
Abstract
Background
MicroRNAs (miRNAs) are a class of small noncoding RNAs that play important roles in the regulation of gene expression. However, the role of miRNAs in bovine mammary gland responses to heat stress is not well understood.
Results
In the present study, we performed a deep RNA sequencing analysis to identify miRNAs associated with the heat stress potential of the bovine mammary gland. We identified 27 miRNAs that were differentially expressed significantly between the mammary tissue of Holstein cattle heat stress and normal conditions. Twenty miRNAs had higher expression in the mammary tissue of heat-stressed Holstein cattle. The seven highest differentially expressed candidate miRNAs (bta-miR-21-5p, bta-miR-99a-5p, bta-miR-146b, bta-miR-145, bta-miR-2285 t, bta-miR-133a, and bta-miR-29c) identified by deep RNA sequencing were additionally evaluated by stem-loop qPCR. Enrichment analyses for targeted genes revealed that the major differences between miRNAs expression in the mammary gland of heat-stressed versus control were associated with the regulation of Wnt, TGF-β, MAPK, Notch, and JAK-STAT.
Conclusions
These data indicated that the differentially expressed miRNAs identified in this study may act as dominant regulators during heat stress. We might reduce heat stress damage of Holstein cows by up-regulating or down-regulating these differentially expressed miRNAs.
Similar content being viewed by others


Background
Stress can be defined as an external condition that produces a “strain” in a biological system [1]. The environmental stress may be measured by changes in body temperature, metabolic rate, or productivity. Heat stress negatively impacts all features of dairy cattle production including milk composition and mammary gland pathogens. It substantially influences a cow’s growth and development [2]. Reduction in reproductive performance of lactating cows during summer is associated with decreased thermoregulatory competence [3]. Heat stress causes cow metabolic disorders and a reduction in milk production [4, 5], and it also decreases immunity and increases susceptibility to mastitis, endometritis disease and even death in severe cases [6,7,8]. Diminished milk yield and reproductive losses during summer months seriously affect the economic potential of the dairy industry. In addition, global warming may boost the occurrence of heat stress [9]. Thus, for the dairy industry, heat stress has been a bottleneck which limits the efficiency of the dairy supply throughout the year.
The heat stress response is a complex molecular process that involves the transcriptional and post-transcriptional regulation of stress-related genes. Acute environmental change initiates the heat stress response at the cellular level. Changes in gene expression are associated with a reaction to an environmental stressor as well as changes across a variety of organs and tissues associated with the acclimation response. Functional genomics establishes a verifiable link between gene expression and phenotype. Endogenous noncoding small RNAs known as microRNAs (miRNAs) are increasingly being recognized as important modulators of gene expression at the post-transcriptional level and have been shown to be involved in diverse biological processes such as differentiation, development, apoptosis, and viral infection. RNA-Seq, in particular, allows a global analysis of gene expression responses to environmental change.
Some miRNAs have been shown to be involved in plant stress responses by down-regulating the respective target genes encoding regulatory and functional proteins [10]. Differential expression of miRNAs is also associated with thermal stress in cattle. Down-regulation of miR-181a can reduce heat stress damage in peripheral blood mononuclear cells (PBMCs) of Holstein cows [11]. miRNA profiles in bovine mammary tissue infected with Staphylococcus [12, 13] and miRNA profiles in serum or PBMC cell of heat-stressed cattle have been studied [14,15,16]; however, miRNA profiles in bovine mammary gland of stressed lactating cattle have not been compared to normal lactating cattle. Therefore, it was our objective to profile miRNA expression under heat stress using bovine mammary glands. To investigate the role of miRNA in the heat stress response, miRNA expression in bovine mammary glands were characterized by next-generation sequencing during summer and spring with and without a heat stress challenge.
Methods
Tissue collection
Samples were collected from eight mammary gland tissues of the four lactating Chinese Holstein cows from Yucheng dairy farm, China. The samples were collected in two different environmental seasons, viz. spring and summer, with the temperature ranges between 15 and 20 °C (March; designated as non-heat stressed, NHS) and 30–38 °C (July; designated as heat stressed, HS), respectively. The temperature humidity index (THI) was used as a heat stress indicator. The temperature probing procedure has been described in detail elsewhere [17]. Dry-bulb and wet-bulb temperatures were recorded using a dry and wet bulb thermometer consisting of two thermometers, a dry bulb thermometer and a wet bulb thermometer. THI was calculated as THI = 0.72 (Td + Tw) + 40.6 where Td is the dry-bulb temperature and Tw is the wet-bulb temperature. Stress response of the animals was characterized by recording physiological parameters. The rectal temperature (RT) and heat tolerance coefficient (HTC) were measured for both NHS and HS groups according to a method previously described, HTC = 100–10 (RT-38.3) [18]. Four cows were designated as biological replicates for NHS and HS. In each case, one complete mammary gland was removed after excision of the intramammary lymph node. The samples were frozen in liquid nitrogen until RNA extraction.
Total RNA extraction
Total RNA from the mammary gland was isolated using Trizol Reagent (Life Technologies, USA) according to manufacturer’s instructions. RQ1 DNase (Promega, USA) was used to treat the total RNA to digest the DNA. The concentration and purity of the extracted RNA were determined with a NanoDrop 1000 Spectrophotometer (NanoDrop Technologies, USA). The quality of RNA was assessed through the Agilent 2100 Bioanalyzer (Agilent Technologies, USA). All eight RNA samples had an RNA integrity number (RIN) of ≥8 and were stored at − 80 °C for further analysis.
Small RNA library construction
Equal amounts of total RNA (330 ng) from each mammary gland sample of both NHS and HS groups were used to construct a miRNA library through a TruSeq Small RNA Sample Preparation kit (Illumina, USA) following the manufacturer’s protocol. PCR amplification was performed including 13 cycles. Eight small RNA libraries were constructed in equal amounts for gel purification. Quality and quantity of purified small RNA were estimated using an Agilent 2100 Bioanalyzer (Agilent Technologies, USA). Sequencing was carried out on a HiSeq-2000 system (Illumina, USA). Real-Time Analysis (RTA) and base calling were performed by HiSeq Control Software Version 1.4.8 (Illumina, USA).
Small RNA sequence analysis
Low-quality reads were removed from the raw data. After trimming the 3′ adaptor sequence, small RNA reads with the length of 18–30 nt from all libraries were extracted using miRDeep2 (version 2.0.0.5) with the default parameters to identify known and novel miRNAs [19]. Each library was processed separately. Subsequently, all reads were mapped to the bovine genome with Bowtie [20]. The reads that mapped to bovine tRNAs, rRNAs, and snoRNAs in the Rfam RNA family database [21] were discarded. Small RNAs that only mapped to genomic repeat loci were removed. Novel miRNA and precursors were identified by the core module miRDeep2.pl. Datasets of novel miRNA and precursors were created through adding miRNA predicted with a miRDeep2 score > 0 to known miRNA. The expression of detected miRNAs for each library was estimated by the Quantifier module of miRDeep2 [19]. To investigate the regulation of miRNAs in mammary gland in response to heat stress, miRNA expression under heat stress (summer) were compared to expression in controls (spring) using DEseq [22].
Target gene prediction and pathway analysis
The target genes of differentially expressed miRNAs were predicted through TargetScan (http://www.targetscan.org) and miRanda (http://www.microrna.org/microrna/home.do) according to a previous study [23]. To comprehensively describe the properties of genes and gene products, we executed gene ontology (GO) annotation and enrichment analysis from three ontologies: molecular function, cellular component and biological process. Functional analysis of predicted gene targets was performed using the Database for Annotation, Visualization and Integrated Discovery (DAVID version 6.7, http://david.abcc.ncifcrf.gov/) for pathway analysis.
Stem-loop quantitative PCR and data analysis
Seven differentially expressed miRNAs (bta-miR-21-5, bta-miR-99a-5p, bta-miR-146b, bta-miR-145, bta-miR-2285 t, bta-miR-133a, and bta-miR-29c) were validated through a standardized and reliable stem-loop qRT-PCR procedure. The RT reaction mixture contained a 4 μg aliquot of total RNA and a mixture of 1 μL of each RT primers (10 μM) for all of the mature miRNAs and U6, which was chosen as a reference control (Additional file 1), 10 μL 2 × reaction mix, 1.5 μL RT enzyme mix (Sangon, China). The mixture was incubated at 16 °C for 30 min, 37 °C for 30 min, 85 °C for 5 min, subsequently, frozen on ice for at least 5 min. SYBR® Green Realtime PCR Master Mix (Sangon, China) was used to detect miRNA expression by a Bio-Rad IQ5 System (USA). Briefly, cDNAs were diluted 10 times and a 1 μL diluted sample was used as a template in a 20 μL PCR reaction, which contained 10 μL 2 × SYBR Green Realtime PCR Master Mix, 0.25 μM of a miRNA-specific forward primer and universal reverse primer, respectively. The quantitative PCR was conducted in triplicate for 1 min at 95 °C, followed by 40 cycles of 15 s at 95 °C and 30 s at 60 °C. For each PCR, dissociation curve analysis was carried out to discriminate specific products from primer dimers. The fold changes of miRNA in different samples were calculated by ΔCt method.
The relative expression of target miRNA was calculated by the following formula: ΔCt (target miRNA) = Ct (target miRNA)–Ct (internal reference). U6 was used as an internal reference. The NHS group at spring was selected as a calibrator for the relative quantification. The relative expression of miRNA normalized to internal control and relative to the calibrator was calculated as follows: Relative expression of target miRNA = 2-ΔΔCt, where ΔΔCt = ΔCt (target miRNA, sample) –ΔCt (target miRNA, calibrator). The difference between the two groups was compared using T-test (SAS version 9.2, 2008). Statistical difference was at P < 0.05.
Results
Characterization of the stress response
Rectal temperature and HTC at two different environmental temperature ranges are presented in Table 1. A THI of above 72 and a rectal temperature of above 39 °C were regarded as an indication of heat stress. The physiological parameters of HS were significantly different from the NHS group of cows (P < 0.05). The average rectal temperature of HS group was higher, and a significant decrease of HTC as compared with the control. Rectal temperature and HTC data showed that the cattle were under heat stress.
miRNA sequencing
Eight small RNA libraries were constructed and sequenced. A total of 20,818,666 high-quality reads were generated. Among them, 19,859,218 sequences ranging from 18 to 30 nucleotides were obtained after adaptor trimming, accounting for 95.39% of all small RNA (sRNA) sequences. A summary of the data is provided in Table 2. Alignment with miRBase (Release 21) indicated that miRNAs were highly enriched in all libraries. To assess the efficiency of high-throughput sequencing for sRNA detection, all sequence reads were annotated and classified by alignment with Rfam databases. Of the 18 to 30 nucleotide sRNA fraction, four out of five (86.72%) were identified as other noncoding sRNA, while only a small number (13.28%) aligned to rRNAs, tRNAs, snRNAs and snoRNAs in bovine (Fig. 1). The major reads from sRNAs were 20 to 24 nucleotides in length (accounting for 91.14% of total number). Dominant reads of sRNAs were 22 nucleotides in length (41.38%), followed by 21 and 23 nucleotides, and lastly 20 and 24 nucleotides (Fig. 2).

A pie graph showed relative abundance of different classes of small RNAs. Of the 18 to 30 nucleotide sRNA fraction, four out of five (86.72%) were identified as other noncoding sRNA, while only a small number (13.28%) aligned to rRNAs, tRNAs, snRNAs and snoRNAs in bovine

Frequency distribution of detected small RNAs (18–30 nt)based on all reads. The major reads from small RNAs were 20 to 24 nucleotides in length (accounting for 91.14% of total number). Dominant reads of small RNAs were 22 nucleotides in length (41.38%), followed by 21 and 23 nucleotides, and lastly 20 and 24 nucleotides
We identified 483 known bovine miRNAs (counts per million, CPM > 10 in at least one library). Among them, eight highly expressed miRNAs accounted for 35.41% of the total reads of identified known miRNAs (Table 3). The four highest expressed miRNAs were bta-miR-21-5p, bta-let-7a-5p, bta-miR-26a, and bta-miR-148a, accounting for 9.15, 5.55, 5.46, and 4.45% of total known miRNA reads, respectively.
Identification of novel miRNAs and miRNA candidates
To determine whether these small RNA sequences are genuine bovine miRNAs, we scanned the bovine genome for hairpin structures comprising the candidate miRNA with miRDeep2 software (version 2.0.0.5), which can be used to identify both known and novel miRNAs from deep sequenced sRNA libraries. In total, 483 loci possessed the typical stem-loop structures matching the known miRNA hairpins (808 miRNA, miRbase 21) in the mammary gland. A total of 139 novel miRNA hairpins were identified (Additional file 2).
Analyses of the first nucleotide bias of the 18–30 nt miRNAs candidates revealed that uridine (U) was the most common at the 5′ end of the 20–24 nt miRNAs in the mammary gland (Fig. 3). Moreover, miRNA nucleotide bias at each position also showed that U was the dominant first nucleotide (Fig. 4).

First nucleotides bias of 18–30 nt miRNAs candidates. Uridine (U) was the most common at the 5′ end of the 20–24 nt miRNAs in the mammary gland

Nucleotides bias at each position of miRNA. The U nucleotide was the dominant first nucleotide, while the G content was very low at position 1. The U nucleotide had a low frequency in the 2th, 3th, and 4th positions
Effects of heat stress on miRNA expression in mammary gland
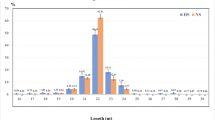
Given that miRNAs play important roles in many biological processes, we speculated that the expression of miRNAs might be regulated in the mammary glands of Holstein cattle under heat stress. The global expression of miRNAs under normal condition and heat-stressed condition were profiled and the correlations between libraries were performed with the normalized counts of all detected miRNAs. Only a small number of miRNAs were significantly regulated in mammary glands of Holstein cattle under heat stress. DEseq, an R/Bioconductor package [22] method was used to analyze differentially expressed miRNAs between different conditions based on sequence counts. We observed that 24 miRNAs were differentially expressed (P < 0.05) in the heat stress condition when compared to the control (Additional file 3). The highest differentially expressed miRNAs (bta-miR-21-5p, bta-miR-99a-5p, bta-miR-146b, bta-miR-145, bta-miR-2285 t, bta-miR-133a, and bta-miR-29c) are shown in Fig. 5. The expression of bta-miR-145, bta-miR-2285 t, bta-miR-133a, and bta-miR-29c was increased and reached a 3.50, 3.24, 4.30, and 4.03-fold increase (P < 0.05), respectively. Conversely, the expression of bta-miR-21-5p, bta-miR-99a-5p, and bta-miR-146b were decreased under heat stress condition (P < 0.05). Furthermore, we also verified the expression of these miRNAs using qRT-PCR (P < 0.05) and the expression pattern was consistent with sequencing results (Additional file 4).

Expression of differentially expressed miRNAs between heat stressed and normal conditions detected by qRT-PCR and RNA-seq. a Expression of bta-miR-21-5p; b Expression of bta-miR-99a-5p; c Expression of bta-miR-146b; d Expression of bta-miR-145; e Expression of bta-miR-2285 t; f Expression of bta-miR-133a; g Expression of bta-miR-29c. Lines on the top represented miRNA expression from qRT-PCR and values were showed on the right vertical axis as relative abundance. Bars on the bottom represented miRNA expression from RNA-seq and values were showed on the left vertical axis as log2RPM (normalized reads number). * on the top of lines or bars indicated significant difference (P < 0.05 or FDR < 0.05) between heat stressed and normal condition. Data were presented as Mean ± Standard Error
Biological function enrichment analysis of predicted target genes of differentially expressed miRNAs in mammary glands of Holstein cattle under heat stress
Target gene prediction of 24 miRNAs differentially regulated by heat stress indicates that about 26,824 genes that were enriched in 59 GO terms may be regulated by these miRNAs (Fig. 6). GO analysis showed that the predicted targets of differentially expressed miRNAs were significantly enriched (P < 0.05) in different functional groups, namely, biological regulation, cellular process, metabolic process, multicellular organismal process, regulation of biological process, response to stimulus and single-organism process (Fig. 6, Table 4). One of the GO terms included more than 2000 responses to stimulus-related genes. Interestingly, several KEGG (Kyoto encyclopedia of genes and genomes) pathways were significantly enriched (P < 0.05) by target genes of the 24 differentially expressed miRNAs (Table 5). Notably, pathways of the RNA degradation, mTOR signaling pathway, immune system and pathways in human diseases, especially cancer were significantly enriched by target genes. This indicated that the differentially expressed miRNAs identified in this study might act as a dominant regulator during heat stress.

GO analysis of predicted target genes of differentially expressed miRNAs during heat stressed challenge of bovine mammary gland. X axis: GO classification (biological process, cellular component, and molecular function). Y axis: Left, percentage of genes; Right, number of genes in this term
Discussion
Heat stress triggers a dramatic and complex program of altered gene expression in mammary glands similar to patterns investigated in other cell types exposed to thermal stress. As reported by Sonna et al. [24], these changes include inhibition of DNA synthesis, RNA transcription, and translation, disruption of cytoskeletal components, and alterations in metabolism. In the present study, thermal stress induced changes in the miRNA expression in dairy cattle mammary glands. Although the miRNA expression profiles in dairy cattle have been studied, there is still a limited understanding of their role in heat-stressed bovine mammary glands and the influence of this on dairy potential. In this study, 483 known miRNAs were detected in dairy cattle mammary glands by RNA-Seq.
Several studies have uncovered the roles of the highly abundant miRNAs in mammary glands. For example, two of the eight miRNAs with the highest expression in the dairy cattle mammary gland in this study, bta-miR-21-5p, the other arm of bta-miR-21, and bta-miR-143, were shown to promote adipogenesis [25, 26], which accounts for 12.70% of the miRNAs expressed in the dairy cattle mammary gland. The expression of miR-21-5p was strongly induced at 7 d postpartum compared with the dry period suggesting that it might promote mammary cell proliferation during early lactation [27]. MiR-21 decreases the expression of Tgfbr2 by targeting TGF b receptor II (Tgfbr2) and eventually enhances adipogenic differentiation [26]. Moreover, miR-21 is associated with thermal stress in Frieswal crossbred dairy cattle [15]. Our data showed that the expression of bta-miR-21-5p was lower in heat stressed group than that in control, which is consistent with bta-miR-21 expression in PBMC of Sahiwal cows [16]. The up-regulation of miR-143 decreased the expression of pleiotrophin and increased some adipocyte-important genes, enhancing the rate of adipocyte differentiation at early stages of adipogenesis [25]. Decreased expression of miR-143 by its antisense sequence suppressed differentiation of preadipocytes through repressing ERK5, suggesting this miRNA may play a key role in adipocyte differentiation [28].
One of the other eight most abundant miRNAs in this study is miR-148a, orthologs of miR-148. MiR-148 was highly abundant in the lactating mammary gland of mouse and goat [29, 30]. MiR-148a and miR-26a show the similar expression patterns during the lactation period in cow milk [31]. Accumulating evidence indicates that miR-148a induce cell proliferation and differentiation [32, 33]. MiR-148a promotes adipogenesis by repressing Wnt signaling [34]. In addition, the expression of bta-miR-145, bta-miR-133a, and bta-miR-29c was increased in the heat-stressed group. MiR-145 is associated with cow mastitis caused by Staphylococcus aureus [12]. Moreover, the expression of miR-145 during differentiation can regulate the insulin receptor substrate 1 to inhibit adipogenesis [35]. Inhibition of miR-133 led to the expression of GLUT4 and insulin-mediated glucose uptake attenuation in cardiomyocytes [36]. Overexpression of miR-29a-c reduced the protein levels of PGC-1a and G6 Pase in primary hepatocytes and mouse livers [37]. These researches indicate that the highly expressed miRNAs may be related to the mammary gland biology, milk synthesis, and lactation process.
Bta-miR-2285 t was significantly increased in the mammary tissue of dairy cattle (Holstein-Friesian) compared with beef (Limousin) postpubertal heifers [38]. However, its role in the mammary gland remains unclear. The expression of bta-miR-2285 t and bta-miR-146b was decreased in the heat-stressed group compared with the control. Similarly, miR-146b has a lower expression in the serum of heat-stressed Holstein cows [14]. It has been suggested that miR-146b can modulate Sirtuin1, suppressing the negative regulators of adipogenesis, and eventually promoting adipogenesis [39]. Up-regulation of miR-146b is found during pregnancy, especially in the luminal progenitors compared to the basal/stem cells, suggesting it is involved in the differentiation of mammary epithelial cells [40]. Furthermore, the expression of miR-146b was upregulated in the luminal progenitors in pregnant mice, which indicates that miR-146b is involved in the differentiation of the mammary stem cells [40].
Interestingly, we found that the expression of bta-miR-21-5p and bta-miR-146b tended to decrease, and bta-miR-145 tended to increase in the heat-stressed group compared with control group. These results are consistent with the phenomenon that, acute heat stress affects the lipolysis and the rate-limiting enzyme of lipogenesis in bovine adipocytes [41]. Considering that adipocytes in the mammary gland can regulate the growth and biological function of the mammary epithelium [42], we speculated that these miRNAs might have a role in the regulation of milk fat synthesis.
A total of 139 novel miRNA hairpins were detected. High-throughput sequencing and bioinformatics analysis have become the main methods to identify the potentially novel miRNAs since 2008 [43]. Given the number of known miRNAs (808 miRNAs) in the miRBase (Release 21), our data may enrich the miRNA resources in bovine.
To further understand and provide some molecular insight into the physiological functions and biological processes involving these miRNAs in heat-stressed mammary glands, target genes were predicted based on miRNA/mRNA interactions. The predicted target genes were classified using KEGG function annotations to character the pathways that were actively regulated by miRNAs in the mammary gland. In the present study, pathway analysis of miRNA targets revealed that Wnt, TGF-β, MAPK, Notch, and JAK-STAT signaling pathways may play key roles in the mammary gland in the process of heat stress. Wnt, EGFR, TGF-β, and insulin signaling pathways are known to play a key role in normal development of the mammary gland [44,45,46,47]. The MAPK pathway is an important regulator of mammary epithelial cell differentiation and function [48]. These reports are supported by our findings. In addition, MAPK signaling, PI3k-Akt signaling, and immune-regulatory are greatly influenced by miRNA-mediated regulation in Frieswal cattle [15]. The function analysis of differentially expressed miRNAs and their target genes suggested the effects of heat stress on signaling mechanisms. Although many target gene candidates were predicted by bioinformatics methods, structural verification and signaling pathways analysis in vitro need to be further performed to validate the relationship between miRNAs and mRNA.
Conclusions
In this study, we characterized miRNAs expressed in dairy cattle mammary gland under heat stress and identified 483 known bovine miRNAs and 139 novel miRNAs, and the heat-dependent differential modulation of miRNAs. The results showed that significant enrichment of predicted target genes of differentially expressed miRNAs in several biological processes, including developmental process, cellular process, biological regulation, cell death, focal adhesion, and biosynthesis of secondary metabolites. Moreover, our data provides the valuable information of the role of miRNAs in heat response and may be helpful for developing miRNA-based biomarkers for the control of heat stress in cows. We might reduce heat stress damage of Holstein cows by up-regulating or down-regulating these differentially expressed miRNAs.
Abbreviations
- miR, miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
References
Lee DH. Climatic stress indices for domestic animals. Int J Biometeorol. 1965;9(1):29–35.
Tao S, Bubolz JW, do Amaral BC, Thompson IM, Hayen MJ, Johnson SE, dahl GE. Effect of heat stress during the dry period on mammary gland development. J Dairy Sci. 2011;94(12):5976–86.
Flamenbaum I, Galon N. Management of heat stress to improve fertility in dairy cows in Israel. J Reprod Dev. 2010;56(Suppl):S36–41.
Shwartz G, Rhoads ML, VanBaale MJ, Rhoads RP, Baumgard LH. Effects of a supplemental yeast culture on heat-stressed lactating Holstein cows. J Dairy Sci. 2009;92(3):935–42.
Tao S, Monteiro AP, Thompson IM, Hayen MJ, Dahl GE. Effect of late-gestation maternal heat stress on growth and immune function of dairy calves. J Dairy Sci. 2012;95(12):7128–36.
Biffani S, Bernabucci U, Vitali A, Lacetera N, Nardone A. Short communication: effect of heat stress on nonreturn rate of italian Holstein cows. J Dairy Sci. 2016;99(7):5837–43.
Ravagnolo O, Misztal I. Effect of heat stress on nonreturn rate in Holstein cows: genetic analyses. J Dairy Sci. 2002;85(11):3092–100.
Carroll JA, Forsberg NE. Influence of stress and nutrition on cattle immunity. Vet Clin North Am Food Anim Pract. 2007;23(1):105–49.
Renaudeau D, Collin A, Yahav S, de Basilio V, Gourdine JL, Collier RJ. Adaptation to hot climate and strategies to alleviate heat stress in livestock production. Animal. 2012;6(5):707–28.
Ding Y, Tao Y, Zhu C. Emerging roles of micrornas in the mediation of drought stress response in plants. J Exp Bot. 2013;64(11):3077–86.
Chen KL, Fu YY, Shi MY, Li HX. Down-regulation of mir-181a can reduce heat stress damage in pbmcs of Holstein cows. In Vitro Cell Dev Biol Anim. 2016;52(8):864–71.
Li R, Zhang CL, Liao XX, Chen D, Wang WQ, Zhu YH, Geng XH, Ji DJ, Mao YJ, Gong YC, et al. Transcriptome microrna profiling of bovine mammary glands infected with staphylococcus aureus. Int J Mol Sci. 2015;16(3):4997–5013.
Pu J, Li R, Zhang C, Chen D, Liao X, Zhu Y, Geng X, Ji D, Mao Y, Gong Y, et al. Expression profiles of mirnas from bovine mammary glands in response to streptococcus agalactiae-induced mastitis. J Dairy Res. 2017;84(3):300–8.
Zheng Y, Chen KL, Zheng XM, Li HX, Wang GL. Identification and bioinformatics analysis of micrornas associated with stress and immune response in serum of heat-stressed and normal Holstein cows. Cell Stress Chaperones. 2014;19(6):973–81.
Sengar GS, Deb R, Singh U, Raja TV, Kant R, Sajjanar B, Alex R, Alyethodi RR, Kumar A, Kumar S, et al. Differential expression of micrornas associated with thermal stress in frieswal (bos taurus x bos indicus) crossbred dairy cattle. Cell Stress Chaperones. 2018;23(1):155–70
Sengar GS, Deb R, Singh U, Junghare V, Hazra S, Raja TV, Alex R, Kumar A, Alyethodi RR, Kant R, et al. Identification of differentially expressed micrornas in sahiwal (bos indicus) breed of cattle during thermal stress. Cell Stress Chaperones. 2018;23:1019–32.
Li Q, Han J, Du F, Ju Z, Huang J, Wang J, Li R, Wang C, Zhong J. Novel snps in hsp70a1a gene and the association of polymorphisms with thermo tolerance traits and tissue specific expression in chinese Holstein cattle. Mol Biol Rep. 2011;38(4):2657–63.
Li QL, Ju ZH, Huang JM, Li JB, Li RL, Hou MH, Wang CF, Zhong JF. Two novel snps in hsf1 gene are associated with thermal tolerance traits in chinese Holstein cattle. DNA Cell Biol. 2011;30(4):247–54.
Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N. Mirdeep2 accurately identifies known and hundreds of novel microrna genes in seven animal clades. Nucleic Acids Res. 2012;40(1):37–52.
Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25.
Burge SW, Daub J, Eberhardt R, Tate J, Barquist L, Nawrocki EP, Eddy SR, Gardner PP, Bateman A. Rfam 11.0: 10 years of rna families. Nucleic Acids Res. 2013;41(Database issue):D226–32.
Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11(10):R106.
Liang G, Malmuthuge N, McFadden TB, Bao H, Griebel PJ, Stothard P, Guan le L. Potential regulatory role of micrornas in the development of bovine gastrointestinal tract during early life. PLoS One. 2014;9(3):e92592.
Sonna LA, Fujita J, Gaffin SL, Lilly CM. Invited review: Effects of heat and cold stress on mammalian gene expression. J Appl Physiol (1985). 2002;92(4):1725–42.
Yi C, Xie WD, Li F, Lv Q, He J, Wu J, Gu D, Xu N, Zhang Y. Mir-143 enhances adipogenic differentiation of 3t3-l1 cells through targeting the coding region of mouse pleiotrophin. FEBS Lett. 2011;585(20):3303–9.
Kim YJ, Hwang SJ, Bae YC, Jung JS. Mir-21 regulates adipogenic differentiation through the modulation of tgf-beta signaling in mesenchymal stem cells derived from human adipose tissue. Stem Cells. 2009;27(12):3093–102.
Wang M, Moisa S, Khan MJ, Wang J, Bu D, Loor JJ. Microrna expression patterns in the bovine mammary gland are affected by stage of lactation. J Dairy Sci. 2012;95(11):6529–35.
Esau C, Kang X, Peralta E, Hanson E, Marcusson EG, Ravichandran LV, Sun Y, Koo S, Perera RJ, Jain R, et al. Microrna-143 regulates adipocyte differentiation. J Biol Chem. 2004;279(50):52361–5.
Avril-Sassen S, Goldstein LD, Stingl J, Blenkiron C, Le Quesne J, Spiteri I, Karagavriilidou K, Watson CJ, Tavare S, Miska EA, et al. Characterisation of microrna expression in post-natal mouse mammary gland development. BMC Genomics. 2009;10:548.
Ji Z, Wang G, Xie Z, Wang J, Zhang C, Dong F, Chen C. Identification of novel and differentially expressed micrornas of dairy goat mammary gland tissues using solexa sequencing and bioinformatics. PLoS One. 2012;7(11):e49463.
Chen X, Gao C, Li H, Huang L, Sun Q, Dong Y, Tian C, Gao S, Dong H, Guan D, et al. Identification and characterization of micrornas in raw milk during different periods of lactation, commercial fluid, and powdered milk products. Cell Res. 2010;20(10):1128–37.
Guo SL, Peng Z, Yang X, Fan KJ, Ye H, Li ZH, Wang Y, Xu XL, Li J, Wang YL, et al. Mir-148a promoted cell proliferation by targeting p27 in gastric cancer cells. Int J Biol Sci. 2011;7(5):567–74.
Zhang J, Ying ZZ, Tang ZL, Long LQ, Li K. Microrna-148a promotes myogenic differentiation by targeting the rock1 gene. J Biol Chem. 2012;287(25):21093–101.
Qin L, Chen Y, Niu Y, Chen W, Wang Q, Xiao S, Li A, Xie Y, Li J, Zhao X, et al. A deep investigation into the adipogenesis mechanism: profile of micrornas regulating adipogenesis by modulating the canonical wnt/beta-catenin signaling pathway. BMC Genomics. 2010;11:320.
Guo Y, Chen Y, Zhang Y, Zhang Y, Chen L, Mo D. Up-regulated mir-145 expression inhibits porcine preadipocytes differentiation by targeting irs1. Int J Biol Sci. 2012;8(10):1408–17.
Horie T, Ono K, Nishi H, Iwanaga Y, Nagao K, Kinoshita M, Kuwabara Y, Takanabe R, Hasegawa K, Kita T, et al. Microrna-133 regulates the expression of glut4 by targeting klf15 and is involved in metabolic control in cardiac myocytes. Biochem Biophys Res Commun. 2009;389(2):315–20.
Liang J, Liu C, Qiao A, Cui Y, Zhang H, Cui A, Zhang S, Yang Y, Xiao X, Chen Y, et al. Microrna-29a-c decrease fasting blood glucose levels by negatively regulating hepatic gluconeogenesis. J Hepatol. 2013;58(3):535–42.
Wicik Z, Gajewska M, Majewska A, Walkiewicz D, Osinska E, Motyl T. Characterization of microrna profile in mammary tissue of dairy and beef breed heifers. J Anim Breed Genet. 2016;133(1):31–42.
Ahn J, Lee H, Jung CH, Jeon TI, Ha TY. Microrna-146b promotes adipogenesis by suppressing the sirt1-foxo1 cascade. EMBO Mol Med. 2013;5(10):1602–12.
Elsarraj HS, Hong Y, Valdez K, Carletti M, Salah SM, Raimo M, Taverna D, Prochasson P, Bharadwaj U, Tweardy DJ, et al. A novel role of microrna146b in promoting mammary alveolar progenitor cell maintenance. J Cell Sci. 2013;126(Pt 11):2446–58.
Faylon MP, Baumgard LH, Rhoads RP, Spurlock DM. Effects of acute heat stress on lipid metabolism of bovine primary adipocytes. J Dairy Sci. 2015;98(12):8732–40.
Gregor MF, Misch ES, Yang L, Hummasti S, Inouye KE, Lee AH, Bierie B, Hotamisligil GS. The role of adipocyte xbp1 in metabolic regulation during lactation. Cell Rep. 2013;3(5):1430–9.
Kozomara A, Griffiths-Jones S. Mirbase: integrating microrna annotation and deep-sequencing data. Nucleic Acids Res. 2011;39(Database issue):D152–7.
Akers RM, Ellis SE, Berry SD. Ovarian and igf-i axis control of mammary development in prepubertal heifers. Domest Anim Endocrinol. 2005;29(2):259–67.
Musters S, Coughlan K, McFadden T, Maple R, Mulvey T, Plaut K. Exogenous tgf-beta1 promotes stromal development in the heifer mammary gland. J Dairy Sci. 2004;87(4):896–904.
Roarty K, Serra R. Wnt5a is required for proper mammary gland development and tgf-beta-mediated inhibition of ductal growth. Development. 2007;134(21):3929–39.
Mukhopadhyay C, Zhao X, Maroni D, Band V, Naramura M. Distinct effects of egfr ligands on human mammary epithelial cell differentiation. PLoS One. 2013;8(10):e75907.
Whyte J, Bergin O, Bianchi A, McNally S, Martin F. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res. 2009;11(5):209.
Acknowledgements
We thank Zhengfeng Zhang for his great technical assistance.
Funding
This work was supported by National Natural Science Funds (31402056, 31340067) in designing the study and sample collection, Youth Talent Project in Hebei Province (BJ2014048) in analysis and interpretation of data, and the Doctoral Foundation of Langfang Teachers University (LSLB201404) in writing the manuscript in China.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its Additional files.
Author information
Authors and Affiliations
Contributions
QLL, CFW, and JFZ designed the experiments and drafted the manuscript. CHY, JD, BGZ, and YH collected the samples, provided advices, and revised the manuscript. QMH, MRL, CHY, JD, and YMZ carried out the experiments and organized the research team. QLL, BGZ, and CFW analyzed the data. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Animal studies were approved by the Animal Care and Use Committee of Langfang Normal University, Langfang, Hebei, China. We obtained written permission from the owner of the cattle to take samples.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
The primers of real-time quantitative RT-PCR used in this study. (XLS 31 kb)
Additional file 2:
Novel miRNAs identified in this study. (XLS 45 kb)
Additional file 3:
The differential expression of miRNAs in heat stressed and non-heat stressed groups. (XLS 33 kb)
Additional file 4:
P-values for the differentially expressed miRNAs. (XLS 23 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Li, Q., Yang, C., Du, J. et al. Characterization of miRNA profiles in the mammary tissue of dairy cattle in response to heat stress. BMC Genomics 19, 975 (2018). https://doi.org/10.1186/s12864-018-5298-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5298-1