
Abstract
Seven classes of mitogen-activated protein kinase (MAPK) intracellular signalling cascades exist, four of which are implicated in breast disease and function in mammary epithelial cells. These are the extracellular regulated kinase (ERK)1/2 pathway, the ERK5 pathway, the p38 pathway and the c-Jun N-terminal kinase (JNK) pathway. In some forms of human breast cancer and in many experimental models of breast cancer progression, signalling through the ERK1/2 pathway, in particular, has been implicated as being important. We review the influence of ERK1/2 activity on the organised three-dimensional association of mammary epithelial cells, and in models of breast cancer cell invasion. We assess the importance of epidermal growth factor receptor family signalling through ERK1/2 in models of breast cancer progression and the influence of ERK1/2 on its substrate, the oestrogen receptor, in this context. In parallel, we consider the importance of these MAPK-centred signalling cascades during the cycle of mammary gland development. Although less extensively studied, we highlight the instances of signalling through the p38, JNK and ERK5 pathways involved in breast cancer progression and mammary gland development.
Similar content being viewed by others


Introduction
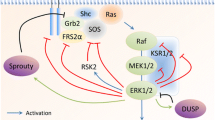
Four dominating mitogen-activated protein kinase (MAPK) signalling cascades are implicated in breast disease in mammalian cells: the extracellular regulated kinase (ERK)1/2 pathway, the c-Jun N-terminal kinase (JNK) pathway, the p38 pathway and the ERK5 pathway [1, 2]. In some forms of human breast cancer and in many experimental models of breast cancer progression, signalling through the ERK1/2 pathway, in particular, has been implicated as being important [3]. Signalling through each pathway involves sequential activation of a MAPK kinase kinase (MAPKKK), a MAPK kinase (MAPKK) and the MAPK. Considering the ERK1/2 pathway, the primary input activator is activated Ras, a small GTPase. It activates Raf1 (MAPKKK), which then phosphorylates and activates MEK1/2 (MAPKK), which finally activates ERK1/2 [1]. In its active form, ERK1/2 phosphorylates a wide range of protein substrates (on serine or threonine). MAPKKKs and MAPKKs are dual Thr/Tyr kinases. Signalling through the core to activate the MAPK is supported on a protein scaffold [4]. However, activated ERK1/2 is translocated from the scaffold to, for example, the nucleus, where it phosphorylates its substrates. The activity of the core Raf1-MEK1/2-ERK1/2 cascade is auto-regulated by ERK1/2 inactivation of MEK1 or by ERK1/2 upregulation of ERK1/2-specific phosphatases, for example, MKP2 and 3 (MAPK dual-specificity phosphatases) [5, 6]. In this way, ERK1/2 itself can exert control over the intensity and duration of ERK1/2 signalling.
The ERK1/2 cascade is triggered by growth factors and cytokines acting through receptor tyrosine kinases, G-protein-coupled receptors, and non-nuclear activated steroid hormone receptors [4, 7–13]. ERK1/2 substrates include transcriptional regulators, apoptosis regulators and steroid hormone receptors (for example, oestrogen receptor (ER)α). The biological consequences of ERK1/2 substrate phosphorylation include pro-proliferative [14], pro-differentiation [14], pro-survival [14], pro-angiogenic [15], pro-motility [16] and pro-invasive effects [17]. We review the contribution of MAPK signalling events to breast cancer progression as reflected in experimental models of progression (contrasting this to their role in normal mammary gland development).
ERK1/2 signalling in models of breast cancer progression
In approximately 30% of human breast cancers, mutations are found in the ERK1/2 MAPK pathway; however, the nature of the deregulation varies between individual tumours and cancer subtypes [18, 19]. Dys-regulated ERK1/2 signalling alone is usually not sufficient to cause cancer. Additional mutations and changes in the expression levels of other genes is commonly observed alongside (for example, those encoding ErbB2 receptor, c-myc, p53 and hyaluronan-mediated motility receptor (RHAMM)) [20, 21]. Furthermore, other major signalling networks (such as phosphoinositide 3-kinase (PI3K)/phosphatase and tensin homolog (PTEN)/AKT) crosstalk with ERK1/2 signalling, thereby enhancing cell proliferation, preventing apoptosis and, in some cases, inducing drug resistance [22]. ERK1/2 is hyper-activated in a large subset of mammary tumours, and downstream ERK1/2 targets are also hyper-phosphorylated there [23]. There is a trend for higher ERK1/2 activity in primary tumours of 'node-positive' patients than in those from 'node-negative' patients, suggesting higher ERK1/2 activity impacts metastasis [23]. A survival analysis demonstrated that low ERK1/2 activity in primary breast tumours is prognostic for relapse-free survival of patients [23]. The expression and activity of ERK1/2 can also impact a patient's response to treatment [3, 24]. Over-expression of upstream ERK1/2 modulators (such as MEK1) in normal mammary epithelial cells induces neoplastic transformation, highlighting the relevance of this signalling network to breast cancer [25]. Although balanced against the above are a series of clinical studies in three cohorts of patients that link high ERK1/2 and/or high activated (phosphorylated) ERK1/2 levels to good disease outcome [26–28]; the apparent contradictory nature of the two streams of data remains to be resolved.
Immortalized mammary epithelial cell lines with normal phenotype (including MCF10A/12A, S1, NMuMG, epH4, human mammary epithelial cell and HB2), breast cancer cell lines (including MCF-7, T47D, MDA-MB-231, T-42, SKBR-3, BT549) and primary mammary epithelial cells (of both normal and tumour origin) can be cultured and are commonly used to model the molecular contribution of MAPK signalling in normal epithelia and in breast cancer. We describe a range of such studies. In addition, we present a summary in Table 1.
Sustained ERK1/2 signalling undermines epithelial cell organisation
Primary cells and various phenotypically normal mammary epithelial cell lines, when supported on a reconstituted basement membrane (extracellular matrix (ECM)) mimic in vivo alveolus cell organisation and form three-dimensional spherical acini. Firstly, the outer layer of cells in contact with the ECM polarise and exit the cell cycle. Lumen clearance then occurs (at least in part by apoptosis) to generate the acinus in which the mammary epithelial cells (MECs) are organised in organotypic fashion [29]. Of particular interest is that transformed MECs do not organise in this fashion and their failure to do so is often determined by persistent ERK1/2 signalling (reviewed in [30]). For instance, tumourigenic HMT-3522 T-42 MECs fail to organise as acini because of a persistent β1-integrin-epidermal growth factor receptor (EGFR)-ERK1/2 drive. However, they 'revert' and form acini (like their isogenic 'normal' S-1 cell counterparts) if β1-integrin, EGFR or ERK1/2 function is inhibited [31]. Importantly, this bidirectional cross-modulation of β1-integrin and EGFR signalling via the MAPK pathway is dependent upon a three-dimensional context. Wang and colleagues [31] showed how partial 'reversion' in functional phenotype of a range of breast cancer cells could be achieved by inhibiting ERK1/2. A more complete 'reversion' was possible with multiple signalling inhibitors (for example, of ERK1/2 plus PI3K pathways). Thus, persistent ERK1/2 signalling contributes to, but need not necessarily be the only, intracellular signalling change that is driving loss of epithelial characteristics in breast cancer cells. Finally, in breast cancer SKBR-3 cells grown in monolayer culture, ErbB2 and ErbB3 form heterodimers, whereas in multicellular spheroids ErbB2 homodimers are formed, leading to enhanced activation of ErbB2 and a switch in signalling pathways from PI3K to ERK1/2 [32]. Thus, comparisons of three-dimensional and monolayer cultures emphasise the use of three-dimensional systems to better reflect some in vivo aspects of ErbB and ERK1/2 signalling.
Expression of an oncogene was found to be sufficient to impair acinus formation by normal MECs while in parallel triggering activation of ERK1/2 signalling [33]. Persistent activation of fibroblast frowth factor (FGF) receptor in HC11 MECs caused ERK1/2 activation, impaired acinus formation and led to an invasive phenotype [34]. Likewise, persistent activation of ErbB2 in MCF-10A cells, which is associated with activation of ERK1/2, caused aberrant acinus maturation but the cells were not invasive [35]. However, addition of transforming growth factor (TGF)β1 induced a migratory and invasive behaviour in these cells. Significantly, blocking ERK1/2 activation inhibited the effect of TGFβ1 and ErbB2 on migration.
Nolan and colleagues [36] show that, in three-dimensional assemblies, activated ErbB2 recruits the cell polarity regulators Partitioning defect (Par)6-atypical protein kinase C (aPKC) from their association with Par3. This disrupted cell polarisation and caused luminal cell survival (but not additional cell proliferation) and the formation of dysmorphic, multi-acinar spheres. In contrast, if Par6 is over-expressed, it acts in a complex with cdc42 and aPKC to cause hyper-proliferation and generate multi-acinar structures in an ERK1/2-dependent fashion [36]. Thus, disrupting the cell polarisation machinery can affect control of cell proliferation and/or cell survival and so impact lumen clearance.
The Bcl-2 homology domain (BH3)-only pro-apoptotic proteins Bim and Bmf (Bcl-2 modifying factor) selectively trigger apoptosis of central cells, leading to lumen formation in the acinus [37, 38]. Oncogenes that drive luminal filling, such as ErbB2, suppress Bim expression and drive Bim inactivation by phosphorylation in an ERK1/2-dependent manner. Activation of the ERK1/2 pathway is sufficient to block Bim activity and correlates with protection from luminal apoptosis and inhibition of lumen formation [37]. In a parallel fashion, in pubertal mammary glands of Bim-/- mice, terminal end buds and ducts have filled lumena, presumably due to a deficit in pro-apoptotic signalling [39]. Thus, there is ample evidence supporting the identification of the ERK1/2 MAPK node as a signalling driver that must be tightly controlled if mammary epithelial cells are to successfully organise, organotypically.
Invasion - a role for ERK1/2 signalling
Metastatic breast tumours represent the final lethal stage of the disease, with cancer cells from the primary mammary gland site having 'spread' to a secondary site (for example, bone, lung or liver). 'Invasive' cells with de-regulated growth properties and an acquired ability to degrade basement membrane invade the local tissue environment, eventually reaching and circulating through the lymph and vascular systems before entering other tissues and forming secondary tumours. Epithelial cells that have undergone an epithelial-to-mesenchymal transition (EMT) display many of the characteristics of invasive cancer cells, in that they have lost their adhesions and polarised phenotype, have down-regulated epithelial-specific proteins (such as tight junction components) and have gained a proliferative and migratory/invasive phenotype with an ability to degrade and migrate through ECM. A range of growth factors and cytokines (for example, human growth factor, FGF and TGFβ) have been found to trigger EMT [40–45]. But in particular, cooperation between TGFβ and sustained ERK1/2 signalling induces EMT and an invasive and metastatic potential in cultured mammary epithelial cells [40]. TGFβ signals through trans-membrane receptor kinases and activates p38 and ERK1/2 through both Smad-dependent and Smad-independent mechanisms and the MAPK4 pathway [46, 47]. TGFβ inhibits growth of normal epithelial cells but induces proliferation and EMT in immortalised, phenotypically normal MCF10A cells and in cells from advanced carcinomas [48].
During normal mammary gland development, TGFβ isoforms seem required for pubertal ductal morphogenesis [49], pregnancy-induced alveolar morphogenesis (in part by mediating the action of progesterone) [49, 50], for normal transition from pregnancy to lactation and to drive involution [51]. In pubertal mammary gland development, TGFβ inhibits ductal growth and its role may be in sculpting the newly developing mammary tree: branches form at sites of lowest local TGFβ concentration [52]. Little is known of intracellular signalling networks that support ductal branching; however, in a pubertal mammary explant culture it was shown that sustained ERK1/2 activation induced by TGFα caused branching, while transient ERK1/2 activation induced by FGF7 did not. It was hypothesised that the former would trigger necessary matrix-metalloproteinase (MMP) expression and the latter not [7].
Invasive cells synthesise MMPs, which degrade type IV collagen (and other matrix components) and confer on cells the ability to migrate and invade neighbouring tissue. ERK1/2 signalling induces MMP expression/activity, thereby impacting extracellular re-modelling [53]. In breast cancer cell cultures, invasiveness and MMP2 and MMP9 activity are induced in response to TGFβ stimulation and ErbB2 over-expression [54, 55] and requires p38 activity in some circumstances. The serine protease uPA (Plasminogen activator, Urokinase) and its receptor (uPAR) also play a crucial role in breast cancer cell invasion and metastasis alongside MMPs. uPA induces cell proliferation in MDA-MB-231 breast cancer cells via ERK1/2 activation and binding of uPA to uPAR activates ERK1/2 in MCF-7 cells [8, 56]. Endogenously produced uPA was found to be a major determinant of the basal level of activated ERK1/2 and prevented apoptosis in metastatic MDA-MB-231 cells [57]. ERK1/2 signalling in MDA-MB-231 breast carcinoma cells is adapted to obtain high urokinase expression and rapid cell proliferation; there, inhibition of ERK1/2 activity leads to decreased cell proliferation and decreased cyclin D1 expression [58]. In the normal mammary gland, MMPs orchestrate extensive reductive re-modelling in the later phase of involution via degradation of ECM, and MMP2, 3, 7, 9 and 11 are important to this processing [59], which is probably driven by cytokine-triggered ERK1/2 activity (see below) [60, 61].
ERK1/2 activity has been highlighted in several studies of invasion (reviewed in [62]). Cell-cell adhesion molecules (including E-cadherins and tight junction proteins such as ZO-1 (zona occludens 1)) play a pivotal role in maintaining normal breast epithelial architecture. A hallmark of EMT is the loss of E-cadherin expression and an increase in the expression of N-cadherin and vimentin. EGF-induced EGFR activation destabilises E-cadherin adhesions [63]. However, it is known from several studies that re-expression of E-cadherin alone may not be sufficient to restore regulated cell growth and epithelial-like behaviour. For example, in MCF10A cells over-expressing activated Ras, restoration of an epithelial cell phenotype required both the over-expression of E-cadherin and the suppression of ERK1/2 activity [64]. Overexpression of N-cadherin in MCF-7 cells induces cell migration in vitro and invasion and metastasis in vivo by an ERK1/2-dependent mechanism [65, 66].
Polarity proteins (such as Par-, Crumbs- and Scribble-complex proteins) determine the establishment and maintenance of cell polarity in epithelial cells (as reflected, for instance, by the asymmetric distribution of cadherin and tight junctions). But they have been shown to redeploy and cooperate in a sustained ERK1/2 signalling environment to induce abnormal cell growth and invasive potential [36]. And, suppressed expression of the polarity regulator Scribble in MCF10A cells was found to co-operate with mutations in Ras and Raf to induce a migratory phenotype via induction of ERK1/2 (but not p38 or JNK) signalling [67].
Alongside a loss of cell-cell adhesions and polarity, cytoskeleton re-modelling and the formation of cellular protrusions are a feature of breast cancer cell migration and early metastasis. The dynamics and regulation of the actin cytoskeleton is central to migratory processes and may be corrupted at several levels in invasive cancers (reviewed in [68]). Induced ERK1/2 signalling can inactivate integrin-mediated cell adhesion and activate myosin light chain kinase, thereby impacting cell migration [69]. Also central to migratory processes are focal adhesion components, for instance Focal adhesion kinase, which is activated by integrins and by growth factor receptors, including the EGFR family [70]. Focal adhesion kinase is hyper-activated during the early stages of invasion responses in cultured breast cancer cells and was found to both stimulate cell proliferation [60] and inhibit apoptosis [71]. It is also hyper-activated in ErbB2-positive breast cancers [72]. Interestingly, mice with conditional Focal adhesion kinase knockout in the mammary gland display decreased alveolar epithelial cell proliferative capacity during late pregnancy, with reduced cyclin D1 expression and reduced ERK1/2 phosphorylation [73].
Another environmental cue, tissue rigidity (due to a stiffer ECM), can promote tumour progression (reviewed in [74]). ECM changes impact integrin signalling and can promote mitogenic signalling through EGFR-ERK1/2 signalling and also cell contractility through Rho, which can further increase matrix stiffness [75], contributing to the cycle of tumour growth and progression.
Key ERK1/2 substrates, the Ets-1 transcription factors, have been shown to induce EMT and invasiveness in a series of human breast cancer cell lines. The Ets-factor-ESE-1 transforms MCF-12A mammary cells in culture [76]. Ets-1 activity promotes breast cancer cell metastasis (and ERK1/2 signalling) by driving the expression of MMPs, FGF, uPA, uPAR and EGF [77, 78].
'Cancer stem cells' are potentially rare cells with both stem and tumourigenic properties. They have an indefinite potential for self-renewal and are hypothesised to drive tumourigenesis [79]. The existence of a subpopulation of breast cancer stem cells was demonstrated recently when they were isolated from breast cancer pleural effusions by limiting dilution transplantation of the cells into the mammary fat pad of immuno-compromised mice [80]. 'Tumour-initiating cells' can be derived from cultured mammary cells following the activation of the ERK1/2 pathway and induction of EMT and such cells are found in ErbB2-induced mammary tumours [81, 82]. While the capability of stimuli that drive EMT to enhance the abundance of breast cancer stem cells is most interesting and will provide a ready experimental model in which to study their generation, the relevance of the phenomenon to breast tumour progression remains to be established.
ErbB/EGFR family signalling to ERK1/2
Of the growth factor receptors that input signals to ERK1/2 in breast tumour cells, the ErbB/EGFR family of receptor tyrosine kinases is one of the major classes: they are commonly over-expressed (for example, ErbB2 gene amplification occurs in 20 to 30% of tumours [19]) or persistently signal (for example, EGFR in tamoxifen resistance [83]) in human breast tumours. An anti-receptor antibody, trastuzumab, is used with some success in treating disease with amplified ErbB2; gefitinib, an EGFR kinase inhibitor, is used to treat patients exhibiting tamoxifen resistance [84]. A family of ten ligands, including EGF, amphiregulin and heregulin, bind the ErbB receptors, induce receptor dimerization, trigger kinase activity, autophosphorylation and downstream activation of ERK1/2 and PI3K-AKT signalling. There are four closely related ErbB receptors (ErbB1/EGFR, ErbB2/Her-2/Neu, ErbB3 and ErbB4) that form hetero- or homodimers. ErbB receptor signalling can increase cell proliferation, decrease apoptosis and affect the survival and motility of primary and metastatic breast tumour cells. ErbB2 action also enhances signalling interactions with the cellular microenvironment and affects cell adhesion (reviewed in [85]).
The potential contribution of the ErbB receptor-ERK1/2 signalling has been widely studied in experimental model systems of progression (reviewed in [19]). In a range of breast cancer cell lines hyper-proliferation is mediated by EGFR- or ErbB2-dependent ERK1/2 activation (reviewed in [86]). ErbB2-ERK1/2 signalling may affect breast cancer cell behaviour through influencing cell survival as much as through affecting proliferation: expression of ErbB2 in MCF-7 cells induces the anti-apoptotic proteins Survivin and Bcl-2 via ERK1/2 and PI3K signalling [87], and experimentally triggered ErbB2 activation protects against apoptosis and disrupts MCF10A epithelial cell three-dimensional organisation in an ERK1/2-dependent manner [88, 89]. Over-expression of ErbB2 induces EMT through ERK1/2 activation in MCF10A human breast epithelial cells [90]. While EGFR-ERK1/2 signalling is conventionally ligand-activated, this may often arise through an autocrine mechanism; MMP9 partially acts to drive invasiveness through induction of EGF expression [91]. Interestingly, in MCF10A and a range of breast cancer cell lines, activation or over-expression of ErbB2 has been shown to increase cell growth in vitro, to initiate tumours in vivo, to activate p38 and to induce cell migration via up-regulation of MMP-9 (which may involve autocrine EGFR activation). Common also is activation of this signalling by EGFR trans-activation, which has been shown to be induced by G-protein-coupled receptors [92, 93]. Furthermore, in a single cell line insulin-like growth factor (IGF)-1, vascular epidermal growth factor (VEGF), growth hormone and a range of ligands all require EGFR to induce ERK1/2 activation [91]. In addition, the progesterone receptor was recently found to upregulate WNT-1 and to induce EGFR transactivation and a sustained activation of ERK1/2 in T47D cells growing in an adhesion-independent fashion in soft agar [94].
Promoters that selectively drive gene expression in the mammary gland (that is, those for mouse mammary tumour virus (MMTV) and whey acidic protein) have allowed the creation of useful mouse models for studying breast cancer in vivo. The best characterised models are those expressing the viral oncogene encoding the polyoma middle T antigen (MMTV-PyV MT) as paradigm of viral transforming potential, and that expressing ErbB2 (MMTV-Neu) [95, 96]. A complementation study that crossed tyrosine phosphatase PTP1B knockout mice into this ErbB2 over-expression background led to down-regulation of ERK1/2 signalling and delayed tumour development, suggesting a role for the ERK1/2 pathway in ErbB2-induced mammary tumourigenesis [97, 98]. A direct role for ERK1/2 in induction of mammary tumour progression has been recently shown in vivo, using cells transplanted into the mammary fat pad [99]. The authors showed that ERK1/2 directly modulates the phosphorylation and degradation of the tumour suppressor FOXO3a (Forkhead box 3), leading to a decrease in the anti-tumourigenic activity of this protein in the mammary gland. Lastly, the EphA2 receptor tyrosine kinase, a regulator of mammary gland branching morphogenesis [100], which drives Ras-ERK1/2 and RhoA GTPase signalling, has been shown to enhance tumour formation in MMTV-Neu mice, but not in MMTV-PyV MT mice [101], again potentially linking the ERK1/2 pathway to ErbB2-induced tumourigenesis.
Thus, in breast cancer, EGFR/ErbB receptors are commonly over-expressed [19] or persistently signal (in tamoxifen resistance [83]), and an anti-receptor antibody is being used with success in treating disease with amplified ErbB2, and an EGFR kinase inhibitor is being used to treat tamoxifen-resistant disease [84]. In cell culture-based experimental model systems it is clear, as outlined above, that a functional link can be made between EGFR/ErbB receptor activation, induction of ERK1/2 pathway signalling and relevant outcomes such as increased proliferation [102], cell survival [103] and induction of motility [16] and invasiveness [17]. But does this qualify the MEK1-ERK1/2 signalling pathway as a useful therapeutic target in breast cancer, generally? Available clinical evidence linking high activated ERK1/2 levels to a good disease outcome [26–28] would say no. But, for instance, in a subset of cases where anti-EGFR therapy fails there may be some potential [84]. But perhaps the most useful experimental endeavour would be to develop additional cell-based models to more accurately mimic progression of particular disease subtypes in order to define the dominant supporting intracellular signalling pathways and, thus, new key therapeutic targets.
Oestrogen receptor, tamoxifen resistance and ERK1/2 signalling
The biological actions of oestrogen are mediated both by transcriptional regulation (involving ERα and ERβ in the nucleus) and by non-genomic actions via ligand binding to ERα at the plasma membrane [104–106]. In addition, ERK1/2 phosphorylates ERα on sites in its amino-terminal ligand-independent transcriptional activating domain (Ser118 and Ser167) [83, 107]. Ser167 is phosphorylated by AKT, p90 Ribosomal S6 kinase, and casein kinase II as well as by ERK1/2 [108]. Phosphorylation on Ser118 by ERK1/2 increases the transcriptional activation function, thus providing a means by which ERα can be activated in a ligand-independent manner through growth factor receptor signalling. It may also facilitate blocking of inhibitory actions of anti-oestrogens/selective ER modulators on ERα in breast cancer cells and, hence, contribute to tamoxifen resistance; the phosphorylation event may act to reduce the binding of the high affinity ligand, 4-hydroxy-tamoxifen, to ERα [83]. It has been shown that ErbB2-over-expressing MCF-7 cells are tamoxifen resistant; they have high levels of activated ERK1/2 and high levels of phosphorylation of ERα at Ser118 [109]. MEK inhibitors reverse this resistance [110]. But interestingly, several studies have linked elevated levels of phosphorylation at Ser118 of ERα to a positive survival outcome for breast cancer patients [26, 111]. One study positively correlates elevated levels of ERK1/2 activation with elevated phosphorylation of ERα at Ser118 and with a good survival outcome [26]; here also phosphorylation of ERα at Ser118 inversely correlated with ErbB2 expression, suggesting that ErbB2 is not primarily signalling through ERK1/2 in this context [83]. The relationship of phosphorylation at the other ERK1/2 target, Ser167, to outcome for patients and to tamoxifen resistance is less than clear; suffice it to say that AKT rather than ERK1/2 would seem to be its major generator [83].
ERK1 and ERK2 are also activated via oestrogen signalling through the G-protein-coupled receptor GPR30, which results in transactivation of EGFR in MCF-7 cells and in SKBR3 breast cancer cells [10]. However, GPR30 may play a broader role in balancing ERK1/2 activation as oestrogen signalling via the G-protein-coupled receptor inhibits ERK1/2 activity by a cAMP-dependent mechanism, which results in Raf-1 inactivation in breast carcinoma cells [112]. It is implied that oestrogen may drive cellular responses that are growth factor dependent via GPR30 in ER-negative breast tumours [10].
Splice isoforms of ER exist that account for the heterogeneity of ER gene expression, which is also tissue-specific [113, 114]. Post-translational modification by palmitoylation recruits an abundant amino-terminally truncated ERα (46 kDa) to the plasma membrane in human endothelial cells, osteoblasts and MCF-7 cells [113, 115, 116]. ER46 mediates rapid membrane-initiated oestrogen responses, including activation of endothelial nitric oxide synthetase [116, 117]. A functional signalling node composed of ER46, c-Src and p85 (the regulatory subunit of PI3K) has been identified in the oestrogen-induced activation of endothelial cells, and is directed by c-Src [118]. The importance of the presence of membrane associated isoforms of ER lie in their ability to illicit rapid and non-transcriptional responses to oestrogen [114].
Anti-hormone treatment with selective ER modulators, most commonly tamoxifen, act to inhibit ER activity in breast cancer cells and their metastases; aromatase inhibitors block oestrogen biosynthesis and reduce endogenous ligand-dependent ER activation [83, 119]. In suitable patient cohorts both therapies prolong survival, but resistance to these therapeutics limits their use [83]. In cell culture studies in which EGFR or ErbB2 have been over-expressed and anti-oestrogen resistance induced [120], or resistance induced by prolonged exposure to anti-oestrogens [9, 121], resistance correlated with high ERK1/2 and AKT activity. There is accumulating evidence that growth factor-triggered ERK1/2 signalling contributes significantly to tamoxifen resistance but it may do so by multiple mechanisms. Firstly, both tamoxifen-resistant tumours and cell lines tend to be rich in EGFR and ErbB2 receptors and their activation of ERK1/2 and PI3K-AKT would have significant anti-apoptotic effects. Secondly, ERK1/2 phosphorylation of Ser118 in ERα may also contribute to resistance [83]. And finally, non-nuclear ER activity may contribute: the IGF receptor (IGF-1R) acts upstream of ErbB1 in a signalling pathway in MCF-7 cells, which mediates oestrogen activation of ERK1/2, with subsequent cell growth stimulation and protection against apoptosis. Oestrogen stimulated the phosphorylation of both IGF-1R and ErbB1, and inhibitor studies that blocked this pathway also blocked ERK1/2 activation and the mitogenic and anti-apoptotic effects of oestrogen [9].
The tamoxifen resistance phenotype is predominantly associated with tumour re-growth rather than metastasis [83]. However, Santen and colleagues [3] comment that the phenotype (potentially sustained by EGFR/ErbB2-ERK1/2/PI3K-AKT signalling) is that that would promote invasion and metastasis. Interestingly, when over-expressing Ras mutants in NIH 3T3 fibroblasts to activate downstream effectors (Raf-ERK1/2, PI3K or RalGEF) and, thereby, tumour growth in nude mice, it was found that only the mutant that activated ERK1/2 (V12S35 Ras expressed in NIH 3T3 fibroblasts) also induced lung metastasis [122], highlighting the potential importance of sustained ERK1/2 signalling in metastasis.
Pubertal mammary gland ductal morphogenesis in the mouse requires epithelial cell ERα-induced EGFR ligand (amphiregulin) expression, as indicated by knockout/mammary fat pad transplantation studies [123]. Additional knockout/transplantation studies indicated that the target for the amphiregulin was stromal cell EGFR, without which the phased epithelial proliferation and branching of mammary tree development fails [124]. This relationship and its morphological outcome contrasts with the ER-EGFR-family-ERK1/2 signalling interactions seen in 'ErbB2 amplified' breast tumours and tamoxifen-resistant disease, where we suppose a dominant and sustained ERK1/2 signalling contributes. Additional studies have demonstrated the presence of an ER-controlled Src-ERK1/2 phosphorylation cascade functioning during pubertal ductal morphogenesis [125].
Cell survival and cell death
Both ERK1/2 activation and its inhibition can induce apoptosis, with the output dependent on the cellular context and input signalling. ERK1/2 plays a role in apoptosis induced by a range of therapeutics in breast cancer cells. ERK1/2 and p38 signalling was found to be required for resveratrol-induced apoptosis in MDA-MB-231 breast cancer cells [126, 127] and activation of ERK1/2 and JNK, but not p38, was found to be required for vitamin E succinate-induced apoptosis of MDA-MB-435 breast cancer cells [128]. Genistein, Oligonol, kaempferol, and linoleic acid all induce apoptosis in breast cancer cells in an ERK1/2-dependant manner [129–131].
ERK1/2 phosphorylates the pro-apoptotic BCL-2 family member BimEL (a BH3-only domain protein) [132], leading to its degradation by the proteasome [133]. Bim initiates apoptosis, but after survival factor-induced ERK1/2-phosphorylation, Bim's association with Bax is inhibited, as is its pro-apoptotic activity, leading to cell survival [134]. As outlined earlier, Bim drives cell death during lumen clearance in the ductal terminal end buds of the developing pubertal mammary tree and during acinus formation [37, 39]. But importantly, it drives the cell death of epithelial anoikis, whose inhibition is permissive for invasion and metastasis [135]. ERK1/2 action, regulated by EGFR or coordinately by EGFR and β1-integrin, negatively regulates Bim activity both at a transcriptional level and by phosphorylation; the former may be the principal mode of regulation [37]. The related BH3-only domain protein Bmf is expressed during involution, and is active in epithelial cell anoikis (monolayer culture) and in apoptosis/lumen clearance during acinus formation. Sustained ERK1/2 or PI3K activation suppresses this Bmf up-regulation, suggesting that, in this instance, active ERK1/2 was pro-survival [38]. Growth and survival factor-activated ERK1/2 signalling can also regulate Hdm2 expression (a negative regulator of p53), contributing to a pro-survival effect [136]. p53 itself can activate ERK1/2 signalling via transcriptional activation of the EGF gene [137]. Expression of p53 in other circumstances can reduce phospho-ERK1/2 levels and this inhibition of ERK1/2 signalling accompanies apoptosis inducing factor-mediated apoptosis in MCF-7 cells in response to metals [138]. The authors also found that suppression of p53 in MDA-MB231 and MCF7-E6 cells resulted in strong ERK1/2 phosphorylation.
Normal mammary gland involution is characterized by extensive alveolar epithelial cell apoptosis in its initial stages, and later by matrix degradation and tissue re-modelling directed by matrix-degrading MMPs 2, 3, 7 and 11, the plasminogen activation pathway and cathepsins (reviewed in [59]). In conditional Stat3-/- mice involution is delayed [139]. Stat3 is thus pro-apoptotic. IL6 expression increases with the onset of involution, and parallels the activation of Stat3 and ERK1/2 [61, 140]. Leukemia inhibitory factor also mediates ERK1/2 activation at this time; however, sustained activation of ERK1/2 through to late involution requires Oncostatin M. It is suggested that the controlled re-modelling of mammary tissue at involution is regulated by IL-6, leukemia inhibitory factor and Oncostatin M, acting via ERK1/2 and by driving expression of pro-apoptotic Bax [61].
p38, JNK and ERK5
p38
p38 MAPK plays roles in cell differentiation, growth inhibition and apoptosis [141, 142]. p38 MAPK is activated in cells in response to stress signals, growth factors, inflammatory cytokines, UV, heat and osmotic shock [142]. Its activation often leads to cell death. In addition, a new role for p38 in 'checkpoint control' during cell division and cell survival is now recognized [143]. Four isoforms of p38 exist (p38α, β, γ and δ), although p38α is the most widely expressed. MKK3/6 (MAPKKK) and SEK (MAPKK) activate p38. p38 is also activated via interactions with the scaffolding protein TAB1 [144]. Activated p38 phosphorylates and activates many transcription factors (including Activating transcription factor-2, Max, Myocyte enhancer factor-2, Mac, p53 and Stat1) [145, 146].
Links between p38 and invasiveness in cultured cells have been reported: phospho-p38 is elevated in cultured invasive breast cancer cells and constitutive p38 activity induces the overproduction of the pro-invasive uPA [147]. Also, treatment of the invasive BT549 breast cancer cells with a p38 MAPK inhibitor diminished both uPA and uPAR expression and inhibited the ability of these cells to invade matrigel [147, 148]. Furthermore, activated Src over-expression was shown to necessarily activate p38 during TGFβ-induced breast cancer cell proliferation and invasion [149]. Studies on mammary cells indicate that WAVE-3 (a regulator of cytoskeletal dynamics and cell motility) regulates breast cancer progression, invasion and metastasis through the p38 pathway and MMP production. Knockdown of WAVE3 using small interfering RNA in MDA-MB-231 cells decreases p38 activity but not AKT, ERK1/2 or JNK [150]. Elevated phospho-p38 levels have been associated with high expression of EGFR and ErbB2 as well in tamoxifen-resistant xenografts [151]. p38 would not be seen as driving apoptosis in this context; maybe it acts to support nuclear functions of ER [152]. And interestingly, a relationship between p38 and phospho-p38 levels and lymph node metastasis was identified in human breast cancer samples [151]. Most interestingly, in a study of matched primary and recurrent tamoxifen-resistant tumours (and a parallel study of a mouse xenograph model of tamoxifen resistance) an association of phospho-p38 and elevated ErbB2 with tamoxifen resistance was found [151].
JNK
JNKs 1, 2 and 3 are activated by MKK 4 and 7, which are substrates of MAPKKKs (for example, MEKK1/2 or TGFβ-activated kinase 1). JNK has a wide range of opposing functions within cells but is best known for its role in triggering apoptosis after exposure to cellular and environmental stresses [153]. Classically, JNK phosphorylates c-Jun, leading to the binding of c-Jun to c-Fos and the activation of activator protein-1 (AP-1), which can ultimately promote cell proliferation [154].
JNK-supported proliferation has been reported for a number of cultured breast cancer cell model systems; inhibition of JNK resulted in cell cycle arrest during G2/M in three different breast cancer cell lines, leading to DNA endoreduplication (cellular DNA content greater than 4N), decreased proliferation, and apoptosis [155]. In MCF-7 cells, JNK was found to promote cell growth after induction by IGF-1 and a presumed autocrine stimulation [156]. In both a clinical study of matched pairs of ER-positive tamoxifen-sensitive and tamoxifen-resistant human breast tumours [157] and a xenograph model of tamoxifen resistance [158], a positive association of activated/phosphorylated JNK with tamoxifen resistance has been observed. In contrast, JNK action has been reported to contribute in a number of independent ways to the three-dimensional organisation of normal mammary epithelial cells (that is, during acinus development). The actions of glucocorticoids in supporting the patency of mammary epithelial cell tight junction adhesions is well described [159]. Murtagh and colleagues [160] showed that glucocorticoids act to support such activities during mammary epithelial acinus formation by activating JNK signalling in a BRCA1-dependent manner. Inhibition of JNK action during acinus formation blocks the establishment of cell polarisation, the formation of tight junctions, the AP-1-dependent expression of tight junction component proteins and lumen clearance, allowing the formation of tumour-like disordered assemblies of cells; the latter cascade of events is ERK1/2-dependent and reversible with a MEK1 inhibitor (E McArdle, unpublished observation). Interestingly, it has been subsequently shown that c-Myc induces cell death in MCF-10A cells during acinus formation by inducing formation of a Scribble-bPIX-GIT1 complex that activates a Rac-JNK-c-Jun-Bim apoptotic pathway. Inhibition of ERK1/2 in this context does not inhibit c-Myc-induced apoptosis, suggesting that c-Myc specifically uses the JNK pathway to induce cell death in ordered mammary epithelial cells [161]. These studies emphasise the tumour-suppressor-like activities of the JNK signalling pathway, at least in these cell culture model systems.
ERK5
The fourth MAP kinase of interest in this context is ERK5 [2]. Relatively, it is of large molecular size [162], with its activation being carried out by the MAPKK MEK5 [163], which is itself activated by MEKK2 or 3 [164]. While distantly related to, and sharing its mechanism of molecular action with, ERK1/2, gene knockout studies in mice show ERK5 to have non-redundant functions in heart development [165], vasculogenesis and angiogenesis [166] and endothelial cell survival [166]. Cell culture based experiments have pointed to ERK5 functioning at a molecular level in mitosis [167] and cell survival [168]. ERK5 is activated by growth factors [169], integrin engagement [170] and cell stress [167], and its important molecular targets would seem to include the induction of transcription of components of the transcription factor AP-1 (cJun [171] and Fos [172]) and activation of transcription factors of the myocyte enhancer family group (for example, MEF2C, a well characterised target [173]), and cMyc [174]. Serum and glucocorticod kinase is an interesting target kinase of ERK5 [175]; it may mediate cell survival influences of ERK5 to cellular stress [176]. Interestingly, in 20% of a human breast tumour set analysed, ERK5 levels were elevated and activated ERK5 (phospho-ERK5) was detected in many of the tumours. High levels of ERK5 were found to be an independent predictor of disease-free survival in this cohort and was associated with poor disease outcome [177]. Complementing these findings are, firstly, studies on ERK5 knockdown (using short hairpin RNA) in a human breast cancer cell line, which showed a sensitisation to the effects of ErbB 2 inhibitors [177]; secondly, studies in human breast cancer cell lines that associate constitutively active ERK5 with induced ErbB2, 3 and 4 activation [178]; and, thirdly, that such cells harbouring a dominant defective ERK5 show reduced proliferation rates, which may be due to failure to activate ErbB2 [178].
Future perspectives
We still do not have a full understanding of the key contributions made by MAPK signalling to the different stages of mammary gland development. But the clues we have suggest interesting possibilities: during pubertal ductal morphogenesis, quantitative temporal differences in ERK1/2 activity may trigger the decision to branch [7]; ERK1/2 signalling may be the final mediator of the proliferation associated with alveogenesis, of which β1-integrin is a significant driver [179]; glucocorticoid-JNK-cJun signalling may be the primary driver of tight junction 'closure' required at the transition from late pregnancy to lactation [160]; and JNK may also play a role in ductal development in the terminal end bud at puberty as judged from its role in other developmental epithelial systems [180, 181]. We await developments with interest. At the other end of the scale is the challenge of benefiting from the prognostic measurement of 'activated MAPK' (most immediately activated ERK1/2 and ERK5) levels in breast cancer. With anti-phospho-MAPK antibodies of good quality available, robust measurements of 'activated/phosphorylated-MAPK' can be made. However, this assesses potential rather than actual MAPK activity. Future analyses where phospho-MAPK levels are co-assessed with known molecular outputs of MAPK action will provide a true measure of activity and will be useful both from a prognostic point of view and in defining therapeutic targets. Of great importance also are therapeutic strategies that target the contribution of intracellular signalling networks that support disease progression in human breast cancer. It is suspected that signalling from over-expressed ErbB2 through ERK1/2 contributes to disease progression, but some studies suggest that signalling pathways other than the ERK1/2 pathway may be of equal or greater importance in tumours exhibiting ErbB2 over-expression [26]. EGFR-ERK1/2 signalling plays a role in disease progression in some forms of tamoxifen-resistant disease [84], and VEGF receptor-ERK1/2 signalling has also been linked to disease progression [26]. Currently, mono-therapies that target the receptor or ERK1/2 activity are predicted to be of limited value. Instead, combination therapies that target growth factor receptors, multiple signalling pathways and signalling targets point the way forward [83, 84]; an example of this would be an EGFR/ErbB2 inhibitor plus a multi-signalling pathway inhibitor (for example, the Ras farensyltransferase inhibitor, which blocks ERK1/2 activation and mTor (mammalian target of rapamycin) signals (or a Src inhibitor [182]) plus a 'full oestrogen antagonist' (for example, fulvestrant)). Rapid development of successful strategies in this area is keenly awaited.
Note
This article is part of a review series onKey signalling nodes in mammary gland development and cancer , edited by Adrian Lee and Charles Streuli.
Other articles in the series can be found online at http://breast-cancer-research.com/series/bcr_signalling_nodes
Abbreviations
- AP-1:
-
Activator protein-1
- aPKC:
-
atypical PKC
- BH3:
-
Bcl-2 homology domain 3
- ECM:
-
extracellular matrix
- EGF:
-
epidermal growth factor
- EGFR:
-
EGF receptor
- EMT:
-
epithelial-to-mesenchymal transition
- ER:
-
oestrogen receptor
- ERK:
-
extracellular regulated kinase
- FGF:
-
fibroblast growth factor
- IGF:
-
insulin-like growth factor
- IGF-1R:
-
IGF receptor
- IL:
-
interleukin
- JNK:
-
c-Jun N-terminal kinase
- MAPK:
-
mitogen-activated protein kinase
- MAPKK:
-
MAPK kinase
- MAPKKK:
-
MAPK kinase kinase
- MEC:
-
mammary epithelial cell
- MMP:
-
matrix-metalloproteinase
- MMTV:
-
mouse mammary tumour virus
- Par:
-
Partitioning defect
- PI3K:
-
phosphoinositide 3-kinase
- PKC:
-
protein kinase C
- TGF:
-
transforming growth factor
- uPA:
-
Plasminogen activator, Urokinase
- uPAR:
-
Urokinase receptor
- VEGF:
-
vascular epidermal growth factor.
References
Krishna M, Narang H: The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci. 2008, 65: 3525-3544. 10.1007/s00018-008-8170-7.
Wang X, Tournier C: Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal. 2006, 18: 753-760. 10.1016/j.cellsig.2005.11.003.
Santen RJ, Song RX, McPherson R, Kumar R, Adam L, Jeng MH, Yue W: The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002, 80: 239-256. 10.1016/S0960-0760(01)00189-3.
May LT, Hill SJ: ERK phosphorylation: spatial and temporal regulation by G protein-coupled receptors. Int J Biochem Cell Biol. 2008, 40: 2013-2017. 10.1016/j.biocel.2008.04.001.
Eblen ST, Slack-Davis JK, Tarcsafalvi A, Parsons JT, Weber MJ, Catling AD: Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol Cell Biol. 2004, 24: 2308-2317. 10.1128/MCB.24.6.2308-2317.2004.
Ramos JW: The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int J Biochem Cell Biol. 2008, 40: 2707-2719. 10.1016/j.biocel.2008.04.009.
Fata JE, Mori H, Ewald AJ, Zhang H, Yao E, Werb Z, Bissell MJ: The MAPK(ERK-1,2) pathway integrates distinct and antagonistic signals from TGFalpha and FGF7 in morphogenesis of mouse mammary epithelium. Dev Biol. 2007, 306: 193-207. 10.1016/j.ydbio.2007.03.013.
Nguyen DH, Webb DJ, Catling AD, Song Q, Dhakephalkar A, Weber MJ, Ravichandran KS, Gonias SL: Urokinase-type plasminogen activator stimulates the Ras/Extracellular signal-regulated kinase (ERK) signalling pathway and MCF-7 cell migration by a mechanism that requires focal adhesion kinase, Src, and Shc. Rapid dissociation of GRB2/Sps-Shc complex is associated with the transient phosphorylation of ERK in urokinase-treated cells. J Biol Chem. 2000, 275: 19382-19388. 10.1074/jbc.M909575199.
Song RX, Zhang Z, Chen Y, Bao Y, Santen RJ: Estrogen signalling via a linear pathway involving insulin-like growth factor I receptor, matrix metalloproteinases, and epidermal growth factor receptor to activate mitogen-activated protein kinase in MCF-7 breast cancer cells. Endocrinology. 2007, 148: 4091-4101. 10.1210/en.2007-0240.
Filardo EJ, Quinn JA, Bland KI, Frackelton AR: Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via transactivation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol. 2000, 14: 1649-1660. 10.1210/me.14.10.1649.
Liu ZX, Yu CF, Nickel C, Thomas S, Cantley LG: Hepatocyte growth factor induces ERK-dependent paxillin phosphorylation and regulates paxillin-focal adhesion kinase association. J Biol Chem. 2002, 277: 10452-10458. 10.1074/jbc.M107551200.
Boonstra J, Rijken P, Humbel B, Cremers F, Verkleij A, van Bergen en Henegouwen P: The epidermal growth factor. Cell Biol Int. 1995, 19: 413-430. 10.1006/cbir.1995.1086.
Schlessinger J: Cell signalling by receptor tyrosine kinases. Cell. 2000, 103: 211-225. 10.1016/S0092-8674(00)00114-8.
Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH: Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev. 2001, 22: 153-183. 10.1210/er.22.2.153.
Pagès G, Milanini J, Richard DE, Berra E, Gothié E, Viñals F, Pouysségur J: Signaling angiogenesis via p42/p44 MAP kinase cascade. Ann N Y Acad Sci. 2000, 902: 187-200.
Joslin EJ, Opresko LK, Wells A, Wiley HS, Lauffenburger DA: EGF-receptor-mediated mammary epithelial cell migration is driven by sustained ERK signalling from autocrine stimulation. J Cell Sci. 2007, 120: 3688-3699. 10.1242/jcs.010488.
Price DJ, Avraham S, Feuerstein J, Fu Y, Avraham HK: The invasive phenotype in HMT-3522 cells requires increased EGF receptor signalling through both PI 3-kinase and ERK 1,2 pathways. Cell Commun Adhes. 2002, 9: 87-102. 10.1080/15419060214147.
Cobleigh MA, Vogel CL, Tripathy D, Robert NJ, Scholl S, Fehren-bacher L, Wolter JM, Paton V, Shak S, Lieberman G, Slamon DJ: Multinational study of the efficacy and safety of humanized anti-HER2 monoclonal antibody in women who have HER2-overexpressing metastatic breast cancer that has progressed after chemotherapy for metastatic disease. J Clin Oncol. 1999, 17: 2639-2648.
Navolanic PM, Steelman LS, McCubrey JA: EGFR family signalling and its association with breast cancer development and resistance to chemotherapy (Review). Int J Oncol. 2003, 22: 237-252.
Wang C, Thor AD, Moore DH, Zhao Y, Kerschmann R, Stern R, Watson PH, Turley EA: The overexpression of RHAMM, a hyaluronan-binding protein that regulates ras signalling, correlates with overexpression of mitogen-activated protein kinase and is a significant parameter in breast cancer progression. Clin Cancer Res. 1998, 4: 567-576.
Marampon F, Ciccarelli C, Zani BM: Down-regulation of c-Myc following MEK/ERK inhibition halts the expression of malignant phenotype in rhabdomyosarcoma and in non muscle-derived human tumors. Mol Cancer. 2006, 5: 31-10.1186/1476-4598-5-31.
McCubrey JA, Steelman LS, Abrams SL, Lee JT, Chang F, Bertrand FE, Navolanic PM, Terrian DM, Franklin RA, D'Assoro AB, Salisbury JL, Mazzarino MC, Stivala F, Libra M: Roles of the RAF/MEK/ERK and PI3K/PTEN/AKT pathways in malignant transformation and drug resistance. Adv Enzyme Regul. 2006, 46: 249-279. 10.1016/j.advenzreg.2006.01.004.
Mueller H, Flury N, Eppenberger-Castori S, Kueng W, David F, Eppenberger U: Potential prognostic value of mitogen-activated protein kinase activity for disease-free survival of primary breast cancer patients. Int J Cancer. 2000, 89: 384-388. 10.1002/1097-0215(20000720)89:4<384::AID-IJC11>3.0.CO;2-R.
McGlynn LM, Kirkegaard T, Edwards J, Tovey S, Cameron D, Twelves C, Bartlett JM, Cooke TG: Ras/Raf-1/MAPK pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin Cancer Res. 2009, 15: 1487-1495. 10.1158/1078-0432.CCR-07-4967.
Pinkas J, Leder P: MEK1 signalling mediates transformation and metastasis of EpH4 mammary epithelial cells independent of an epithelial to mesenchymal transition. Cancer Res. 2002, 62: 4781-4790.
Bergqvist J, Elmberger G, Ohd J, Linderholm B, Bjohle J, Hellborg H, Nordgren H, Borg AL, Skoog L, Bergh J: Activated ERK1/2 and phosphorylated oestrogen receptor alpha are associated with improved breast cancer survival in women treated with tamoxifen. Eur J Cancer. 2006, 42: 1104-1112. 10.1016/j.ejca.2006.01.028.
Svensson S, Jirström K, Rydén L, Roos G, Emdin S, Ostrowski MC, Landberg G: ERK phosphorylation is linked to VEGFR2 expression and Ets-2 phosphorylation in breast cancer and is associated with tamoxifen treatment resistance and small tumours with good prognosis. Oncogene. 2005, 24: 4370-4379. 10.1038/sj.onc.1208626.
Milde-Langosch K, Bamberger AM, Rieck G, Grund D, Hemminger G, Müller V, Löning T: Expression and prognostic relevance of activated extracellular-regulated kinases (ERK1/2) in breast cancer. Br J Cancer. 2005, 92: 2206-2215. 10.1038/sj.bjc.6602655.
Blatchford DR, Quarrie LH, Tonner E, McCarthy C, Flint DJ, Wilde CJ: Influence of microenvironment on mammary epithelial cell survival in primary culture. J Cell Physiol. 1999, 181: 304-311. 10.1002/(SICI)1097-4652(199911)181:2<304::AID-JCP12>3.0.CO;2-5.
Debnath J, Brugge JS: Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005, 5: 675-688. 10.1038/nrc1695.
Wang F, Hansen RK, Radisky D, Yoneda T, Barcellos-Hoff MH, Petersen OW, Turley EA, Bissell MJ: Phenotypic reversion or death of cancer cells by altering signalling pathways in three-dimensional contexts. J Natl Cancer Inst. 2002, 94: 1494-1503.
Pickl M, Ries CH: Comparison of 3D and 2D tumor models reveals enhanced HER2 activation in 3D associated with an increased response to trastuzumab. Oncogene. 2009, 28: 461-468. 10.1038/onc.2008.394.
Brummer T, Schramek D, Hayes VM, Bennett HL, Caldon CE, Musgrove EA, Daly RJ: Increased proliferation and altered growth factor dependence of human mammary epithelial cells overexpressing the Gab2 docking protein. J Biol Chem. 2006, 281: 626-637. 10.1074/jbc.M509567200.
Xian W, Schwertfeger KL, Rosen JM: Distinct roles of fibroblast growth factor receptor 1 and 2 in regulating cell survival and epithelial-mesenchymal transition. Mol Endocrinol. 2007, 21: 987-1000. 10.1210/me.2006-0518.
Seton-Rogers SE, Lu Y, Hines LM, Koundinya M, LaBaer J, Muthuswamy SK, Brugge JS: Cooperation of the ErbB2 receptor and transforming growth factor beta in induction of migration and invasion in mammary epithelial cells. Proc Natl Acad Sci USA. 2004, 101: 1257-1262. 10.1073/pnas.0308090100.
Nolan ME, Aranda V, Lee S, Lakshmi B, Basu S, Allred DC, Muthuswamy SK: The polarity protein Par6 induces cell proliferation and is overexpressed in breast cancer. Cancer Res. 2008, 68: 8201-8209. 10.1158/0008-5472.CAN-07-6567.
Reginato MJ, Mills KR, Becker EB, Lynch DK, Bonni A, Muthuswamy SK, Brugge JS: Bim regulation of lumen formation in cultured mammary epithelial acini is targeted by oncogenes. Mol Cell Biol. 2005, 25: 4591-4601. 10.1128/MCB.25.11.4591-4601.2005.
Schmelzle T, Mailleux AA, Overholtzer M, Carroll JS, Solimini NL, Lightcap ES, Veiby OP, Brugge JS: Functional role and oncogene-regulated expression of the BH3-only factor Bmf in mammary epithelial anoikis and morphogenesis. Proc Natl Acad Sci USA. 2007, 104: 3787-3792. 10.1073/pnas.0700115104.
Mailleux AA, Overholtzer M, Schmelzle T, Bouillet P, Strasser A, Brugge JS: BIM regulates apoptosis during mammary ductal morphogenesis, and its absence reveals alternative cell death mechanisms. Dev Cell. 2007, 12: 221-234. 10.1016/j.devcel.2006.12.003.
Janda E, Nevolo M, Lehmann K, Downward J, Beug H, Grieco M: Ras and TGF[beta] cooperatively regulate epithelial cell plasticity and metastasis: dissection of Ras signalling pathways. J Cell Biol. 2002, 156: 299-313. 10.1083/jcb.200109037.
Smith AP, Verrecchia A, Fagà G, Doni M, Perna D, Martinato F, Guccione E, Amati B: A positive role for Myc in TGFbeta-induced Snail transcription and epithelial-to-mesenchymal transition. Oncogene. 2009, 28: 422-430. 10.1038/onc.2008.395.
Katoh M, Katoh M: Crosstalk of WNT and FGF signalling pathways at GSK3beta to regulate beta-catenin and SNAIL signalling cascades. Cancer Biol Ther. 2006, 5: 1059-1064.
Horn G, Gaziel A, Wreschner DH, Smorodinsky NI, Ehrlich M: ERK and PI3K regulate different aspects of the epithelial to mesenchymal transition of mammary tumor cells induced by truncated MUC1. Exp Cell Res. 2009, 315: 1490-1504. 10.1016/j.yexcr.2009.02.011.
Yu M, Smolen GA, Zhang J, Wittner B, Schott BJ, Brachtel E, Ramaswamy S, Maheswaran S, Haber DA: A developmentally regulated inducer of EMT, LBX1, contributes to breast cancer progression. Genes Dev. 2009, 23: 1737-1742. 10.1101/gad.1809309.
Dhasarathy A, Kajita M, Wade PA: The transcription factor snail mediates epithelial to mesenchymal transitions by repression of estrogen receptor-alpha. Mol Endocrinol. 2007, 21: 2907-2918. 10.1210/me.2007-0293.
Lee MK, Pardoux C, Hall MC, Lee PS, Warburton D, Qing J, Smith SM, Derynck R: TGF-beta activates Erk MAP kinase signalling through direct phosphorylation of ShcA. EMBO J. 2007, 26: 3957-3967. 10.1038/sj.emboj.7601818.
Takekawa M, Tatebayashi K, Itoh F, Adachi M, Imai K, Saito H: Smad-dependent GADD45beta expression mediates delayed activation of p38 MAP kinase by TGF-beta. EMBO J. 2002, 21: 6473-6482. 10.1093/emboj/cdf643.
Kim ES, Kim MS, Moon A: TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signalling in MCF10A human breast epithelial cells. Int J Oncol. 2004, 25: 1375-1382.
Ingman WV, Robertson SA: Mammary gland development in transforming growth factor beta1 null mutant mice: systemic and epithelial effects. Biol Reprod. 2008, 79: 711-717. 10.1095/biolreprod.107.067272.
Monks J: TGFbeta as a potential mediator of progesterone action in the mammary gland of pregnancy. J Mammary Gland Biol Neoplasia. 2007, 12: 249-257. 10.1007/s10911-007-9056-2.
Gorska AE, Jensen RA, Shyr Y, Aakre ME, Bhowmick NA, Moses HL: Transgenic mice expressing a dominant-negative mutant type II transforming growth factor-beta receptor exhibit impaired mammary development and enhanced mammary tumor formation. Am J Pathol. 2003, 163: 1539-1549.
Nelson CM, Vanduijn MM, Inman JL, Fletcher DA, Bissell MJ: Tissue geometry determines sites of mammary branching morphogenesis in organotypic cultures. Science. 2006, 314: 298-300. 10.1126/science.1131000.
McCawley LJ, Li S, Wattenberg EV, Hudson LG: Sustained activation of the mitogen-activated protein kinase pathway. A mechanism underlying receptor tyrosine kinase specificity for matrix metalloproteinase-9 induction and cell migration. J Biol Chem. 1999, 274: 4347-4353. 10.1074/jbc.274.7.4347.
Kim MS, Lee EJ, Kim HR, Moon A: p38 kinase is a key signalling molecule for H-Ras-induced cell motility and invasive phenotype in human breast epithelial cells. Cancer Res. 2003, 63: 5454-5461.
Kim ES, Kim MS, Moon A: Transforming growth factor (TGF)-beta in conjunction with H-ras activation promotes malignant progression of MCF10A breast epithelial cells. Cytokine. 2005, 29: 84-91. 10.1016/j.cyto.2004.10.001.
Gandhari M, Arens N, Majety M, Dorn-Beineke A, Hildenbrand R: Urokinase-type plasminogen activator induces proliferation in breast cancer cells. Int J Oncol. 2006, 28: 1463-1470.
Ma Z, Webb DJ, Jo M, Gonias SL: Endogenously produced urokinase-type plasminogen activator is a major determinant of the basal level of activated ERK/MAP kinase and prevents apoptosis in MDA-MB-231 breast cancer cells. J Cell Sci. 2001, 114: 3387-3396.
Seddighzadeh M, Zhou JN, Kronenwett U, Shoshan MC, Auer G, Sten-Linder M, Wiman B, Linder S: ERK signalling in metastatic human MDA-MB-231 breast carcinoma cells is adapted to obtain high urokinase expression and rapid cell proliferation. Clin Exp Metastasis. 1999, 17: 649-654. 10.1023/A:1006741228402.
Green KA, Lund LR: ECM degrading proteases and tissue remodelling in the mammary gland. Bioessays. 2005, 27: 894-903. 10.1002/bies.20281.
Zhao J, Pestell R, Guan JL: Transcriptional activation of cyclin D1 promoter by FAK contributes to cell cycle progression. Mol Biol Cell. 2001, 12: 4066-4077.
Kritikou EA, Sharkey A, Abell K, Came PJ, Anderson E, Clarkson RW, Watson CJ: A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development. 2003, 130: 3459-3468. 10.1242/dev.00578.
Viala E, Pouyssegur J: Regulation of tumor cell motility by ERK mitogen-activated protein kinases. Ann N Y Acad Sci. 2004, 1030: 208-218. 10.1196/annals.1329.027.
Hazan RB, Norton L: The epidermal growth factor receptor modulates the interaction of E-cadherin with the actin cytoskeleton. J Biol Chem. 1998, 273: 9078-9084. 10.1074/jbc.273.15.9078.
Li Q, Mattingly RR: Restoration of E-cadherin cell-cell junctions requires both expression of E-cadherin and suppression of ERK MAP kinase activation in Ras-transformed breast epithelial cells. Neoplasia. 2008, 10: 1444-1458.
Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA: Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000, 148: 779-790. 10.1083/jcb.148.4.779.
Hulit J, Suyama K, Chung S, Keren R, Agiostratidou G, Shan W, Dong X, Williams TM, Lisanti MP, Knudsen K, Hazan RB: N-cad-herin signalling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007, 67: 3106-3116. 10.1158/0008-5472.CAN-06-3401.
Dow LE, Elsum IA, King CL, Kinross KM, Richardson HE, Humbert PO: Loss of human Scribble cooperates with H-Ras to promote cell invasion through deregulation of MAPK signalling. Oncogene. 2008, 27: 5988-6001. 10.1038/onc.2008.219.
Jiang P, Enomoto A, Takahashi M: Cell biology of the movement of breast cancer cells: Intracellular signalling and the actin cytoskeleton. Cancer Lett. 2009, 284: 122-130. 10.1016/j.canlet.2009.02.034.
Huang C, Jacobson K, Schaller MD: MAP kinases and cell migration. J Cell Sci. 2004, 117: 4619-4628. 10.1242/jcs.01481.
Vadlamudi RK, Adam L, Nguyen D, Santos M, Kumar R: Differential regulation of components of the focal adhesion complex by heregulin: role of phosphatase SHP-2. J Cell Physiol. 2002, 190: 189-199. 10.1002/jcp.10054.
Lunn JA, Jacamo R, Rozengurt E: Preferential phosphorylation of focal adhesion kinase tyrosine 861 is critical for mediating an anti-apoptotic response to hyperosmotic stress. J Biol Chem. 2007, 282: 10370-10379. 10.1074/jbc.M607780200.
Behmoaram E, Bijian K, Jie S, Xu Y, Darnel A, Bismar TA, Alaoui-Jamali MA: Focal adhesion kinase-related proline-rich tyrosine kinase 2 and focal adhesion kinase are co-overexpressed in early-stage and invasive ErbB-2-positive breast cancer and cooperate for breast cancer cell tumorigenesis and invasiveness. Am J Pathol. 2008, 173: 1540-1550. 10.2353/ajpath.2008.080292.
Nagy T, Wei H, Shen TL, Peng X, Liang CC, Gan B, Guan JL: Mammary epithelial-specific deletion of the focal adhesion kinase gene leads to severe lobulo-alveolar hypoplasia and secretory immaturity of the murine mammary gland. J Biol Chem. 2007, 282: 31766-31776. 10.1074/jbc.M705403200.
Huang S, Ingber DE: Cell tension, matrix mechanics, and cancer development. Cancer Cell. 2005, 8: 175-176. 10.1016/j.ccr.2005.08.009.
Wang F, Weaver VM, Petersen OW, Larabell CA, Dedhar S, Briand P, Lupu R, Bissell MJ: Reciprocal interactions between beta1-integrin and epidermal growth factor receptor in three-dimensional basement membrane breast cultures: a different perspective in epithelial biology. Proc Natl Acad Sci USA. 1998, 95: 14821-14826. 10.1073/pnas.95.25.14821.
Prescott JD, Koto KS, Singh M, Gutierrez-Hartmann A: The ETS transcription factor ESE-1 transforms MCF-12A human mammary epithelial cells via a novel cytoplasmic mechanism. Mol Cell Biol. 2004, 24: 5548-5564. 10.1128/MCB.24.12.5548-5564.2004.
Gilles C, Polette M, Birembaut P, Brünner N, Thompson EW: Expression of c-ets-1 mRNA is associated with an invasive, EMT-derived phenotype in breast carcinoma cell lines. Clin Exp Metastasis. 1997, 15: 519-526. 10.1023/A:1018427027270.
Watabe T, Yoshida K, Shindoh M, Kaya M, Fujikawa K, Sato H, Seiki M, Ishii S, Fujinaga K: The Ets-1 and Ets-2 transcription factors activate the promoters for invasion-associated uroki-nase and collagenase genes in response to epidermal growth factor. Int J Cancer. 1998, 77: 128-137. 10.1002/(SICI)1097-0215(19980703)77:1<128::AID-IJC20>3.0.CO;2-9.
Rosen JM, Jordan CT: The increasing complexity of the cancer stem cell paradigm. Science. 2009, 324: 1670-1673. 10.1126/science.1171837.
Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF: Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA. 2003, 100: 3983-3988. 10.1073/pnas.0530291100.
Morel AP, Lièvre M, Thomas C, Hinkal G, Ansieau S, Puisieux A: Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE. 2008, 3: e2888-10.1371/journal.pone.0002888.
Liu JC, Deng T, Lehal RS, Kim J, Zacksenhaus E: Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res. 2007, 67: 8671-8681. 10.1158/0008-5472.CAN-07-1486.
Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH: Pathways to tamoxifen resistance. Cancer Lett. 2007, 256: 1-24. 10.1016/j.canlet.2007.03.016.
Gee JM, Howell A, Gullick WJ, Benz CC, Sutherland RL, Santen RJ, Martin LA, Ciardiello F, Miller WR, Dowsett M, Barrett-Lee P, Robertson JF, Johnston SR, Jones HE, Wakeling AE, Duncan R, Nicholson RI: Consensus statement. Workshop on therapeutic resistance in breast cancer: impact of growth factor signalling pathways and implications for future treatment. Endocr Relat Cancer. 2005, 12 (Suppl 1): S1-7. 10.1677/erc.1.01054.
Freudenberg JA, Wang Q, Katsumata M, Drebin J, Nagatomo I, Greene MI: The role of HER2 in early breast cancer metastasis and the origins of resistance to HER2-targeted therapies. Exp Mol Pathol. 2009, 87: 1-11. 10.1016/j.yexmp.2009.05.001.
McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA: Roles of the Raf/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochim Biophys Acta. 2007, 1773: 1263-1284. 10.1016/j.bbamcr.2006.10.001.
Siddiqa A, Long LM, Li L, Marciniak RA, Kazhdan I: Expression of HER-2 in MCF-7 breast cancer cells modulates anti-apoptotic proteins Survivin and Bcl-2 via the extracellular signal-related kinase (ERK) and phosphoinositide-3 kinase (PI3K) signalling pathways. BMC Cancer. 2008, 8: 129-10.1186/1471-2407-8-129.
Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS: ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001, 3: 785-792. 10.1038/ncb0901-785.
Aranda V, Haire T, Nolan ME, Calarco JP, Rosenberg AZ, Fawcett JP, Pawson T, Muthuswamy SK: Par6-aPKC uncouples ErbB2 induced disruption of polarized epithelial organization from proliferation control. Nat Cell Biol. 2006, 8: 1235-1245. 10.1038/ncb1485.
Kim IY, Yong HY, Kang KW, Moon A: Overexpression of ErbB2 induces invasion of MCF10A human breast epithelial cells via MMP-9. Cancer Lett. 2009, 275: 227-233. 10.1016/j.canlet.2008.10.013.
Rodland KD, Bollinger N, Ippolito D, Opresko LK, Coffey RJ, Zangar R, Wiley HS: Multiple mechanisms are responsible for transactivation of the epidermal growth factor receptor in mammary epithelial cells. J Biol Chem. 2008, 283: 31477-31487. 10.1074/jbc.M800456200.
Daub H, Weiss FU, Wallasch C, Ullrich A: Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996, 379: 557-560. 10.1038/379557a0.
Prenzel N, Zwick E, Daub H, Leserer M, Abraham R, Wallasch C, Ullrich A: EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature. 1999, 402: 884-888.
Faivre EJ, Lange CA: Progesterone receptors upregulate Wnt-1 to induce epidermal growth factor receptor transactivation and c-Src-dependent sustained activation of Erk1/2 mitogen-activated protein kinase in breast cancer cells. Mol Cell Biol. 2007, 27: 466-480. 10.1128/MCB.01539-06.
Ursini-Siegel J, Schade B, Cardiff RD, Muller WJ: Insights from transgenic mouse models of ERBB2-induced breast cancer. Nat Rev Cancer. 2007, 7: 389-397. 10.1038/nrc2127.
Marcotte R, Muller WJ: Signal transduction in transgenic mouse models of human breast cancer--implications for human breast cancer. J Mammary Gland Biol Neoplasia. 2008, 13: 323-335. 10.1007/s10911-008-9087-3.
Julien SG, Dubé N, Read M, Penney J, Paquet M, Han Y, Kennedy BP, Muller WJ, Tremblay ML: Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat Genet. 2007, 39: 338-346. 10.1038/ng1963.
Demidov ON, Kek C, Shreeram S, Timofeev O, Fornace AJ, Appella E, Bulavin DV: The role of the MKK6/p38 MAPK pathway in Wip1-dependent regulation of ErbB2-driven mammary gland tumorigenesis. Oncogene. 2007, 26: 2502-2506. 10.1038/sj.onc.1210032.
Yang JY, Zong CS, Xia W, Yamaguchi H, Ding Q, Xie X, Lang JY, Lai CC, Chang CJ, Huang WC, Huang H, Kuo HP, Lee DF, Li LY, Lien HC, Cheng X, Chang KJ, Hsiao CD, Tsai FJ, Tsai CH, Sahin AA, Muller WJ, Mills GB, Yu D, Hortobagyi GN, Hung MC: ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nat Cell Biol. 2008, 10: 138-148. 10.1038/ncb1676.
Vaught D, Chen J, Brantley-Sieders DM: Regulation of mammary gland branching morphogenesis by EphA2 receptor tyrosine kinase. Mol Biol Cell. 2009, 20: 2572-2581. 10.1091/mbc.E08-04-0378.
Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM, Coffman K, Jackson D, Bruckheimer E, Muraoka-Cook RS, Chen J: The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signalling. J Clin Invest. 2008, 118: 64-78. 10.1172/JCI33154.
Ono M, Kuwano M: Molecular mechanisms of epidermal growth factor receptor (EGFR) activation and response to gefitinib and other EGFR-targeting drugs. Clin Cancer Res. 2006, 12: 7242-7251. 10.1158/1078-0432.CCR-06-0646.
Arany I, Megyesi JK, Nelkin BD, Safirstein RL: STAT3 attenuates EGFR-mediated ERK activation and cell survival during oxidant stress in mouse proximal tubular cells. Kidney Int. 2006, 70: 669-674. 10.1038/sj.ki.5001604.
Paech K, Webb P, Kuiper GG, Nilsson S, Gustafsson J, Kushner PJ, Scanlan TS: Differential ligand activation of estrogen receptors ERalpha and ERbeta at AP1 sites. Science. 1997, 277: 1508-1510. 10.1126/science.277.5331.1508.
Green S, Chambon P: Oestradiol induction of a glucocorticoid-responsive gene by a chimaeric receptor. Nature. 1987, 325: 75-78. 10.1038/325075a0.
Nilsson S, Gustafsson JA: Estrogen receptor transcription and transactivation: Basic aspects of estrogen action. Breast Cancer Res. 2000, 2: 360-366. 10.1186/bcr81.
Murphy LC, Weitsman GE, Skliris GP, Teh EM, Li L, Peng B, Davie JR, Ung K, Niu YL, Troup S, Tomes L, Watson PH: Potential role of estrogen receptor alpha (ERalpha) phosphorylated at Serine118 in human breast cancer in vivo. J Steroid Biochem Mol Biol. 2006, 102: 139-146. 10.1016/j.jsbmb.2006.09.021.
Yamashita H, Nishio M, Kobayashi S, Ando Y, Sugiura H, Zhang Z, Hamaguchi M, Mita K, Fujii Y, Iwase H: Phosphorylation of estrogen receptor alpha serine 167 is predictive of response to endocrine therapy and increases postrelapse survival in metastatic breast cancer. Breast Cancer Res. 2005, 7: R753-764. 10.1186/bcr1285.
Likhite VS, Stossi F, Kim K, Katzenellenbogen BS, Katzenellenbogen JA: Kinase-specific phosphorylation of the estrogen receptor changes receptor interactions with ligand, deoxyribonucleic acid, and coregulators associated with alterations in estrogen and tamoxifen activity. Mol Endocrinol. 2006, 20: 3120-3132. 10.1210/me.2006-0068.
Britton DJ, Hutcheson IR, Knowlden JM, Barrow D, Giles M, McClelland RA, Gee JM, Nicholson RI: Bidirectional cross talk between ERalpha and EGFR signalling pathways regulates tamoxifen-resistant growth. Breast Cancer Res Treat. 2006, 96: 131-146. 10.1007/s10549-005-9070-2.
Sarwar N, Kim JS, Jiang J, Peston D, Sinnett HD, Madden P, Gee JM, Nicholson RI, Lykkesfeldt AE, Shousha S, Coombes RC, Ali S: Phosphorylation of ERalpha at serine 118 in primary breast cancer and in tamoxifen-resistant tumours is indicative of a complex role for ERalpha phosphorylation in breast cancer progression. Endocr Relat Cancer. 2006, 13: 851-861. 10.1677/erc.1.01123.
Filardo EJ, Quinn JA, Frackelton AR, Bland KI: Estrogen action via the G protein-coupled receptor, GPR30: stimulation of adenylyl cyclase and cAMP-mediated attenuation of the epidermal growth factor receptor-to-MAPK signalling axis. Mol Endocrinol. 2002, 16: 70-84. 10.1210/me.16.1.70.
Flouriot G, Brand H, Denger S, Metivier R, Kos M, Reid G, Sonntag-Buck V, Gannon F: Identification of a new isoform of the human estrogen receptor-alpha (hER-alpha) that is encoded by distinct transcripts and that is able to repress hER-alpha activation function 1. EMBO J. 2000, 19: 4688-4700. 10.1093/emboj/19.17.4688.
Figtree GA, McDonald D, Watkins H, Channon KM: Truncated estrogen receptor alpha 46-kDa isoform in human endothelial cells: relationship to acute activation of nitric oxide synthase. Circulation. 2003, 107: 120-126. 10.1161/01.CIR.0000043805.11780.F5.
Denger S, Reid G, Kos M, Flouriot G, Parsch D, Brand H, Korach KS, Sonntag-Buck V, Gannon F: ERalpha gene expression in human primary osteoblasts: evidence for the expression of two receptor proteins. Mol Endocrinol. 2001, 15: 2064-2077. 10.1210/me.15.12.2064.
Li L, Haynes MP, Bender JR: Plasma membrane localization and function of the estrogen receptor alpha variant (ER46) in human endothelial cells. Proc Natl Acad Sci USA. 2003, 100: 4807-4812. 10.1073/pnas.0831079100.
Rosen EM, Fan S, Ma Y, Levin ER: Role of plasma membrane ER protein in breast cancer. Breast Cancer Online. 2006, 9: 1-5. 10.1017/S1470903105003366.
Haynes MP, Li L, Sinha D, Russell KS, Hisamoto K, Baron R, Collinge M, Sessa WC, Bender JR: Src kinase mediates phosphatidylinositol 3-kinase/Akt-dependent rapid endothelial nitric-oxide synthase activation by estrogen. J Biol Chem. 2003, 278: 2118-2123. 10.1074/jbc.M210828200.
Henderson IC, Piccart-Gebhart MJ: The evolving role of aro-matase inhibitors in adjuvant breast cancer therapy. Clin Breast Cancer. 2005, 6: 206-215. 10.3816/CBC.2005.n.022.
Kurokawa H, Arteaga CL: Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer. Clin Cancer Res. 2001, 7: 4436s-4442s. discussion 4411s-4412s.
Knowlden JM, Hutcheson IR, Jones HE, Madden T, Gee JM, Harper ME, Barrow D, Wakeling AE, Nicholson RI: Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology. 2003, 144: 1032-1044. 10.1210/en.2002-220620.
Webb CP, Van Aelst L, Wigler MH, Woude GF: Signaling pathways in Ras-mediated tumorigenicity and metastasis. Proc Natl Acad Sci USA. 1998, 95: 8773-8778. 10.1073/pnas.95.15.8773.
Ciarloni L, Mallepell S, Brisken C: Amphiregulin is an essential mediator of estrogen receptor alpha function in mammary gland development. Proc Natl Acad Sci USA. 2007, 104: 5455-5460. 10.1073/pnas.0611647104.
Wiesen JF, Young P, Werb Z, Cunha GR: Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development. 1999, 126: 335-344.
Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ: Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with Src/Erk phosphorylation cascade. Proc Natl Acad Sci USA. 2002, 99: 14783-14788. 10.1073/pnas.192569699.
Nguyen TH, Mustafa FB, Pervaiz S, Ng FS, Lim LH: ERK1/2 activation is required for resveratrol-induced apoptosis in MDA-MB-231 cells. Int J Oncol. 2008, 33: 81-92.
She QB, Bode AM, Ma WY, Chen NY, Dong Z: Resveratrol-induced activation of p53 and apoptosis is mediated by extra-cellular-signal-regulated protein kinases and p38 kinase. Cancer Res. 2001, 61: 1604-1610.
Yu W, Sanders BG, Kline K: RRR-alpha-tocopheryl succinate-induced apoptosis of human breast cancer cells involves Bax translocation to mitochondria. Cancer Res. 2003, 63: 2483-2491.
Li Z, Li J, Mo B, Hu C, Liu H, Qi H, Wang X, Xu J: Genistein induces cell apoptosis in MDA-MB-231 breast cancer cells via the mitogen-activated protein kinase pathway. Toxicol In Vitro. 2008, 22: 1749-1753. 10.1016/j.tiv.2008.08.001.
Jo EH, Lee SJ, Ahn NS, Park JS, Hwang JW, Kim SH, Aruoma OI, Lee YS, Kang KS: Induction of apoptosis in MCF-7 and MDA-MB-231 breast cancer cells by Oligonol is mediated by Bcl-2 family regulation and MEK/ERK signalling. Eur J Cancer Prev. 2007, 16: 342-347. 10.1097/01.cej.0000236247.86360.db.
Kim BW, Lee ER, Min HM, Jeong HS, Ahn JY, Kim JH, Choi HY, Choi H, Kim EY, Park SP, Cho SG: Sustained ERK activation is involved in the kaempferol-induced apoptosis of breast cancer cells and is more evident under 3-D culture condition. Cancer Biol Ther. 2008, 7: 1080-1089. 10.1158/1535-7163.MCT-07-0526.
Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R, Wagner EF, Cook SJ: Activation of ERK1/2 by deltaRaf-1:ER* represses Bim expression independently of the JNK or PI3K pathways. Oncogene. 2003, 22: 1281-1293. 10.1038/sj.onc.1206261.
Ley R, Balmanno K, Hadfield K, Weston C, Cook SJ: Activation of the ERK1/2 signalling pathway promotes phosphorylation and proteasome-dependent degradation of the BH3-only protein, Bim. J Biol Chem. 2003, 278: 18811-18816. 10.1074/jbc.M301010200.
Harada H, Quearry B, Ruiz-Vela A, Korsmeyer SJ: Survival factor-induced extracellular signal-regulated kinase phosphorylates BIM, inhibiting its association with BAX and proapoptotic activity. Proc Natl Acad Sci USA. 2004, 101: 15313-15317. 10.1073/pnas.0406837101.
Kochetkova M, Kumar S, McColl SR: Chemokine receptors CXCR4 and CCR7 promote metastasis by preventing anoikis in cancer cells. Cell Death Differ. 2009, 16: 664-673. 10.1038/cdd.2008.190.
Phelps M, Phillips A, Darley M, Blaydes JP: MEK-ERK signalling controls Hdm2 oncoprotein expression by regulating hdm2 mRNA export to the cytoplasm. J Biol Chem. 2005, 280: 16651-16658. 10.1074/jbc.M412334200.
Wu GS: The functional interactions between the p53 and MAPK signalling pathways. Cancer Biol Ther. 2004, 3: 156-161.
Ostrakhovitch EA, Cherian MG: Inhibition of extracellular signal regulated kinase (ERK) leads to apoptosis inducing factor (AIF) mediated apoptosis in epithelial breast cancer cells: the lack of effect of ERK in p53 mediated copper induced apoptosis. J Cell Biochem. 2005, 95: 1120-1134. 10.1002/jcb.20484.
Chapman RS, Lourenco PC, Tonner E, Flint DJ, Selbert S, Takeda K, Akira S, Clarke AR, Watson CJ: Suppression of epithelial apoptosis and delayed mammary gland involution in mice with a conditional knockout of Stat3. Genes Dev. 1999, 13: 2604-2616. 10.1101/gad.13.19.2604.
Zhao L, Melenhorst JJ, Hennighausen L: Loss of interleukin 6 results in delayed mammary gland involution: a possible role for mitogen-activated protein kinase and not signal transducer and activator of transcription. Mol Endocrinol. 2002, 16: 2902-2912. 10.1210/me.2001-0330.
Hui L, Bakiri L, Stepniak E, Wagner EF: p38alpha: a suppressor of cell proliferation and tumorigenesis. Cell Cycle. 2007, 6: 2429-2433.
Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ: Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995, 270: 7420-7426. 10.1074/jbc.270.13.7420.
Thornton TM, Rincon M: Non-classical p38 map kinase functions: cell cycle checkpoints and survival. Int J Biol Sci. 2009, 5: 44-51.
Ge B, Gram H, Di Padova F, Huang B, New L, Ulevitch RJ, Luo Y, Han J: MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science. 2002, 295: 1291-1294. 10.1126/science.1067289.
Morooka H, Bonventre JV, Pombo CM, Kyriakis JM, Force T: Ischemia and reperfusion enhance ATF-2 and c-Jun binding to cAMP response elements and to an AP-1 binding site from the c-jun promoter. J Biol Chem. 1995, 270: 30084-30092. 10.1074/jbc.270.50.30084.
Zhao M, New L, Kravchenko VV, Kato Y, Gram H, di Padova F, Olson EN, Ulevitch RJ, Han J: Regulation of the MEF2 family of transcription factors by p38. Mol Cell Biol. 1999, 19: 21-30.
Huang S, New L, Pan Z, Han J, Nemerow GR: Urokinase plasminogen activator/urokinase-specific surface receptor expression and matrix invasion by breast cancer cells requires constitutive p38alpha mitogen-activated protein kinase activity. J Biol Chem. 2000, 275: 12266-12272. 10.1074/jbc.275.16.12266.
Han Q, Leng J, Bian D, Mahanivong C, Carpenter KA, Pan ZK, Han J, Huang S: Rac1-MKK3-p38-MAPKAPK2 pathway promotes urokinase plasminogen activator mRNA stability in invasive breast cancer cells. J Biol Chem. 2002, 277: 48379-48385. 10.1074/jbc.M209542200.
Galliher AJ, Schiemann WP: Src phosphorylates Tyr284 in TGF-beta type II receptor and regulates TGF-beta stimulation of p38 MAPK during breast cancer cell proliferation and invasion. Cancer Res. 2007, 67: 3752-3758. 10.1158/0008-5472.CAN-06-3851.
Sossey-Alaoui K, Ranalli TA, Li X, Bakin AV, Cowell JK: WAVE3 promotes cell motility and invasion through the regulation of MMP-1, MMP-3, and MMP-9 expression. Exp Cell Res. 2005, 308: 135-145. 10.1016/j.yexcr.2005.04.011.
Gutierrez MC, Detre S, Johnston S, Mohsin SK, Shou J, Allred DC, Schiff R, Osborne CK, Dowsett M: Molecular changes in tamoxifen-resistant breast cancer: relationship between estrogen receptor, HER-2, and p38 mitogen-activated protein kinase. J Clin Oncol. 2005, 23: 2469-2476. 10.1200/JCO.2005.01.172.
Lee H, Bai W: Regulation of estrogen receptor nuclear export by ligand-induced and p38-mediated receptor phosphorylation. Mol Cell Biol. 2002, 22: 5835-5845. 10.1128/MCB.22.16.5835-5845.2002.
Davis RJ: Signal transduction by the c-Jun N-terminal kinase. Biochem Soc Symp. 1999, 64: 1-12.
Jochum W, Passegue E, Wagner EF: AP-1 in mouse development and tumorigenesis. Oncogene. 2001, 20: 2401-2412. 10.1038/sj.onc.1204389.
Mingo-Sion AM, Marietta PM, Koller E, Wolf DM, Berg Van Den CL: Inhibition of JNK reduces G2/M transit independent of p53, leading to endoreduplication, decreased proliferation, and apoptosis in breast cancer cells. Oncogene. 2004, 23: 596-604. 10.1038/sj.onc.1207147.
Monno S, Newman MV, Cook M, Lowe WL: Insulin-like growth factor I activates c-Jun N-terminal kinase in MCF-7 breast cancer cells. Endocrinology. 2000, 141: 544-550. 10.1210/en.141.2.544.
Johnston SR, Lu B, Scott GK, Kushner PJ, Smith IE, Dowsett M, Benz CC: Increased activator protein-1 DNA binding and c-Jun NH2-terminal kinase activity in human breast tumors with acquired tamoxifen resistance. Clin Cancer Res. 1999, 5: 251-256.
Schiff R, Reddy P, Ahotupa M, Coronado-Heinsohn E, Grim M, Hilsenbeck SG, Lawrence R, Deneke S, Herrera R, Chamness GC, Fuqua SA, Brown PH, Osborne CK: Oxidative stress and AP-1 activity in tamoxifen-resistant breast tumors in vivo. J Natl Cancer Inst. 2000, 92: 1926-1934. 10.1093/jnci/92.23.1926.
Buse P, Woo PL, Alexander DB, Reza A, Firestone GL: Glucocorticoid-induced functional polarity of growth factor responsiveness regulates tight junction dynamics in transformed mammary epithelial tumor cells. J Biol Chem. 1995, 270: 28223-28227. 10.1074/jbc.270.47.28223.
Murtagh J, McArdle E, Gilligan E, Thornton L, Furlong F, Martin F: Organization of mammary epithelial cells into 3D acinar structures requires glucocorticoid and JNK signalling. J Cell Biol. 2004, 166: 133-143. 10.1083/jcb.200403020.
Zhan L, Rosenberg A, Bergami KC, Yu M, Xuan Z, Jaffe AB, Allred C, Muthuswamy SK: Deregulation of scribble promotes mammary tumorigenesis and reveals a role for cell polarity in carcinoma. Cell. 2008, 135: 865-878. 10.1016/j.cell.2008.09.045.
Lee JD, Ulevitch RJ, Han J: Primary structure of BMK1: a new mammalian map kinase. Biochem Biophys Res Commun. 1995, 213: 715-724. 10.1006/bbrc.1995.2189.
Yan C, Luo H, Lee JD, Abe J, Berk BC: Molecular cloning of mouse ERK5/BMK1 splice variants and characterization of ERK5 functional domains. J Biol Chem. 2001, 276: 10870-10878. 10.1074/jbc.M009286200.
English JM, Vanderbilt CA, Xu S, Marcus S, Cobb MH: Isolation of MEK5 and differential expression of alternatively spliced forms. J Biol Chem. 1995, 270: 28897-28902. 10.1074/jbc.270.48.28897.
Wang X, Merritt AJ, Seyfried J, Guo C, Papadakis ES, Finegan KG, Kayahara M, Dixon J, Boot-Handford RP, Cartwright EJ, Mayer U, Tournier C: Targeted deletion of mek5 causes early embryonic death and defects in the extracellular signal-regulated kinase 5/myocyte enhancer factor 2 cell survival pathway. Mol Cell Biol. 2005, 25: 336-345. 10.1128/MCB.25.1.336-345.2005.
Hayashi M, Kim SW, Imanaka-Yoshida K, Yoshida T, Abel ED, Eliceiri B, Yang Y, Ulevitch RJ, Lee JD: Targeted deletion of BMK1/ERK5 in adult mice perturbs vascular integrity and leads to endothelial failure. J Clin Invest. 2004, 113: 1138-1148.
Pi X, Yan C, Berk BC: Big mitogen-activated protein kinase (BMK1)/ERK5 protects endothelial cells from apoptosis. Circ Res. 2004, 94: 362-369. 10.1161/01.RES.0000112406.27800.6F.
Suhara T, Kim HS, Kirshenbaum LA, Walsh K: Suppression of Akt signalling induces Fas ligand expression: involvement of caspase and Jun kinase activation in Akt-mediated Fas ligand regulation. Mol Cell Biol. 2002, 22: 680-691. 10.1128/MCB.22.2.680-691.2002.
Kato Y, Tapping RI, Huang S, Watson MH, Ulevitch RJ, Lee JD: Bmk1/Erk5 is required for cell proliferation induced by epidermal growth factor. Nature. 1998, 395: 713-716. 10.1038/27234.
Sawhney RS, Liu W, Brattain MG: A novel role of ERK5 in integrin-mediated cell adhesion and motility in cancer cells via Fak signalling. J Cell Physiol. 2009, 219: 152-161. 10.1002/jcp.21662.
Kayahara M, Wang X, Tournier C: Selective regulation of c-jun gene expression by mitogen-activated protein kinases via the 12-o-tetradecanoylphorbol-13-acetate-responsive element and myocyte enhancer factor 2 binding sites. Mol Cell Biol. 2005, 25: 3784-3792. 10.1128/MCB.25.9.3784-3792.2005.
Kamakura S, Moriguchi T, Nishida E: Activation of the protein kinase ERK5/BMK1 by receptor tyrosine kinases. Identification and characterization of a signalling pathway to the nucleus. J Biol Chem. 1999, 274: 26563-26571. 10.1074/jbc.274.37.26563.
Kato Y, Kravchenko VV, Tapping RI, Han J, Ulevitch RJ, Lee JD: BMK1/ERK5 regulates serum-induced early gene expression through transcription factor MEF2C. EMBO J. 1997, 16: 7054-7066. 10.1093/emboj/16.23.7054.
English JM, Pearson G, Baer R, Cobb MH: Identification of substrates and regulators of the mitogen-activated protein kinase ERK5 using chimeric protein kinases. J Biol Chem. 1998, 273: 3854-3860. 10.1074/jbc.273.7.3854.
Hayashi M, Tapping RI, Chao TH, Lo JF, King CC, Yang Y, Lee JD: BMK1 mediates growth factor-induced cell proliferation through direct cellular activation of serum and glucocorticoid-inducible kinase. J Biol Chem. 2001, 276: 8631-8634. 10.1074/jbc.C000838200.
Leong ML, Maiyar AC, Kim B, O'Keeffe BA, Firestone GL: Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003, 278: 5871-5882. 10.1074/jbc.M211649200.
Montero JC, Ocaña A, Abad M, Ortiz-Ruiz MJ, Pandiella A, Esparís-Ogando A: Expression of Erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One. 2009, 4: e5565-10.1371/journal.pone.0005565.
Esparís-Ogando A, Díaz-Rodríguez E, Montero JC, Yuste L, Crespo P, Pandiella A: Erk5 participates in neuregulin signal transduction and is constitutively active in breast cancer cells overexpressing ErbB2. Mol Cell Biol. 2002, 22: 270-285. 10.1128/MCB.22.1.270-285.2002.
Li N, Zhang Y, Naylor MJ, Schatzmann F, Maurer F, Wintermantel T, Schuetz G, Mueller U, Streuli CH, Hynes NE: Beta1 integrins regulate mammary gland proliferation and maintain the integrity of mammary alveoli. EMBO J. 2005, 24: 1942-1953. 10.1038/sj.emboj.7600674.
Xia Y, Karin M: The control of cell motility and epithelial morphogenesis by Jun kinases. Trends Cell Biol. 2004, 14: 94-101. 10.1016/j.tcb.2003.12.005.
Suzanne M, Perrimon N, Noselli S: The Drosophila JNK pathway controls the morphogenesis of the egg dorsal appendages and micropyle. Dev Biol. 2001, 237: 282-294. 10.1006/dbio.2001.0384.
Finn RS: Targeting Src in breast cancer. Ann Oncol. 2008, 19: 1379-1386. 10.1093/annonc/mdn291.
Acknowledgements
Limitations due to space and the extensive relevant literature have curtailed the numbers of published works that we could cite. We apologize to researchers whose work in this area we have failed to cite. Research in our own laboratory has been funded by Science Foundation Ireland and The Health Research Board, Ireland.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
About this article
Cite this article
Whyte, J., Bergin, O., Bianchi, A. et al. Key signalling nodes in mammary gland development and cancer. Mitogen-activated protein kinase signalling in experimental models of breast cancer progression and in mammary gland development. Breast Cancer Res 11, 209 (2009). https://doi.org/10.1186/bcr2361
Published:
DOI: https://doi.org/10.1186/bcr2361