
Abstract
Background
Soybean is not only an important oil crop, but also an important source of edible protein and industrial raw material. Yield-traits and quality-traits are increasingly attracting the attention of breeders. Therefore, fine mapping the QTLs associated with yield-traits and quality-traits of soybean would be helpful for soybean breeders. In the present study, a high-density linkage map was constructed to identify the QTLs for the yield-traits and quality-traits, using specific length amplified fragment sequencing (SLAF-seq).
Results
SLAF-seq was performed to screen SLAF markers with 149 F8:11 individuals from a cross between a semi wild soybean, ‘Huapidou’, and a cultivated soybean, ‘Qihuang26’, which generated 400.91 M paired-end reads. In total, 53,132 polymorphic SLAF markers were obtained. The genetic linkage map was constructed by 5111 SLAF markers with segregation type of aa×bb. The final map, containing 20 linkage groups (LGs), was 2909.46 cM in length with an average distance of 0.57 cM between adjacent markers. The average coverage for each SLAF marker on the map was 81.26-fold in the male parent, 45.79-fold in the female parent, and 19.84-fold average in each F8:11 individual. According to the high-density map, 35 QTLs for plant height (PH), 100-seeds weight (SW), oil content in seeds (Oil) and protein content in seeds (Protein) were found to be distributed on 17 chromosomes, and 14 novel QTLs were identified for the first time. The physical distance of 11 QTLs was shorter than 100 Kb, suggesting a direct opportunity to find candidate genes. Furthermore, three pairs of epistatic QTLs associated with Protein involving 6 loci on 5 chromosomes were identified. Moreover, 13, 14, 7 and 9 genes, which showed tissue-specific expression patterns, might be associated with PH, SW, Oil and Protein, respectively.
Conclusions
With SLAF-sequencing, some novel QTLs and important QTLs for both yield-related and quality traits were identified based on a new, high-density linkage map. Moreover, 43 genes with tissue-specific expression patterns were regarded as potential genes in further study. Our findings might be beneficial to molecular marker-assisted breeding, and could provide detailed information for accurate QTL localization.
Similar content being viewed by others


Background
Soybean is not only an important oil crop, but also an important source of edible protein and industrial raw material [1]. Agronomic traits, such as yield, plant height (PH), lodging and seed weight (SW), have been the primary focus of breeders for many years. As the major factors of the market price of soybean, seed quality traits are increasingly attracting the attention of breeders. However, the negative correlation between yield and quality of crops makes it much difficult to select for these traits [2]. Therefore, simultaneous improvement of yield and quality has become a major problem for soybean breeders.
Molecular marker-assisted selection (MAS) might be an alternative to fit the increasing global demand for soybean products [3]. A number of QTLs underlying important agronomic traits and seed quality traits have been reported over the past decades. So far, at least 196, 265, 297 and 221 QTLs controlling PH, SW, Oil and Protein have been identified respectively (www.soybase.org), based on the different genetic backgrounds, environments and statistical methods. Furthermore, large confidence intervals around QTLs make the causative gene identification difficult.
With the development of next generation sequencing technology, several methods for single nucleotide polymorphisms (SNP) discovery, such as restriction-site associated DNA sequencing (RADseq) [4, 5], genotyping-by-sequencing (GBS) [6], specific length amplified fragment sequencing (SLAF-seq) [7] have been produced, which make it possible to obtain thousands of SNPs suitable for high-density genetic map throughout the genome. SLAF markers, which have the properties of being present in large amount, being evenly distributed and avoiding repeated sequences, has been used for genetic analysis in plants, such as sesame [8], walnut [9], rice [10], sorghum [11], wax gourd [12], grape [13] and soybean [14,15,16,17]. Since the first high-density map was constructed by SLAF-seq [7], there have been several maps reported so far. Qi et al. constructed a map, including 5308 markers with 2655.68 cM in length, using a RIL population derived from a cross between ‘Charleston’ and ‘Dongnong594’ [16]. Li et al. constructed a high-density map, using a F5:8 population of 110 RILs from a cross between ‘Luheidou2’ and ‘Nanhuizao’, which was used to identified QTLs associated the isoflavone content and fatty acid composition in soybean [15, 18]. Zhang et al. reported 20 QTLs associated with phosphorus efficiency-related traits based on a high-density map constructed by SLAF-seq [17]. Cao et al. mapped QTL associated with plant height and flowering time according the map constructed by SLAF-seq using a population of 236 RILs derived from a cross between two summer planting varieties, ‘ZXD’ and ‘NN1138–2’ [14]. Nevertheless, based on the high-density map, little QTLs related to seed weight and/or protein have been reported. Therefore, we reported a high-density genetic linkage map using the SLAF-seq approach, which was based on an F8:11 RIL population with 149 individuals. Moreover, the QTLs associated with plant height, seed weight, oil and protein content were located and analyzed. The results presented here will aid molecular marker-assisted breeding and provide detailed information for accurate QTL localization.
Results
Analysis of SLAF-seq and SLAF markers
DNA sequencing generated about 400.91 M pair-end reads. The Q30 (indicating a 0.1% chance of error) was 90.69% and guanine-cytosine (GC) content was 40.3%. The numbers of SLAFs in the female and male parents were 312,740 and 275,046, respectively. The numbers of SLAFs in each individual ranged from 167,933 to 237,666, with an average of 207,105. Among the 391,476 SLAF markers detected, 53,132 markers were polymorphic. All polymorphic SLAFs were then genotyped separately for all individuals. After discarding the SLAF markers lacking parent information, 30,415 markers were genotyped successfully and were classified into eight segregation types (Fig. 1). As the population was derived from a cross between two fully homozygous parents, only 27,472 markers with aa×bb type might be suitable for map construction. After filtering low-quality SLAF markers, segregation distortion markers and makers with the MLOD value ≤3, 5111 markers were used for the map construction (Additional files 1, 2, and 3). The average depth of the markers was 81.26-fold in the female parent, 45.79-fold in the male parent, and 19.84-fold in the offspring.

Number of markers for eight segregation patterns
The basic characteristics of the genetic map
The length of final map was 2909.46 cM, with an average distance of 0.57 cM between adjacent markers (Table 1; Fig. 2). There were 8597 SNP loci among the 5111 markers on the map. For each chromosome, the average distance ranged from 0.24 cM to 2.55 cM (Table 1). The largest linkage group was LG18 (chr18) with 480 markers, a length of 202.52 cM, and an average distance of only 0.42 cM between adjacent markers. The smallest linkage group was LG7 (chr7) with 63 markers, a length of 54.57 cM, and an average distance of 0.87 cM between adjacent markers.

The positions of QTLs for four traits. QTLs for four traits are depicted in different shapes on the right side of each linkage group. 35 QTLs identified by ICIM are colored in black; 18 QTLs identified in 2013 by CIM are colored in red; 21 QTLs identified in 2014 by CIM are colored in green
Phenotypic evaluation
‘Huapidou’ and ‘Qihuang26’ showed a significant difference in PH, SW and Protein, but did not differ from each other in Oil significantly (Table 2; Fig. 3). However, the phenotypic values were all in a condition of continuous distribution approximately (Fig. 3). The coefficients of variation for four traits were about 20%. The heritabilities of four traits ranged from 49.58 to 82.73%. However, the heritability of Protein was only 49.58%, indicating that other factors affected Protein should be considered.

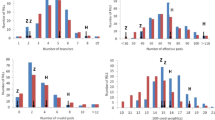
The frequency distribution for soybean traits of the RIL population and parents. F: female parent, ‘Huapidou’; M: male parent, ‘Qihuang26’; (a): the frequency distribution for PH in 2013; (b): the frequency distribution for PH in 2014; (c): the frequency distribution for SW in 2013; (d): the frequency distribution for SW in 2014; (e): the frequency distribution for Oil in 2013; (f): the frequency distribution for Oil in 2014; (g): the frequency distribution for Protein in 2013; (h): the frequency distribution for Protein in 2014
Analyses of additive QTLs
In total, 35 additive QTLs for PH, SW, Oil and Protein were identified on 17 chromosomes by ICIM method (Table 3; Figs. 2, 4). A single QTL explained 2.66% (qPH16–1) to 37.61% (qPH8–1) of phenotypic variance. Among the QTLs, 14 QTLs of them were observed for the first time (Table 3). A total of 21 QTLs were related to the region of the QTLs reported previously, and 19 of them were co-located in the regions with shorter intervals than previously reported, which might provide more detailed information for gene identification.

QTL mapping for the soybean traits with ICIM-ADD method. (a): LOD curve on the genome for PH; (b): LOD curve on the genome for SW; (c): LOD curve on the genome for Oil; (d): LOD curve on the genome for Protein
CIM method was also used to identify QTLs separately for 2 years. In total, 18 QTLs were observed on 11 chromosomes in 2013, and 21 QTLs were found on 15 chromosomes in 2014 (Additional file 4; Fig. 2). Of these QTLs, 31 QTLs were both identified by two methodsMoreover, q2013PH19–1 and q2014PH19–2 with shorter intervals than previously reported [19], which were located in the same confidence intervals, might be stable across both years. It was noteworthy that q2013Oil1–1 was placed in the same confidence intervals as q2013Protein1–1, and q2014Oil10–2 was placed in the same confidence intervals as q2013protein10–1, which might be useful in the coordinated improvement of seed quality for soybean breeding.
Analyses of epistatic effects
A total of 3 pairs of epistatic QTLs involving 6 loci on 5 chromosomes were identified for Protein (Table 4). The epistatic effect explained 5.49%, 4.49% and 4.06% of the PV, respectively. Pair one was composed of 2 QTLs, qProtein2–1 and qProtein3–2, with the PVE of 5.49%. qProtein5–2 was observed to have the epistatic effect with qProtein12–1. Meanwhile, qProtein3–3 showed epistatic interaction with qProtein17–1. However, no epistatic effect was observed for PH, SW and Oil.
Prediction of candidate genes
After filtering QTLs by the PVE and physical distance, 18 QTLs were used to mine candidate genes. According to the physical map, a total of 89, 144, 16 and 64 genes were screened in the interval of the filtered QTLs associated with PH, SW, Oil and Protein, respectively. Based on the expression data of candidate genes from phytozome and soybase (Fig. 5), 43 genes were considered to be potential candidates (Table 5). All genes from specific QTLs intervals were evaluated based on their expression pattern in different organs. In the case of genes from the QTLs associated with PH, 13 genes showed higher expression in stem and shoot apical meristem, indicating it might be considered as candidate genes related to PH (Fig. 5a). A total of 14 genes in the interval of the QTLs associated with SW, expressed in seed development stages (10 to 42 days after flowering), might participate in the pathways affecting SW (Fig. 5b and 5c). As the accumulation of oil and protein in soybean seed was throughout the seed development stage [20, 21], gene expressed sustainably in seed development stage might affect the biological process associated with oil and protein. In the present study, there were 7 genes found in the region of QTLs related to Oil, which expressed stably throughout the seed development stage, suggesting it might be associated with Oil (Fig. 5d). Meanwhile, 9 genes in the interval of QTLs for Protein, showed expression in the seed development stage, might be associated with Protein (Fig. 5e).

Tissue specific expression of candidate genes. RH, root hair; SAM, shoot apical meristem; DAF, day after flowering; (a), expression of candidate gene related to PH; (b), expression of candidate genes related to SW on chr12; (c), expression of candidate genes related to SW on chr5, chr13 and chr15; (d), expression of candidate genes related to Oil; (e), expression of candidate genes related to Protein
Discussion
Construction of a high-density genetic map based on SLAF markers
QTL mapping has been used as an efficient approach to analyze quantitative traits in plants. Parental genetic diversity and marker density are the major factors affecting the efficiency and accuracy of QTL mapping. In this study, the female parent, ‘Huapidou’, was a semi-wild soybean gerplasm, which showed high resistance to whitefly [22]. ‘Qihuang26’, with more than 46% of protein conten in seeds, was a main variety in Huang-Huai-Hai region of China. In the present study, four traits of the RIL population derived from Huapidou and Qihuang26 showed to be continuous with normal or skew normal distributions. Increasing marker density could improve the resolution of genetic map for a given mapping population [23]. SLAF-seq is an effective sequencing-based method for large-scale marker discovery and genotyping, which has been used for genetic analysis in many species [8,9,10,11,12,13, 24]. In the present study, we used 5111 high-quality SLAF markers to construct a high-density map, and a total of 8597 SNP loci were integrated into 20 LGs ultimately. This high-density genetic map, making QTL mapping more accurate and reliable, would be beneficial to MAS breeding.
QTL mapping in soybean using a high-density map
Soybean is a primary source of plant oil and protein for humans due to its high nutritional value. PH and SW were main yield-related traits in soybean. So far, markers associated with the QTL underlying PH, SW, Oil and Protein have been mapped onto all linkage groups. In total, there were 35 QTLs for PH, SW, Oil and Protein observed using a high-density map based on an F8:11 RIL population with 149 individuals from the cross between ‘Huapidou’ and ‘Qihuang26’. Furthermore, there were 14 novel QTLs related to PH, SW, Oil and Protein, indicating the distinct genetic architecture in the population derived from cultivated soybean and semi-wild soybean. Among the novel QTLs, qPH8–1 had the highest PVE value and the highest LOD value might be the major QTL related to PH. It was notable that qSW13–1 explained the hightest PV in the QTLs identified for SW. More remarkably, four novel QTLs for Oil, inculding qOil1–1, qOil1–2, qOil10–1 and qOil10–2 explained up to 72.73% of the PV for Oil, which suggested it might be potential loci to Oil. qProtein1–1, which explained 17.68% of the PV, might be an major QTL for further fine mapping. So many novel QTLs observed in the present study indicated that more germplasms need to be used for revealing the complex genetic basis of soybean.
The stability of QTL is essential for the use in a breeding programme. In the study, 31 QTLs were identified by both ICIM and CIM methods. Furthermore, one QTL for PH was identified by CIM in both experiments from 2013 and 2014. In addition, more than half of the QTLs had been reported. Five QTLs for PH detected in this study, were colocalized as reported [14, 19, 25, 26]. Two major QTLs associated with SW, qSW13–1 and qSW15–1, both with the physical distance of approximate 11 Kb, explained 18.55% and 12.21 of the PV for SW, respectively. qSW13–1 had been reported as being associated with L050–14 [27], Satt144 [28, 29] and Sat_103 [30]. qSW15–1 had been detected in two soybean populations, derived from ‘Young’ and ‘PI416937’ (Pop1), ‘PI97100’ and ‘Coker 237’ (Pop2) [31]. Han et al. also identified the similar QTL on chr15 in the population from a cross between ‘Hefeng25’ and ‘Conrad’ [32]. Therefore, qSW13–1 and qSW15–1 might be considered as major and stable QTLs for further fine mapping and map-based cloning to elucidate the mechanisms of SW. In the present study, four QTLs related to Oil had been reported [28, 33,34,35], inculding qOil6–1, qOil13–1, qOil19–1 and qOil19–2, but none of them explained more than 10% of the PV. Lee et al. [36] reported cr274_1 associated with Protein on chr15 using a population derived from ‘Young’ and ‘PI416937’. The QTL for Protein between Satt173 and Satt581 on chr10 had been identified [37], similar with the result of Liu et al. [38]. Our study detected two QTLs related to Protein, qProtein10–1 and qProtein15–1, with 16.83% and 14.36% of the PVE, respectively, mapped on the same area as previous studies [36,37,38], might be good for MAS breeding and accurate QTL localization.
Several QTLs of various traits can map to the same locus [14, 39]. In this study, two pairs of QTLs, q2013Oil1–1 and q2013Protein1–1 as well as q2014Oil10–2 and q2013protein10–1, with inverse additive effect for Oil and Protein, were located in the same marker interval (Fig. 4; Additional file 4), which implies that q2013Oil1–1 and q2014Oil10–2 not only control oil content in seeds but also affect protein content in seeds. It is consistent with previous reports that an negative correlation is in agreement between protein and oil concentration in soybean seeds [40, 41].
Knowledge of epistasis effect, which is defined as interactions between alleles of two or more genetic loci, is essential to understand the genetic mechanism and the gene networks underlying complex traits. In this study, 3 pairs of epistatic QTLs for Protein were identified by ICIMapping-EPI. However, these epistatic QTLs did not display additive effect alone. It might be considered modifying genes that have no significant effects alone but might affect the expression of Protein related genes through epistatic interactions. Nevertheless, epistatic interaction could not be detected in some map populations [42]. It might be the reason that no epistatic effect observed for PH, SW and Oil in the present study.
Gene mining based on precise QTLs
As the average ratio of gene to physical distance is about 1 gene per 20 Kb in soybean genome [43], the accuracy of QTL mapping is of great benifit to gene localization and identification. The physical distance of 11 QTLs in the current study was shorter than 100 Kb, which might lead to a direct opportunity to find candidate genes by bioinformatics tools. For example, the minimum confidence interval of qPH8–2 was 8.3 Kb, which was much shorter than 0.09 Mb detected previously [26]. Furthermore, Glyma.08 g337400, encoding a transducin/WD40 repeat-like superfamily protein, was predicted in the interval of qPH8–2, which might be a promising target to engineer transgenic plants with higher biomass and improved growth development for plant-based bioenergy production [44]. In the interval of qPH17–1, Glyma.17 g169100, encoding a 2OG-Fe(II) oxygenase superfamily protein, was one of the important gibberellin oxidase genes [45], which might affect plant height directly. Glyma.17 g167700, encoding a growth regulator CYCLIN D3–2, expressed in growing shoot apices preferentially [46,47,48]. In the interval of qSW5–1, Glyma.05 g055700, encoding beta vacuolar processing enzyme, was involved in seed coat formation at the early stage of seed development [49]. In the interval of qSW12–1, ribosomal protein L23AB encoded by Glyma.12 g066700, was required for normal development [50]. Glyma.01 g106000 and Glyma.01 g106100 in the interval of qOil1–2, encoding Glutathione S-transferase TAU 8, might influence Oil by suppressing lipid peroxidation [51]. There were 3 genes found in the confidence interval of qOil10–2, which explained 20.17% of the PV for oil, inculding Glyma.10 g181800, Glyma.10 g181900 and Glyma.10 g182000. Moreover, Glyma.10 g181900, encoding a trigalactosyldiacylglycerol 1 protein (TGD1), affected the metabolic flux of chloroplast lipid synthesis and photosynthetic capacity, which resulted in the change of fatty acid in leaf and seed [52,53,54,55]. It was noteworthy that qOil10–2 was placed in the same confidence intervals as q2013protein10–1. The inverse relationship between oil and protein in soybean seed is well documented in the previous reports [40, 41]. However, little study of TGD1 had been reported on the function of protein accumulation in seeds. In the interval of qProtein10–1, Glyma.10 g183900, encoding peptide transporter 3, contributed to nitrogen allocation and grain Yield [56], Glyma.10 g184900, encoding a ureidoglycolate amidohydrolase, played a key role in nitrogen transport and storage [57,58,59]. In a word, on the basis of the physical position of these precise QTLs detected using a high-density map in the present study, it would be easy to find candidate gene.
Conclusions
In this study, we genotyped a RIL population (Huapidou × Qihuang26) by SLAF-seq. A high-density genetic map for soybean was constructed and used to identify QTLs associated with four traits, including plant height, seed weight, oil content seed and protein content in seed. A total of 35 QTLs related to four traits were identified. Of these QTLs, 21 QTLs were coincident with previous research. Furthermore, three pairs epistatic QTLs involving 6 loci on 5 chromosomes were identified for Protein. In addition, 43 genes with tissue-specific expression patterns were considered to be potential genes in further study. Our findings might be of great useful for MAS breeding, and could provide detailed information for accurate QTL localization.
Methods
Plant material and phenotyping
F8:9 and F8:10 populations of 149 RILs derived from a cross between ‘Huapidou (ZDD09982)’ and ‘Qihuang26 (ZDD23189)’ were planted in the experiment field of Shandong Academy of Agricultural Sciences in Jinan, Shandong Province, China, in 2013 and 2014, respectively. Each individual was planted in one row using single seed sowing; each row was 3 m, with 50 cm row spacing and 10 cm plant spacing, with three replicates. Five plants in each replicate were selected randomly to calculate the plant height (PH). The weight of 100 random filled seeds was measured as seed weight (SW). Oil and protein in soybean seed were detected by DA 7200 NIR food analyzer (Perten, Switzerland). SW, Oil and Protein were repeat 3 times in each replicate. Frequency distribution, descriptive statistics, the broad-sense heritability (h2) and the analysis of variance for RIL population and parents were analyzed with the SPSS statistics 17.0 and Microsoft Excel 2010. The h2 was estimated as described by previous study [60].
DNA extraction and genotyping
Seedlings of the F8:11 population of 149 RILs and parents were planted in 2016. Young healthy leaves from the two parents and RIL individuals were collected and genomic DNA was extracted by the CTAB method [61]. DNA was quantified with NanoDrop and by electrophoresis in 1% agarose gels with a λ DNA standard.
SLAF-seq was used to genotype a total of 151 samples (149 individuals and two parents) as described by previous study [7]. All polymorphic SLAFs were genotyped with consistency in the offspring and parental SNP loci. All SLAF markers should be filtered in quality assessment. A SLAF marker with parental homozygous, which had less than three SNPs, average depths of each sample above 3, was used as a high quality SLAF marker.
Linkage map construction
Before map construction, SLAF marker should be filtered by linkage analysis, markers with the MLOD value > 3 were used to construct genetic linkage map. SLAF markers with high quality were located into 20 LGs. HighMap Strategy was used to order SLAF markers and correct genotyping errors [24]. All LGs should be undergone these procedures: first, markers were arranged by their locations on choromosome; second, genotyping errors or deletions were corrected by SMOOTH [62], according to the relationship between ordered markers; then MSTmap was used to order the map [63]; after that, SMOOTH was used again to corrected the new ordered genotype. High-quality map would be obtained after 4 or more cycles. Map distance was estimated using the Kosambi mapping function.
QTL mapping
Based on the high-density genetic map, the QTLs underlying PH, SW, Oil and Protein were identified by QTL ICIMapping V3.3 software [64]. Inclusive Composite Interval Mapping (ICIM) and Composition interval mapping (CIM) methods were used to identify the QTLs. The threshold of logarithm of odds (LOD) score for evaluating the statistical significance of QTL effect was determined using 1000 permutations at the 5% level of significance. The location of a QTL was described according to its LOD peak location and thesurrounding region with 95% confidence interval [65]. As a result, intervals with a LOD value above 2.5 were detected as effective additive QTLs using ICIM-ADD method; the pairs of QTLs with a LOD value above 5.0 were considered as valid epistatic QTLs using ICIM-EPI method.
Gene mining
According to the marker sequence, QTLs were projected from the genentic map onto Williams 82 physical map (Glyma.Wm82.a2.v1). The QTL with the PVE above 10% and the physical distance less than 1 Mb was used to screen the candidate genes. The QTL with the physical distance less than 100 Kb was also chosen to screen the candidate genes. Gene calls and annotations were retrieved using Glyma.Wm82.a2.v1 gene model from Soybase. The expression data of candidate genes in the seed development stages based on RNA-seq in previous study were obtained from soybase [66]. The expression values of candidate genes in flower, leaf, nodule, pod, root, root hair, seed, shoot apical meristem and stem were downloaded from phytozome (www.phytozome.org). Multiple array viewer (version 4.9.0) was used to construct the heat map to analyze the tissue-specific expression of candidate genes.
Abbreviations
- chr:
-
Chromosome
- CTAB:
-
Hexadecyl trimethyl ammonium bromide
- DAF:
-
Day after flowering
- GBS:
-
Genotyping-by-sequencing
- GC:
-
Guanine-cytosine
- ICIM:
-
Inclusive composite interval mapping
- LGs:
-
Linkage groups
- LOD:
-
Logarithm of odds
- MAS:
-
Molecular marker-assisted selection
- Oil:
-
Oil content in soybean seeds
- PH:
-
Plant height
- Protein:
-
Protein content in soybean seeds
- PV:
-
Phenotypic variance
- PVE:
-
Phenotypic variance explained
- QTL:
-
Quantitative trait locus
- RAD-seq:
-
Restriction-site associated DNA sequencing
- RH:
-
Root hair
- RIL:
-
Recombinant inbred lines
- SAM:
-
Shoot apical meristem
- SLAF-seq:
-
Specific length amplified fragment sequencing
- SNP:
-
Single nucleotide polymorphisms
- SW:
-
100-seeds weight
References
Wilson RF. Soybean: market driven research needs: Springer New York; 2008.
Kibite S, Evans LE. Causes of negative correlations between grain yield and grain protein concentration in common wheat. Euphytica. 1984;33(3):801–10.
Mohan M, Nair S, Bhagwat A, Krishna TG, Yano M, Bhatia CR, Sasaki T. Genome mapping, molecular markers and marker-assisted selection in crop plants. Mol Breed. 1997;3(2):87–103.
Miller MR, Dunham JP, Amores A, Cresko WA, Johnson EA. Rapid and cost-effective polymorphism identification and genotyping using restriction site associated DNA (RAD) markers. Genome Res. 2007;17(2):240.
Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for De novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7(5):e37135.
Poland JA, Brown PJ, Sorrells ME, Jannink JL. Development of high-density genetic maps for barley and wheat using a novel two-enzyme genotyping-by-sequencing approach. PLoS One. 2012;7(2):e32253.
Sun X, Liu D, Zhang X, Li W, Liu H, Hong W, Jiang C, Guan N, Ma C, Zeng H. SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using high-throughput sequencing. PLoS One. 2013;8(3):e58700.
Zhang Y, Wang L, Xin H, Li D, Ma C, Ding X, Hong W, Zhang X. Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 2013;13:141.
Zhu Y, Yin Y, Yang K, Li J, Sang Y, Huang L, Fan S. Construction of a high-density genetic map using specific length amplified fragment markers and identification of a quantitative trait locus for anthracnose resistance in walnut (Juglans regia L.). BMC Genomics. 2015;16:614.
Li D, Huang Z, Song S, Xin Y, Mao D, Lv Q, Zhou M, Tian D, Tang M, Wu Q, et al. Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc Natl Acad Sci U S A. 2016;113(41):E6026–35.
Ji G, Zhang Q, Du R, Lv P, Ma X, Fan S, Li S, Hou S, Han Y, Liu G. Construction of a high-density genetic map using specific-locus amplified fragments in sorghum. BMC Genomics. 2017;18(1):51.
Jiang B, Liu W, Xie D, Peng Q, He X, Lin Y, Liang Z. High-density genetic map construction and gene mapping of pericarp color in wax gourd using specific-locus amplified fragment (SLAF) sequencing. BMC Genomics. 2015;16:1035.
Guo Y, Shi G, Liu Z, Zhao Y, Yang X, Zhu J, Li K, Guo X. Using specific length amplified fragment sequencing to construct the high-density genetic map for Vitis (Vitis vinifera L. x Vitis amurensis Rupr.). Front Plant Sci. 2015;6:393.
Cao Y, Li S, He X, Chang F, Kong J, Gai J, Zhao T. Mapping QTLs for plant height and flowering time in a Chinese summer planting soybean RIL population. Euphytica. 2017;213(2)
Li B, Tian L, Zhang J, Huang L, Han F, Yan S, Wang L, Zheng H, Sun J. Construction of a high-density genetic map based on large-scale markers developed by specific length amplified fragment sequencing (SLAF-seq) and its application to QTL analysis for isoflavone content in Glycine max. BMC Genomics. 2014;15:1086.
Qi Z, Huang L, Zhu R, Xin D, Liu C, Han X, Jiang H, Hong W, Hu G, Zheng H, et al. A high-density genetic map for soybean based on specific length amplified fragment sequencing. PLoS One. 2014;9(8):e104871.
Zhang D, Li H, Wang J, Zhang H, Hu Z, Chu S, Lv H, Yu D. High-density genetic mapping identifies new major loci for tolerance to low-phosphorus stress in soybean. Front Plant Sci. 2016;7:372.
Li B, Fan S, Yu F, Chen Y, Zhang S, Han F, Yan S, Wang L, Sun J. High-resolution mapping of QTL for fatty acid composition in soybean using specific-locus amplified fragment sequencing. Theoretical & Applied Genetics. 2017;130(7):1467–79.
Lark KG, Chase K, Adler F, Mansur LM, Orf JH. Interactions between quantitative trait loci in soybean in which trait variation at one locus is conditional upon a specific allele at another. Proc Natl Acad Sci U S A. 1995;92(10):4656.
Hill JE, Breidenbach RW. Proteins of soybean seeds: II. Accumulation of the major protein components during seed development and maturation. Plant Physiol. 1974;53(5):747–51.
Sale PWG, Campbell LC. Changes in physical characteristics and composition of soybean seed during crop development. Field Crop Res. 1980;3(2):147–55.
Jun Z, Wei L, LiFeng Z, HaiYing D, DunWei C, Ran X. QTL mapping of soybean resistance to whitefly (Bemisia tabaci Gennadius) under multi-environment condition. Aust J Crop Sci. 2013;7(8):1212–8.
Liu BH. Statistical genomics: linkage, mapping, and QTL analysis. Statistical Genomics Linkage Mapping & Qtl Analysis. 1997;
Liu D, Ma C, Hong W, Huang L, Liu M, Liu H, Zeng H, Deng D, Xin H, Song J. Construction and analysis of high-density linkage map using high-throughput sequencing data. PLoS One. 2014;9(6):e98855.
Kabelka EA, Diers BW, Fehr WR, Leroy AR, Baianu IC, You T, Neece DJ, Nelson RL. Putative alleles for increased yield from soybean plant introductions. Crop Sci. 2004;44(3):784–91.
Yin Z, Qi H, Chen Q, Zhang Z, Jiang H, Zhu R, Hu Z, Wu X, Li C, Zhang Y, et al. Soybean plant height QTL mapping and meta-analysis for mining candidate genes. Plant Breed. 2017;136(5):688–98.
Orf JH, Chase K, Jarvik T, Mansur LM, Cregan PB, Adler FR, Lark KG. Genetics of soybean agronomic traits: I. Comparison of three related recombinant inbred populations. Crop Sci. 1999;39(6):1642–51.
Hyten DL, Pantalone VR, Sams CE, Saxton AM, Landau-Ellis D, Stefaniak TR, Schmidt ME. Seed quality QTL in a prominent soybean population. Theoretical & Applied Genetics. 2004;109(3):552–61.
Yan L, Li YH, Yang CY, Ren SX, Chang RZ, Zhang MC, Qiu LJ. Identification and validation of an over-dominant QTL controlling soybean seed weight using populations derived from Glycine max × Glycine soja. Plant Breed. 2015, 133(5):632–7.
Teng W, Han Y, Du Y, Sun D, Zhang Z, Qiu L, Sun G, Li W. QTL analyses of seed weight during the development of soybean (Glycine max L. Merr.). Heredity. 2009;102(4):372.
Mian MAR, Bailey MA, Tamulonis JP, Shipe ER, Jr TEC, Parrott WA, Ashley DA, Hussey RS, Boerma HR. Molecular markers associated with seed weight in two soybean populations. Theoretical & Applied Genetics. 1996;93(7):1011.
Han Y, Li D, Zhu D, Li H, Li X, Teng W, Li W. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theoretical & Applied Genetics. 2012;125(4):671–83.
Qi Z, Wu Q, Han X, Sun Y, Du X, Liu C, Jiang H, Hu G, Chen Q. Soybean oil content QTL mapping and integrating with meta-analysis method for mining genes. Euphytica. 2011;179(3):499–514.
Qi Z-m, Han X, Y-n S, Wu Q, D-p S, X-y D, C-y L, H-w J, Hu G-h, Q-s C. An integrated quantitative trait locus map of oil content in soybean, Glycine max (L.) Merr., generated using a meta-analysis method for mining genes. Agric Sci China. 2011;10(11):1681–92.
Diers BW, Keim P, Fehr WR, Shoemaker RC. RFLP analysis of soybean seed protein and oil content. Theoretical & Applied Genetics. 1992;83(5):608–12.
Lee SH, Bailey MA, Mian MA, Jr CT, Shipe ER, Ashley DA, Parrott WA, Hussey RS, Boerma HR. RFLP loci associated with soybean seed protein and oil content across populations and locations. Theoretical & Applied Genetics. 1996;93(5–6):649–57.
Chen Q, Zhang Z, Liu C. QTL analysis of major agronomic traits in soybean. J Integr Agric. 2007;6(4):399–405.
Liu N, Li M, Hu X, Ma Q, Mu Y, Tan Z, Xia Q, Zhang G, Nian H. Construction of high-density genetic map and QTL mapping of yield-related and two quality traits in soybean RILs population by RAD-sequencing. BMC Genomics. 2017;18(1):466.
Zhang WK, Wang YJ, Luo GZ, Zhang JS, He CY, Wu XL, Gai JY, Chen SY. QTL mapping of ten agronomic traits on the soybean ( Glycine max L. Merr.) genetic map and their association with EST markers. Theoretical & Applied Genetics. 2004;108(6):1131–9.
Ray JD, Fritschi FB, Heatherly LG. Large applications of fertilizer N at planting affects seed protein and oil concentration and yield in the early soybean production system. Field Crop Res. 2006;99(1):67–74.
Hartwig EE, Hinson K. Association between chemical composition of seed and seed yield of soybeans 1. Crop Sci. 1972;12(6):829.
Mao T, Jiang Z, Han Y, Teng W, Zhao X, Li W. Identification of quantitative trait loci underlying seed protein and oil contents of soybean across multi-genetic backgrounds and environments. Plant Breed. 2013;132:630–41.
Schlueter J. Genome sequence of the palaeopolyploid soybean. Nature. 2010;463(7278):178–83.
Gachomo EW, Jimenezlopez JC, Baptiste LJ, Kotchoni SO. GIGANTUS1 (GTS1), a member of Transducin/WD40 protein superfamily, controls seed germination, growth and biomass accumulation through ribosome-biogenesis protein interactions in Arabidopsis thaliana. BMC Plant Biol. 2014;14(1):1–17.
Han F, Zhu B. Evolutionary analysis of three gibberellin oxidase genesin rice, Arabidopsis, and soybean. Gene. 2011;473(1):23.
Horvath DP, Chao WS. Cloning, characterization, and expression of growth regulator CYCLIN D3-2 in leafy spurge (Euphorbia esula). Weed Sci. 2005;53(4):431–7.
Hu Y, Bao F, Li J. Promotive effect of brassinosteroids on cell division involves a distinct CycD3-induction pathway in Arabidopsis. Plant J. 2000;24(5):693–701.
Rhee Y, Hwang K, Cho S, Lee M, Kil E-J, Choi S, Hahn B-S, Kim D, Auh C-K, Lee S. Expression analysis of D-type cyclin in potato (Solanum tuberosum L.) under different culture conditions. Acta Physiol Plant. 2016;38(2)
Nakaune S, Yamada K, Kondo M, Kato T, Tabata S, Nishimura M, Hara-Nishimura I. A vacuolar processing enzyme, deltaVPE, is involved in seed coat formation at the early stage of seed development. Plant Cell. 2005;17(3):876–87.
Degenhardt RF, Bonham-Smith PC. Arabidopsis ribosomal proteins RPL23aA and RPL23aB are differentially targeted to the nucleolus and are disparately required for normal development. Plant Physiol. 2008;147(1):128–42.
Roxas VP, Lodhi SA, Garrett DK, Mahan JR, Allen RD. Stress tolerance in transgenic tobacco seedlings that overexpress glutathione S-transferase/glutathione peroxidase. Plant & cell physiology. 2000;41(11):1229.
Klaus D, Härtel H, Fitzpatrick LM, Froehlich JE, Hubert J, Benning C, Dörmann P. Digalactosyldiacylglycerol synthesis in chloroplasts of the Arabidopsis dgd1 mutant. Plant Physiol. 2002;128(3):885.
Xu C, Fan J, Riekhof W, Froehlich JE, Benning C. A permease-like protein involved in ER to thylakoid lipid transfer in Arabidopsis. EMBO J. 2003;22(10):2370–9.
Xu C, Fan J, Froehlich JE, Awai K, Benning C. Mutation of the TGD1 chloroplast envelope protein affects phosphatidate metabolism in Arabidopsis. Plant Cell. 2005;17(11):3094.
Fan J, Yan C, Zhang X, Xu C. Dual role for phospholipid:diacylglycerol acyltransferase: enhancing fatty acid synthesis and diverting fatty acids from membrane lipids to triacylglycerol in Arabidopsis leaves. Plant Cell. 2013;25(9):3506–18.
Fang Z, Bai G, Huang W, Wang Z, Wang X, Zhang M. The Rice peptide transporter OsNPF7.3 is induced by organic nitrogen, and contributes to nitrogen allocation and grain yield. Front Plant Sci. 2017;8:1338.
Li J, Qin RY, Li H, Xu RF, Yang YC, Ni DH, Ma H, Li L, Wei PC, Yang JB. Low-temperature-induced expression of Rice Ureidoglycolate Amidohydrolase is mediated by a C-repeat/dehydration-responsive element that specifically interacts with Rice C-repeat-binding factor 3. Front Plant Sci. 2015;6:1011.
Werner AK, Medina-Escobar N, Zulawski M, Sparkes IA, Cao FQ, Witte CP. The ureide-degrading reactions of purine ring catabolism employ three amidohydrolases and one aminohydrolase in Arabidopsis, soybean, and rice. Plant Physiol. 2013;163(2):672–81.
Winkler RG, Blevins DG, Randall DD. Ureide catabolism in soybeans. Plant Physiol. 1988;86(4):104–1088.
Wyman ENPD, JBPD R. Estimation of heritability and prediction of selection response in plant populations. Crit Rev Plant Sci. 1991;10(3):235–322.
Del SG, Manfioletti G, Schneider C. The CTAB-DNA precipitation method: a common mini-scale preparation of template DNA from phagemids, phages or plasmids suitable for sequencing. Biotechniques. 1989;7(5):514–20.
Os HV, Stam P, Visser RGF, Eck HJV. SMOOTH: a statistical method for successful removal of genotyping errors from high-density genetic linkage data. Theoretical & Applied Genetics. 2005;112(1):187–94.
Wu Y, Bhat PR, Close TJ, Lonardi S. Efficient and accurate construction of genetic linkage maps from the minimum spanning tree of a graph. PLoS Genet. 2008;4(10):e1000212.
Wang JK. Inclusive composite interval mapping of quantitative trait genes. Acta Agron Sin. 2009;35(2):239–45.
Visscher PM, Thompson R, Haley CS. Confidence intervals in QTL mapping by bootstrapping. Genetics. 1996;143(2):1013–20.
Severin AJ, Woody JL, Bolon YT, Joseph B, Diers BW, Farmer AD, Muehlbauer GJ, Nelson RT, Grant D, Specht JE. RNA-Seq atlas of Glycine max : a guide to the soybean transcriptome. BMC Plant Biol. 2010;10(1):160.
Kato S, Sayama T, Fujii K, Yumoto S, Kono Y, Hwang TY, Kikuchi A, Takada Y, Tanaka Y, Shiraiwa T. A major and stable QTL associated with seed weight in soybean across multiple environments and genetic backgrounds. Theoretical & Applied Genetics. 2014;127(6):1365–74.
Li W, Zheng DH. QTL mapping for major agronomic traits across two years in soybean (Glycine max L. Merr.). In: Jcrop Scibiotechnol; 2008.
Stombaugh SK, Orf JH, Jung HG, Chase K, Lark KG, Somers DA. Quantitative trait loci associated with Cell Wall polysaccharides in soybean seed. Crop Sci. 2004;44(6):2101–6.
Acknowledgements
The authors wish to thank Dr. Xue Zhao for discussion and comments.
Funding
This work was supported by Chinese National Natural Science Foundation (31501329), Shandong Provincial Natural Science Foundation (ZR2015YL070), and Key Research and Development Program of Shandong (2016ZDJS10A03–03). The funding bodies had no role in study design, data collection, analysis and interpretation, decision to public, or writing of the manuscript.
Availability of data and materials
The data sets supporting the results of this study are included in the manuscript and additional supporting files. Soybean seeds are available from the Crop Research Institute, Shandong Academy of Agricultural Sciences, Jinan, China.
Author information
Authors and Affiliations
Contributions
YZ and RX designed the study. YZ, WL, YL, LZ and CW performed the experiments. CW contributed materials/analysis tools. YZ and WL analyzed the data. YZ wrote the manuscript, which was further edited by RX. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
A soybean population of 149 RILs and their parents were used in this study. The field experiment was performed in the experiment field of Shandong Academy of Agricultural Sciences in Jinan, Shandong Province, China, under natural conditions. Collection of plant materials complied with the institutional, national and international guidelines. No specific permits were required.
Consent for publication
Not applicable.
Competing interests
The authors have declared that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Genotype list of 149 RILs. (XLSX 2691 kb)
Additional file 2:
SLAF markers on the 20 linkage groups. (XLSX 129 kb)
Additional file 3:
Information of SLAF markers on the genetic map. (PARTIAL 4956 kb)
Additional file 4:
Additive QTLs identified by CIM in both experiments from 2013 and 2014. (PDF 168 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, Y., Li, W., Lin, Y. et al. Construction of a high-density genetic map and mapping of QTLs for soybean (Glycine max) agronomic and seed quality traits by specific length amplified fragment sequencing. BMC Genomics 19, 641 (2018). https://doi.org/10.1186/s12864-018-5035-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-5035-9