has been reported in previous studies including a joint Chinese-Danish Holstein population [14]. Several genes enriching FA related pathways were detected in the region including APOA1, APOA4, APOA5, and DPAGT1. The apolipoproteins APOA1/4/5 enriched glycerolipid metabolic process (GO:0046486), fat digestion and absorption (bta04975) as well as negative regulation of FA biosynthetic process (GO:0045717) while the DPGAT1 was involved in lipid biosynthetic process (GO:0008610). The strongest associations observed in the region were between C18.0 and variants within the alipoprotein genes, which showed opposite direction of effects on C10:0 and C14:0. Although effects were not significant, the lead SNP in the region also showed moderate effects on the other de novo FAs including C8:0 (−log 10 p-value = 2.96) and C12:0 (−log 10 p-value = 2.96) with direction of effects similar to C10:0 and C14:0. The alipoproteins APOA1/4/5 are thus collectively suggested as the candidates underlying the strong effect on C18:0 observed in the region. The opposing effects on the de novo FAs might be directly through involvement of the alipoproteins in negative regulation of FA biosynthesis or indirectly through the effect on C18:0, which suppresses de novo synthesis.
The two regions detected on BTA 17 are also in agreement with previous findings. The regions detected by Bouwman et al. (2012) [8] (15.0–23.9 Mbp) and Li et al., [10] (19.5–22.5 Mbp) overlap with the first region (17a) detected in our study. In the region, MGST2 significantly enriched GO terms that included FA metabolic (GO:0006631) and biosynthetic (GO:0006633) processes. The MGST2 is previously linked to intramuscular FA composition in pigs [34] and shown to be expressed in all stages of lactation in humans [17]. Therefore, the MGST2 is suggested as the likely candidate gene underlying effects on the first region of BTA 17. Using a subset of the dataset used in the current study to fine map BTA 17, Duchemin et al. [35] suggested the LARP1B as a primary candidate gene in the second region (17b). However, our enrichment analysis did not result in significant enrichment of any of the FA pathways and ontology terms for genes in the region.
Some of the regions detected in our analysis overlap with results from some of the recently published GWAS that are based on IR predicted FA phenotypes [33, 36]. Interestingly, some of the well-established genomic regions in connection to GC-based FA traits, which were also detected in our analysis, have not been found to have significant associations with any of the milk FA phenotypes in these studies. For instance, GWAS by Olsen et al. [33] and Knutsen et al. [36] using the IR predicted FA phenotypes in Nordic Red cattle did not detect any significant association in the DGAT1 and SCD1 regions. Lack of segregation of the A variant of the DGAT1 K232A polymorphism has been suggested as the potential reason for the lack of association in the DGAT1 region. Additionally, Wang et al. [37] showed that the SCD1 polymorphism did not significantly affect any of the milk IR wavenumbers in samples from the Dutch Holstein population. These findings suggest that IR predicted FA phenotypes are not suitable for GWAS. While some FAs can be accurately predicted based on IR [38], low prediction accuracies [39] and low genetic correlations with GC measured FA [40] have been reported for other FAs. Especially FAs found in low concentrations in milk were shown to have low IR prediction accuracies [41]. Apparently, the power to detect QTL can be severely restricted by the IR prediction accuracy.
Novel genomic regions and candidate genes
Of the 56 genomic regions significantly associated with at least one FA trait in this study, regions located on BTA 2, 3 10, 11, 12 and 21 appear to be novel regions that have not been previously connected to milk FA traits. The lead SNPs in these regions explained between 1.4 and 5% of the genetic variation in at least one of the FA traits studied.
BTA 2
Two genes retrieved for region 2a enriched GO terms related to fatty acids. The OSBPL6 gene belonging to the oxysterol-binding protein (OSBP) family, a group of intracellular lipid receptors, is shown to be involved in lipid binding (GO:0008289) and transport (UP_KEYWORDS) processes. The OSBPL6 gene is found to be expressed in the human mammary gland during several stages of lactation [17]. The human OSBPL6 gene is also shown to have a binding site for miR-33a/b [42], which is a microRNA shown to have targeting effects on genes regulating β-oxidation of FAs [43], leading to significantly lower levels of β-hydroxybutyrate [44]. Another gene located in the region (AGPS) also enriched GO terms related to FA synthesis including lipid biosynthesis process (GO:0008610). In the mammalian phenotype database, mutation in the AGPS gene in mice has been linked to abnormal lipid levels (MP:0001547), which is a rather broad term in the database referring to any anomaly in the concentrations of fat-soluble substances in the body, including circulating triglyceride and free FAs. Thus, our enrichment analysis indicate that both the OSBPL6 and AGPS might have roles on de novo synthesis of FAs. Pattern of SNP effects in the region is also in agreement with enrichment analysis such that strongest association was estimated with C8:0 and C10:0 while moderate, but not significant effect was measured for C12:0 and C14:0 (−log 10 p-value = 4.2). Opposing direction of the lead SNP effect were also observed for the de novo synthesized FAs, except C15:0, on the one hand and most of the long chain FAs on the other (Fig. 5a). Therefore, both the OSBPL6 and AGPS are considered as likely candidates in the region. .

Effects of lead SNPs on regions 2a (a), 3 (b), 10b (c), 11b (d), 12b (e) and 21 (f) standardized by dividing the SNP effects with standard deviation of the respective FA trait
BTA 3
On the detected novel region of BTA 3, the prolactin releasing hormone (PRLH) was shown to be involved in FA metabolic process (GO:0006631). Mutations on the PRLH gene in mice have been associated with increased circulating triglyceride levels (MP:0001552) and increased total body fat amount (MP:0010024) in the mammalian phenotype database. In mammals, the PRLH gene is known to stimulate prolactin release and regulate its expression. Prolactin, which is a polypeptide hormone, has been shown to induce lipogenesis in many tissues [45] and stimulate the expression of genes involved in milk protein synthesis and lipid metabolism [46,47,48]. Moreover, prolactin has been shown to have a wide-range of effects on lactation including growth and development of the mammary gland, promotion of milk synthesis and maintenance of milk secretion [49,50,51]. Therefore, the PRLH gene, through regulation of prolactin release might have effects on milk yield. The pattern of SNP effects in the region suggest a connection with the poly-unsaturated fatty acids (PUFAs) with strongest associations observed for C18:3n3 and CLA. The direction of effects of the lead SNP was similar for all unsaturated FAs as well as all the desaturation indexes, while opposing effects were estimated for the de novo synthesized FAs and C16:0 (Fig. 5b). C16:0 is shown to have strong negative genetic correlation with milk yield, while moderate positive correlations were reported for the PUFAs [5]. Therefore, the PRLH is suggested as the candidate gene in the region; the effect of which might be indirect through its effect on milk yield, affecting the concentration of the PUFAs and C16:0.
BTA 10
The second region on BTA 10 contains the solute carrier family 51-beta subunit (SLC51B) genewhich is implicated in lipid transport (UP_KEYWORDS) and localization (GO:0010876) processes. Pattern of effects in the region show strong effect on C14:1 and moderate effects on with C14 index (−log 10 p-value = 4.5) and C18 index (−log 10 p-value = 3.6) in direction opposite to the strong effect on C18:0 (Fig. 5c). This pattern suggests a reduction in desaturation when C18:0 increases. C18:0 in milk is largely derived via direct transport through the blood from the rumen where is it formed from bio-hydrogenation of dietary C18:2n6 and C18:3n3. Therefore, the effect of SLC51B is highly likely through its involvement in the FA transport processes. Dietary poly-unsaturated FAs, such as C18:2n6, are known to suppress SCD1 activity, thereby reducing its desaturation activity [52]. Thus, we hypothesize that SLC51B underlies the effect on C18:0, while observed opposite effects on the unsaturated FAs and desaturation indexes are rather due to the correlation in C18:0 in milk and dietary PUFA, which suppress desaturation.
BTA 11
Among the genes located in the first region of BTA 11, the ATP binding cassette subfamily G5 (ABCG5) and ABCG8 enriched several pathways and processes including fat digestion and absorption pathway (KEGG ~bta04975) and the GO terms of lipid localization (GO:0010876) and transport. In the mammalian phenotype database, the ABCG5 and ABCG8 genes are linked to increased circulating triglyceride level (MP:0001552), abnormal lipid homeostasis (MP:0002118) and abnormal phospholipid level (MP:0004777). In humans, mutations in ABCG5/8 have been linked to conditions characterized by abnormal accumulation of sterols in blood and tissues [53, 54] implicating them in lipid absorption and transport. The KEGG pathway for fat digestion and absorption involves absorption of lipid from the rumen to the blood stream and from the blood stream to the mammary gland. Viturro et al. [55] previously reported high expression levels of both ABCG5 and ABCG8 genes in bovine liver, mammary gland, digestive tract and blood samples. Expression of ABCG5/8 in bovine mammary gland might indicate that apart from absorption and transport of lipids from the digestive tract, ABCG5/8 might also be involved in the secretion of lipids from the mammary gland into the milk. Significant association in the region was limited to C16:0. Although not significant, this region was also associated with C16:1 (−log 10-pvalue = 3.8) and CLA (−log 10-pvalue = 2.2), with directions of effects on CLA opposite to the effects on C16:0, C16:1, and C16 index (Fig. 5d). The GO term of lipid localization and association almost exclusively with C16:0, which is one of the FAs that is highly mobilized from body reserves during negative energy balance, might also indicate a role in the mechanism of body fat reserve mobilization.
BTA 12
The dolichyl-phosphate beta glucosyltransferase (ALG5) gene located on the second region of BTA 12 was shown to enrich the lipid biosynthesis process (GO:0008610) and glycerolipid metabolic process (GO:0046486). The ALG5 gene has previously been shown to be differentially expressed during the different stages of lactation in bovine [16] and human [17]. Significant effects in the region were limited to C14:1. The lead SNP also showed moderate effect on C14 index (−log 10 p-value = 3.07) and C18:0 (−log 10 p-value = 3.43) where opposite direction of effects were observed for C18:0 (Fig. 5e). Therefore, the ALG5 is suggested as promising candidate for further characterization for potential role in desaturation process.
BTA 21
Significant associations were detected on BTA 21 with C10:0, C12:0, C14:0 and C18:1c9. Effects estimated for the lead SNP in the region were generally opposing directions for the de novo synthesized FAs and C16:0 versus for the long-chain FAs and desaturation indexes (Fig. 5f). Significant associations have previously been reported in the region with milk and milk protein yield [56] as well as fertility traits in cows [57]. However, our enrichment analysis show no gene implicated in the significantly enriched pathways and Go terms. Despite lack of genes implicated on FA related pathways, moderate effects observed for multiple traits in the region are of particular interest. QTL detected through GWAS might be located in non-coding regions. Such QTLs might be involved in regulation of expression of other genes affecting the traits of interest. Therefore, the region might be of interest for eQTL based GWAS in milk FA traits.
Regulatory elements within detected genomic regions
Apart from coding genes, retrieved genes from the detected regions included regulatory elements, most commonly microRNAs (miRNAs). MiRNAs are small RNAs that regulate the expression of complementary messenger RNAs [58]. Several studies have reported possible roles of miRNAs in lipid and fatty acid metabolisms and in mammary gland development and lactation in several species [59,60,61]. Some of the miRNAs in the detected genomic regions in our study were previously linked to regulatory roles on genes related to FA metabolism and synthesis. Of these, bta-mir-27b, on BTA 8 (region 8b) was shown to target known FA synthesis genes such as FASN and SCD1 [62] as well as mRNAs involved in lipid metabolism [63] and shown to be highly expressed during different stages of bovine lactation [64]. Among the genes located on BTA 2 (region 2d), the bta-mir-26b was shown to be expressed in bovine milk cells and mammary gland [60]. Wang et al., [61] showed that downregulation of miR-26a/b and their host genes decreased the expression of genes related to fatty acid synthesis, including DGAT1 and SCD1.
Conclusion
Multi-population GWAS for GC-quantified FA traits resulted in the detection of 56 genomic regions significantly associated to at least one of the studied FAs, including novel regions explaining relatively smaller fractions of the genetic variation. Enrichment analysis of genes harbored in detected regions reveals promising candidate genes some of which have not been previously linked to milk FA traits, including OSBPL6 and AGPS on BTA 2, PRLH on BTA 3, SLC51B on BTA 10, ABCG5/8 on BTA 11 and ALG5 on BTA 12. Post-GWAS analyses using multiple data sources on pathways, ontology terms and tissue-specific gene expression status enabled prioritization of highly likely causative candidate genes among several positional candidates on detected regions. Use of such data in combination in combination with analysis of patterns of effects across the milk FA spectrum allowed linking some of the candidates to specific FA synthesis mechanisms. Detection of several novel regions and candidate genes will be contribute to the knowledge base on genetics underlying the bovine milk FA composition.
Methods
Animals and phenotypes
Test-day milk samples were obtained from 784 Chinese, 675 Danish and 1566 Dutch Holstein cows sampled from 18 herds in China, 22 herds across Denmark and 398 herds in the Netherlands. Stages of lactation of sampled cows ranged from 3 to 700 days in milk in the Chinese population, 9 to 481 days in milk in the Danish population and 60 to 278 days in milk in the Dutch Holstein cows. To standardize the samples from the three countries, only cows at days-in-milk of 60 and above were considered for the multi-population GWAS. Thus, 700 Chinese, 614 Danish and 1566 Dutch samples were available for the analysis. The reason to standardize the dataset by lactation stage is that the genetic determination of milk fat composition traits might be different in the early stages of lactation. There is evidence that effects of genes in early lactation differ from those later in lactation [65]. Excluding early lactation records can help eliminate this heterogeneity issue.
FA traits, including C8:0, C10:0, C12:0, C14:0, C14:1, C15:0, C16:0, C16:1, C18:0, C18:1c9, C18:2n6, C18:3n3 and C18:2 cis-9,trans-11 (CLA), were analyzed using the GC method. Details of the sample preparation for the GC analyses are as described by Li et al. [10] for Chinese samples, Poulsen et al. [66] for Danish samples and Stoop et al. [5] for Dutch samples. Genomic regions affecting the saturated FAs might show association to the unsaturated forms because the saturated form available for desaturation determines proportion of the unsaturated FAs. Hence, calculation of the desaturation indexes might allow detection of regions particularly associated with the desaturation process. Accordingly, desaturation indexes were calculated based on the FA measurements as: C14 index = C14:1/(C14:1 + C14:0) * 100; C16 index = C16:1/(C16:1 + C16:0) * 100 and C18 index = C18:1c9/ (C18:1c9 + C18:0) * 100.
Genotypes and imputation
High-density (HD) genotypes, real or imputed, were available for all cows used in the analyses. All cows in the Chinese dataset were initially genotyped using the BovineSNP50 Beadchip (50 K, Illumina). A sample population of 96 Chinese Holstein bulls, genotyped using the BovineHD Beadchip (777 K), was available as reference to impute the 50 K genotypes of the cows to HD.
Part of the Danish cows were genotyped using the BovineSNP50 Beadchip, while the remaining Danish cows were genotyped using the BovineHD Beadchip and used as reference to impute the 50 K genotypes of the first part of the Danish cows to HD as described in Gebreyesus et al. [67]. The Dutch cows were genotyped with a custom 50 K SNP Beadchip and subsequently imputed to HD as presented in detail in Duchemin et al. [68]. SNPs with minor allele frequencies (MAF) less than 0.05 or with a count of one of the genotypes less than 10 in each population were excluded from the association analysis. A total of 464,130 SNPs were available for the association analysis. The SNP positions were based on the bovine genome assembly UMD 3.1 [69].
Genome-wide association
A single-SNP association test was implemented using a mixed linear model in the GCTA program [70]. Association analysis was carried out using the following statistical model:
Where yijkl is the phenotype of cow l; μ is the fixed effect of mean; parityi and herdj are the fixed effects of parity and herd, respectively; b1 is the regression coefficient for DIM, DIMijkl is a covariate of days in milk; b2 is the allele substitution effect for SNP, SNPk is a covariate indicating the number of copies of a specific allele (0, 1 or 2) of the SNP; and animal is the random additive genetic effect. Animal effects were assumed to be distributed as \( \mathrm{N}\left(0,\mathbf{G}{\sigma}_a^2\right), \) where G is the genomic relationship matrix constructed using all HD genotypes but excluding the SNPs on the chromosome on which SNP k is located. Residuals were assumed to be distributed as: \( \mathrm{N}\left(0,\mathbf{I}{\sigma}_e^2\right), \) where I is the identity matrix. Since only cows with more than 60 days-in-milk were included in the analyses, a linear adjustment for DIM was sufficient. For the FA traits C18:2n6, C18:3n3 and CLA, log transformation was applied prior to the association analysis to account for observed heterogeneity of residual variances.
Significance thresholds were determined using a false discovery rate (FDR). Significance thresholds corresponding to FDR of 5% ranged for different FA from –log10 p-value = 3.4 to –log10 p-value = 5.0. We used a –log10 p-value of 5.0 as the genome-wide significance threshold for all FA composition traits.
Determining multiple regions on a chromosome
To determine if a region harbored one or more QTL, iterative approaches fitting the effect of SNPs with the highest –log 10 p-values were employed. In this approach, the SNP with the highest –log 10 p-value for the studied FA trait was considered as the lead SNP. The allelic dosage of such a lead SNP was then fitted as fixed effect for a second round of chromosome-wide analyses. If other SNPs, also significantly associated in the first round GWAS, were still found to have -log 10-pvalue > 5 in the second round analysis, the SNP with the highest –log 10 p-value in the second analysis was taken as the second lead SNP and its allelic dosage fitted as fixed effect for a third round of analysis. This procedure was iterated until no further SNP with -log 10-pvalue > 5 was observed. The SNPs that showed significant association in a round of GWAS but showed –log 10 p-value < 5 upon fitting the allelic dosage of the lead SNP were then considered as part of a region around that lead SNP. The position of the first and last such SNP before and after the lead SNP were considered as the boundaries of the region.
Estimation of genetic variances explained by SNPs
Genetic variance explained by the lead SNP in a region was calculated from the GWAS summary as: 2pqα2, where p and q are the allele frequencies and α is the allele substitution effect [71]. The proportion of total genetic variance explained by such a lead SNP was then calculated as:
where \( {\sigma}_a^2 \) is the additive genetic variance estimated using model 1 but without fitting fixed effects of SNP and using G constructed using all HD SNPs. Computation of genetic variance explained by SNPs from a GWAS summery might lead to overestimation of SNP effects [72] specially for small effect size SNPs that only just reach the significance threshold. Heritability (h2) estimates were computed as:
where \( {\sigma}_e^2 \) is the residual variance.
Gene assignment and enrichment analysis
Genes found within detected genomic regions were retrieved from the ensemble database using the BioMart web interface based on the UMD 3.1 bovine genome assembly (https://www.ensembl.org/biomart/martview). The DAVID functional annotation tool (https://david.ncifcrf.gov) was then used to analyze overrepresented GO biological terms, which included the terms cellular component (CC), molecular function (MF), biological process (BP) and the KEGG (Kyoto Encyclopedia of Genes and Genomes) pathways. Ontologies in the mammalian phenotype database were accessed and searched for genes connected to abnormalities relevant to FA metabolism using the Mouse Genome Informatics (MGI) web platform (http://www.informatics.jax.org/batch).
Abbreviations
- BTA:
-
Bos Taurus Autosome
- FA:
-
Fatty Acid
- FDR:
-
False Discovery Rate
- GC:
-
Gas Chromatography
- GO:
-
Gene Ontology
- GWAS:
-
Genome-Wide Association Study
- HD:
-
High-Density
- IR:
-
Infrared
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LD:
-
Linkage Disequilibrium
- PUFA:
-
Poly-Unsaturated Fatty Acid
- QTL:
-
Quantitative Trait Loci
- SNP:
-
Single Nucleotide Polymorphism
References
Bauman DE, Griinari JM. Nutritional regulation of milk fat synthesis. Annu Rev Nutr. 2003;23:203–27 Epub 2003 Feb 26. Review.
Massart-Leën AM, Roets E, Peeters G, Verbeke R. Propionate for fatty acid synthesis by the mammary gland of the lactating goat. J Dairy Sci. 1983;66(7):1445–54.
Vlaeminck B, Fievez V, Cabrita ARJ, Fonseca AJM, Dewhurst RJ. Factors affecting odd- and branched-chain fatty acids in milk: a review. Anim Feed Sci Technol. 2006;131:389–417.
Chilliard Y, Ferlay A, Mansbridge RM, Doreau M. Ruminant milk fat plasticity: nutritional control of saturated, polyunsaturated, trans and conjugated FA. Ann Zootech. 2000;49:181–205.
Stoop WM, van Arendonk JA, Heck JM, van Valenberg HJ, Bovenhuis H. Genetic parameters for major milk fatty acids and milk production traits of Dutch Holstein-Friesians. J Dairy Sci. 2008;91(1):385–94.
Krag K, Poulsen NA, Larsen MK, Larsen LB, Janns L, Buitenhuis B. Genetic parameters for milk fatty acids in Danish Holstein cattle based on SNP markers using a Bayesian approach. BMC Genet. 2013;14:79.
Schennink A, Heck JM, Bovenhuis H, Visker MH, van Valenberg HJ, van Arendonk JA. Milk fatty acid unsaturation: genetic parameters and effects of stearoyl-CoA desaturase (SCD1) and acyl CoA: diacylglycerol acyltransferase 1 (DGAT1). J Dairy Sci. 2008;91(5):2135–43.
Bouwman AC, Visker MH, van Arendonk JA, Bovenhuis H. Genomic regions associated with bovine milk fatty acids in both summer and winter milk samples. BMC Genet. 2012;13:93. https://doi.org/10.1186/1471-2156-13-93.
Buitenhuis B, Janss LL, Poulsen NA, Larsen LB, Larsen MK, Sørensen P. Genome-wide association and biological pathway analysis for milk-fat composition in Danish Holstein and Danish Jersey cattle. BMC Genomics. 2014;15:1112.
Li C, Sun D, Zhang S, Wang S, Wu X, Zhang Q, Liu L, Li Y, Qiao L. Genome wide association study identifies 20 novel promising genes associated with milk fatty acid traits in Chinese Holstein. PLoS One. 2014;9(5):e96186.
Bouwman AC, Bovenhuis H, Visker MH, van Arendonk JA. Genome-wide association of milk fatty acids in Dutch dairy cattle. BMC Genet. 2011;12:43.
Lund MS, Su S, Janss L, Guldbrandtsen B, Brøndum RF. Genomic evaluation of cattle in a multi-breed context. Livest Sci. 2014;166:101–10.
Zhou L, Ding X, Zhang Q, Wang Y, Lund MS, Su G. Consistency of linkage disequilibrium between Chinese and Nordic Holsteins and genomic prediction for Chinese Holsteins using a joint reference population. Genet Sel Evol. 2013;45:7.
Li X, Buitenhuis AJ, Lund MS, Li C, Sun D, Zhang Q, Poulsen NA, Su G. Joint genome-wide association study for milk fatty acid traits in Chinese and Danish Holstein populations. J Dairy Sci. 2015;98(11):8152–63.
de Roos AP, Hayes BJ, Spelman RJ, Goddard ME. Linkage disequilibrium and persistence of phase in Holstein-Friesian, Jersey and Angus cattle. Genetics. 2008;179(3):1503–12.
Bionaz M, Periasamy K, Rodriguez-Zas SL, Hurley WL, Loor JJ. A novel dynamic impact approach (DIA) for functional analysis of time-course omics studies: validation using the bovine mammary transcriptome. PLoS One. 2012;7(3):e32455.
Lemay DG, Ballard OA, Hughes MA, Morrow AL, Horseman ND, Nommsen-Rivers LA. RNA sequencing of the human milk fat layer transcriptome reveals distinct gene expression profiles at three stages of lactation. PLoS One. 2013;8(7):e67531.
Smith CL, Goldsmith CA, Eppig JT. The mammalian phenotype ontology as a tool for annotating, analyzing and comparing phenotypic information. Genome Biol. 2005;6(1):R7.
Cai Z, Guldbrandtsen B, Lund MS, Sahana G. Dissecting closely linked association signals in combination with the mammalian phenotype database can identify candidate genes in dairy cattle. BMC Genet. 2018;19(1):30.
Schennink A, Stoop WM, Visker MH, van der Poel JJ, Bovenhuis H, van Arendonk JA. Short communication: Genome-wide scan for bovine milk-fat composition. II. Quantitative trait loci for long-chain fatty acids. J Dairy Sci. 2009;92(9):4676–82.
Grisart B, Coppieters W, Farnir F, Karim L, Ford C, Berzi P, Cambisano N, Mni M, Reid S, Simon P, Spelman R, Georges M, Snell R. Positional candidate cloning of a QTL in dairy cattle: identification of a missense mutation in the bovine DGAT1 gene with major effect on milk yield and composition. Genome Res. 2002;12(2):222–31.
Bovenhuis H, Visker MHPW, Poulsen NA, Sehested J, van Valenberg HJF, van Arendonk JAM, Larsen LB, Buitenhuis AJ. Effects of the diacylglycerol o-acyltransferase 1 (DGAT1) K232A polymorphism on fatty acid, protein, and mineral composition of dairy cattle milk. J Dairy Sci. 2016;99(4):3113–23.
Jiang L, Liu J, Sun D, Ma P, Ding X, Yu Y, Zhang Q. Genome wide association studies for milk production traits in Chinese Holstein population. PLoS One. 2010;5(10):e13661.
Arias JA, Keehan M, Fisher P, Coppieters W, Spelman R. A high density linkage map of the bovine genome. BMC Genet. 2009;10:18.
Cole JB, Wiggans GR, Ma L, Sonstegard TS, Lawlor TJ Jr, Crooker BA, Van Tassell CP, Yang J, Wang S, Matukumalli LK, Da Y. Genome-wide association analysis of thirty one production, health, reproduction and body conformation traits in contemporary U.S. Holstein cows. BMC Genomics. 2011;12:408.
Nafikov RA, Schoonmaker JP, Korn KT, Noack K, Garrick DJ, Koehler KJ, Minick-Bormann J, Reecy JM, Spurlock DE, Beitz DC. Association of polymorphisms in solute carrier family 27, isoform A6 (SLC27A6) and fatty acid-binding protein-3 and fatty acid-binding protein-4 (FABP3 and FABP4) with fatty acid composition of bovine milk. J Dairy Sci. 2013;96(9):6007–21. https://doi.org/10.3168/jds.2013-6703.
Marchitelli C, Contarini G, De Matteis G, Crisà A, Pariset L, Scatà MC, Catillo G, Napolitano F, Moioli B. Milk fatty acid variability: effect of some candidate genes involved in lipid synthesis. J Dairy Res. 2013;80(2):165–73.
Zhou H, Cheng L, Azimu W, Hodge S, Edwards GR, Hickford JG. Variation in the bovine FABP4 gene affects milk yield and milk protein content in dairy cows. Sci Rep. 2015;5:10023. https://doi.org/10.1038/srep10023.
Schennink A, Bovenhuis H, Léon-Kloosterziel KM, van Arendonk JAM, Visker MHPW. Effect of polymorphisms in the FASN, OLR1, PPARGC1A, PRL and STAT5A genes on bovine milk-fat composition. Anim Gen. 2009;40:909–16.
Mele M, Conte G, Castiglioni B, Chessa S, Macciotta NP, Serra A, Buccioni A, Pagnacco G, Secchiari P. Stearoyl-coenzyme a desaturase gene polymorphism and milk fatty acid composition in Italian Holsteins. J Dairy Sci. 2007;90(9):4458–65.
Strillacci MG, Frigo E, Canavesi F, Ungar Y, Schiavini F, Zaniboni L, Reghenzani L, Cozzi MC, Samoré AB, Kashi Y, Shimoni E, Tal-Stein R, Soller M, Lipkin E, Bagnato A. Quantitative trait loci mapping for conjugated linoleic acid, vaccenic acid and ∆(9) -desaturase in Italian Brown Swiss dairy cattle using selective DNA pooling. Anim Genet. 2014;45(4):485–99.
Littlejohn MD, Tiplady K, Fink TA, Lehnert K, Lopdell T, Johnson T, Couldrey C, Keehan M, Sherlock RG, Harland C, Scott A, Snell RG, Davis SR, Spelman RJ. Sequence-based association analysis reveals an MGST1 eQTL with pleiotropic effects on bovine Milk composition. Sci Rep. 2016;6:25376. https://doi.org/10.1038/srep25376.
Olsen HG, Knutsen TM, Kohler A, Svendsen M, Gidskehaug L, Grove H, Nome T, Sodeland M, Sundsaasen KK, Kent MP, Martens H, Lien S. Genome-wide association mapping for milk fat composition and fine mapping of a QTL for de novo synthesis of milk fatty acids on bovine chromosome 13. Genet Sel Evol. 2017;49(1):20.
Muñoz M, Rodríguez MC, Alves E, Folch JM, Ibañez-Escriche N, Silió L, Fernández AI. Genome-wide analysis of porcine backfat and intramuscular fat fatty acid composition using high-density genotyping and expression data. BMC Genomics. 2013;14:845.
Duchemin SI, Bovenhuis H, Megens HJ, Van Arendonk JAM, Visker MHPW. Fine-mapping of BTA17 using imputed sequences for associations with de novo synthesized fatty acids in bovine milk. J Dairy Sci. 2017;100(11):9125–35.
Knutsen TM, Olsen HG, Tafintseva V, Svendsen M, Kohler A, Kent MP, Lien S. Unravelling genetic variation underlying de novo-synthesis of bovine milk fatty acids. Sci Rep. 2018;8(1):2179. https://doi.org/10.1038/s41598-018-20476-0.
Wang Q, Hulzebosch A, Bovenhuis H. Genetic and environmental variation in bovine milk infrared spectra. J Dairy Sci. 2016;99(8):6793–803.
Soyeurt H, Dehareng F, Gengler N, McParland S, Wall E, Berry DP, Coffey M, Dardenne P. Mid-infrared prediction of bovine milk fatty acids across multiple breeds, production systems, and countries. J Dairy Sci. 2011;94(4):1657–67.
De Marchi M, Penasa M, Cecchinato A, Mele M, Secchiari P, Bittante G. Effectiveness of mid-infrared spectroscopy to predict fatty acid composition of Brown Swiss bovine milk. Animal. 2011;5(10):1653–8. https://doi.org/10.1017/S1751731111000747.
Poulsen NA, Eskildsen CEA, Skov T, Larsen LB, Buitenhuis AJ. Comparison of genetic parameters estimation of fatty acids from gas chromatography and FT-IR in Holsteins. In: Proceedings: 10th World Congress of Genetics Applied to Livestock Production, Vancouver, BC Canada; 2014. p. 17–22.
Rutten MJ, Bovenhuis H, Hettinga KA, van Valenberg HJ, van Arendonk JA. Predicting bovine milk fat composition using infrared spectroscopy based on milk samples collected in winter and summer. J Dairy Sci. 2009;92(12):6202–9.
Ouimet M, Hennessy EJ, van Solingen C, Koelwyn GJ, Hussein MA, Ramkhelawon B, Rayner KJ, Temel RE, Perisic L, Hedin U, Maegdefessel L, Garabedian MJ, Holdt LM, Teupser D, Moore KJ. miRNA targeting of oxysterol-binding protein-like 6 regulates cholesterol trafficking and efflux. Arterioscler Thromb Vasc Biol. 2016;36(5):942–51.
Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT. Expression of miR-33 from an SREBP2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010;285(44):33652–61.
Goedeke L, Vales-Lara FM, Fenstermaker M, Cirera-Salinas D, Chamorro-Jorganes A, Ramírez CM, Mattison JA, de Cabo R, Suárez Y, Fernández-Hernando C. A regulatory role for microRNA 33* in controlling lipid metabolism gene expression. Mol Cell Biol. 2013;33(11):2339–52. https://doi.org/10.1128/MCB.01714-12.
Barber MC, Finley E, Vernon RG. Mechanisms whereby prolactin modulates lipogenesis in sheep mammary gland. Horm Metab Res. 1991;23(3):143–5.
Houdebine LM, Djiane J, Dusanter-Fourt I, Martel P, Kelly PA, Devinoy E, Servely JL. Hormonal action controlling mammary activity. J Dairy Sci. 1985;68(2):489–500 Review.
Matusik RJ, Rosen JM. Prolactin regulation of casein gene expression: possible mediators. Endocrinology. 1980;106(1):252–9.
Rudolph MC, Russell TD, Webb P, Neville MC, Anderson SM. Prolactin-mediated regulation of lipid biosynthesis genes in vivo in the lactating mammary epithelial cell. Am J Physiol Endocrinol Metab. 2011;300(6):E1059–68.
Shiu RP, Friesen HG. Mechanism of action of prolactin in the control of mammary gland function. Annu Rev Physiol. 1980;42:83–96 Review.
Akers RM, Bauman DE, Capuco AV, Goodman GT, Tucker HA. Prolactin regulation of milk secretion and biochemical differentiation of mammary epithelial cells in periparturient cows. Endocrinology. 1981;109(1):23–30.
Lamberts SW, Macleod RM. Regulation of prolactin secretion at the level of the lactotroph. Physiol Rev. 1990;70(2):279–318 Review.
Jeffcoat R, James AT. The control of stearoyl-CoA desaturase by dietary linoleic acid. FEBS Lett. 1978;85(1):114–8.
Berge KE, Tian H, Graf GA, Yu L, Grishin NV, Schultz J, Kwiterovich P, Shan B, Barnes R, Hobbs HH. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent ABC transporters. Science. 2000;290(5497):1771–5.
Lee MH, Lu K, Hazard S, Yu H, Shulenin S, Hidaka H, Kojima H, Allikmets R, Sakuma N, Pegoraro R, Srivastava AK, Salen G, Dean M, Patel SB. Identification of a gene, ABCG5, important in the regulation of dietary cholesterol absorption. Nat Genet. 2001;27(1):79–83.
Viturro E, Farke C, Meyer HH, Albrecht C. Identification, sequence analysis and mRNA tissue distribution of the bovine sterol transporters ABCG5 and ABCG8. J Dairy Sci. 2006;89(2):553–61.
Kolbehdari D, Wang Z, Grant JR, Murdoch B, Prasad A, Xiu Z, Marques E, Stothard P, Moore SS. A whole genome scan to map QTL for milk production traits and somatic cell score in Canadian Holstein bulls. J Anim Breed Genet. 2009;126(3):216–27.
Nayeri S, Sargolzaei M, Abo-Ismail MK, May N, Miller SP, Schenkel F, Moore SS, Stothard P. Genome-wide association for milk production and female fertility traits in Canadian dairy Holstein cattle. BMC Genet. 2016;17(1):75.
Ambros V. The functions of animal microRNAs. Nature. 2004;431:350–5.
Dávalos A, Goedeke L, Smibert P, Ramírez CM, Warrier NP, Andreo U, Cirera-Salinas D, Rayner K, Suresh U, Pastor-Pareja JC, Esplugues E, Fisher EA, Penalva LO, Moore KJ, Suárez Y, Lai EC, Fernández-Hernando C. miR-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A. 2011;108(22):9232–7.
Li R, Dudemaine PL, Zhao X, Lei C, Ibeagha-Awemu EM. Comparative analysis of the miRNome of bovine Milk fat, Whey and Cells. PLoS One. 2016;11(4):e0154129.
Wang H, Luo J, Zhang T, Tian H, Ma Y, Xu H, Yao D, Loor JJ. MicroRNA-26a/b and their host genes synergistically regulate triacylglycerol synthesis by targeting the INSIG1 gene. RNA Biol. 2016;13(5):500–10. https://doi.org/10.1080/15476286.2016.1164365.
Zhang M, Sun W, Zhou M, Tang Y. MicroRNA-27a regulates hepatic lipid metabolism and alleviates NAFLD via repressing FAS and SCD1. Sci Rep. 2017;7(1):14493.
Vickers KC, Shoucri BM, Levin MG, Wu H, Pearson DS, Osei-Hwedieh D, Collins FS, Remaley AT, Sethupathy P. MicroRNA-27b is a regulatory hub in lipid metabolism and is altered in dyslipidemia. Hepatology. 2013;57(2):533–42. https://doi.org/10.1002/hep.25846.
Do DN, Li R, Dudemaine PL, Ibeagha-Awemu EM. MicroRNA roles in signaling during lactation: an insight from differential expression, time course and pathway analyses of deep sequence data. Sci Rep. 2017;7:44605. https://doi.org/10.1038/srep44605.
Bovenhuis H, Visker MH, van Valenberg HJ, Buitenhuis AJ, van Arendonk JA. Effects of the DGAT1 polymorphism on test-day milk production traits throughout lactation. J Dairy Sci. 2015;98(9):6572–82. https://doi.org/10.3168/jds.2015-9564.
Poulsen NA, Gustavsson F, Glantz M, Paulsson M, Larsen LB, Larsen MK. The influence of feed and herd on fatty acid composition in 3 dairy breeds (Danish Holstein, Danish Jersey, and Swedish red). J Dairy Sci. 2012;95(11):6362–71.
Gebreyesus G, Lund MS, Janss L, Poulsen NA, Larsen LB, Bovenhuis H, Buitenhuis AJ. Short communication: multi-trait estimation of genetic parameters for milk protein composition in the Danish Holstein. J Dairy Sci. 2016;99(4):2863–6.
Duchemin SI, Visker MH, Van Arendonk JA, Bovenhuis H. A quantitative trait locus on Bos taurus autosome 17 explains a large proportion of the genetic variation in de novo synthesized milk fatty acids. J Dairy Sci. 2014;97(11):7276–85.
Zimin AV, Delcher AL, Florea L, Kelley DR, Schatz MC, Puiu D, Hanrahan F, Pertea G, Van Tassell CP, Sonstegard TS, Marçais G, Roberts M, Subramanian P, Yorke JA, Salzberg SL. A whole-genome assembly of the domestic cow, Bos taurus. Genome Biol. 2009;10(4):R42.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88(1):76–82.
Park JH, Wacholder S, Gail MH, Peters U, Jacobs KB, Chanock SJ, Chatterjee N. Estimation of effect size distribution from genome-wide association studies and implications for future discoveries. Nat Genet. 2010;42(7):570–5.
Beavis WD. QTL analyses: power precision and accuracy. In Molecular dissection of complex traits. Edited by: Paterson AH. New York: CRC Press; 1998. p. 145–62.
Acknowledgements
The study used data from the Dutch Milk Genomics Initiative (www.milkgenomics.nl), High Technology R & D Program of China, 2013AA102504 and Beijing Dairy Industry Innovation Team, BAIC06-2018, Danish-Swedish Milk Genomics Initiative and the Milk Levy Fund (Denmark) projects: “Phenotypic and genetic markers for specific milk quality parameters” and “The importance of the metagenome for milk composition and quality”.
Funding
This study was supported by the Center for Genomic Selection in Animals and Plants (GenSAP) funded by the Danish council for strategic research. GG is supported by fellowship from the Erasmus-Mundus joint doctorate European Graduate School in Animal Breeding and Genetics (EGS-ABG) funded by the European commission. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Availability of data and materials
The list and estimated effects on studied FAs of the lead-SNPs in each detected region and gene list generated from the enrichment analyses in the current study can be found in the Additional files 1, 2 and 3. The raw datasets analyzed in the study are not publicly available but can be made available, on reasonable request, from the co-authors responsible for the different datasets (bart.buitenhuis@mbg.au.dk for the Danish dataset, henk.bovenhuis@wur.nl for the Dutch dataset, sundx@cau.edu.cn for the Chinese dataset).
Author information
Authors and Affiliations
Contributions
GG conceived the study, implemented the analyses and drafted the manuscript. HB co-conceived the study and contributed to the discussion of the results. AJB supervised the project and contributed to the discussion of the results. NAP and HJFV collected the milk samples and contributed to the milk analysis and discussion of the results. MHPWV, DS and QZ contributed to the discussion of the results. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
All samples in the Dutch and Chinese datasets were collected in accordance with the guidelines and protocols for the care and use of animals approved by the ethical committee on animal experiments of Wageningen University (protocol: 200523.b) and the Animal Welfare Committee of China Agricultural University (Permit Number: DK996), respectively. All procedures to collect the Danish samples followed the protocols approved by the National Guidelines for Animal Experimentation and the Danish Animal Experimental Ethics Committee, and that all sampling was restricted to routine on-farm procedures that did not cause any inconvenience or stress to the animals, and hence, no specific permission was required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Effects of the lead SNPs in all the detected genomic regions. SNP regression coefficients (SNP effects) in all the FA traits studied, along with the standard errors and –log 10 p-values, for the lead-SNP in each of the detected genomic region. (XLSX 45 kb)
Additional file 2:
List of genes identified as enriched using the DAVID based enrichment analysis. Gene names and the positions in the genome (chromosome, start and end bp) clustered to at least one of the significantly enriched pathways and GO terms. (XLSX 24 kb)
Additional file 3:
Micro-RNAs located within detected genomic regions. List of micro-RNAs (names and ensemble stable IDs) located in detected genomic regions and their positions in the genome (chromosome, start and end bp). (XLSX 11 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Gebreyesus, G., Buitenhuis, A.J., Poulsen, N.A. et al. Multi-population GWAS and enrichment analyses reveal novel genomic regions and promising candidate genes underlying bovine milk fatty acid composition. BMC Genomics 20, 178 (2019). https://doi.org/10.1186/s12864-019-5573-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-019-5573-9