
Abstract
Background
Type strains of the genus Porphyrobacter belonging to the family Erythrobacteraceae and the class Alphaproteobacteria have been isolated from various environments, such as swimming pools, lake water and hot springs. P. cryptus DSM 12079T and P. tepidarius DSM 10594T out of all Erythrobacteraceae type strains, are two type strains that have been isolated from geothermal environments. Next-generation sequencing (NGS) technology offers a convenient approach for detecting situational types based on protein sequence differences between thermophiles and mesophiles; amino acid substitutions can lead to protein structural changes, improving the thermal stabilities of proteins. Comparative genomic studies have revealed that different thermal types exist in different taxa, and few studies have been focused on the class Alphaproteobacteria, especially the family Erythrobacteraceae. In this study, eight genomes of Porphyrobacter strains were compared to elucidate how Porphyrobacter thermophiles developed mechanisms to adapt to thermal environments.
Results
P. cryptus DSM 12079T grew optimally at 50 °C, which was higher than the optimal growth temperature of other Porphyrobacter type strains. Phylogenomic analysis of the genus Porphyrobacter revealed that P. cryptus DSM 12079T formed a distinct and independent clade. Comparative genomic studies uncovered that 1405 single-copy genes were shared by Porphyrobacter type strains. Alignments of single-copy proteins showed that various types of amino acid substitutions existed between P. cryptus DSM 12079T and the other Porphyrobacter strains. The primary substitution types were changes from glycine/serine to alanine.
Conclusions
P. cryptus DSM 12079T was the sole thermophile within the genus Porphyrobacter. Phylogenomic analysis and amino acid frequencies indicated that amino acid substitutions might play an important role in the thermophily of P. cryptus DSM 12079T. Bioinformatic analysis revealed that major amino acid substitutional types, such as changes from glycine/serine to alanine, increase the frequency of α-helices in proteins, promoting protein thermostability in P. cryptus DSM 12079T. Hence, comparative genomic analysis broadens our understanding of thermophilic mechanisms in the genus Porphyrobacter and may provide a useful insight in the design of thermophilic enzymes for agricultural, industrial and medical applications.
Similar content being viewed by others

Background
There are nine species in the genus Porphyrobacter at the time of writing [1,2,3,4,5,6,7,8,9], eight species of which have been validly published (except for Candidatus Porphyrobacter meromictius) according to the List of Prokaryotic Names with Standing in Nomenclature [10]. Porphyrobacter species live in diverse environments, including hot springs [4, 6], fresh water [2, 7], stadium seats [1], seawater [5, 8, 9], and swimming pools [3]. Among all type strains within the family Erythrobacteraceae, P. cryptus and P. tepidarius are the only two species isolated from geothermal environments. Moreover, the genus Porphyrobacter has potential applications, such as hydrocarbon degradation [11], algalytic activity [12] and bioleaching [13]. Type strains isolated from hot springs may provide an insight to how these bacteria have adapted to thermal environments. This information may help us design thermophilic enzymes for many important applications.
Thermophiles are microorganisms whose optimum temperature for growth exceeds 45 °C [14]. Thermophiles are widely distributed [14,15,16] and have been isolated from various geothermal environments, such as artificial hot environments [17,18,19], marine hydrothermal vents [20,21,22] and hot springs [23,24,25]. The thermal stabilities of proteins play an important role in the survival of thermophiles [14, 26]. Amino acid substitutions in protein sequences can improve the thermal stabilities of proteins, which are beneficial changes that help microorganisms adapt to the environment [27,28,29]. Compared with the proteins of mesophiles, the proteins of thermophiles have a larger fraction of residues in α-helices, enhancing the stability of proteins [30]. Moreover, the proteins of thermophiles often have hydrophobic residues in the protein interior, promoting conformational stability in the inner part of proteins [15, 31]. Therefore, it is worth studying amino acid substitutions in the proteins of thermophiles that exhibit a close phylogenetic relationship to elucidate their adaptational mechanisms to geothermal environments and guide site-directed substitutions in the engineering of industrial enzymes to improve their thermostability.
Next-generation sequencing (NGS) technology generates massive data sets, which offer a convenient approach to studying amino acid substitutions in the protein sequences of microbes. Nishio et al. [32] studied the amino acid substitutions of Corynebacterium efficiens with 45 °C as an upper temperature limit for growth (UTLG) by comparing the genomes of C. efficiens (UTLG: 40 °C) and C. glutamicum and pointed out that three substitutions, changing the amino acid from lysine to arginine, serine to alanine or serine to threonine, enhanced the thermostability of proteins. Takami et al. [33] first sequenced the complete genome of a thermophilic Bacillus-related species, Geobacillus kaustophilus HTA426, with 60 °C as an optimal temperature for growth (OTG) and compared its genome with those of five mesophilic (OTG: 20–45 °C) Bacillus-related species, namely, Bacillus anthracis Ames, B. cereus ATCC 14579, B. halodurans C-125, B. subtilis 168 and Oceanobacillus iheyensis HTE831, and reported that arginine, alanine, glycine, proline and valine amino acid substitutions were found at greater than expected frequency in G. kaustophilus proteins, whereas asparagine, glutamine, serine and threonine were found at lower than expected frequency. However, Singer and Hickey [34] analyzed the genomes of thermophilic Aquificae (OTG: 85 °C), Crenarchaeota (OGT: 57.5–97.5 °C), Euryarchaeota (OTG: 65–95 °C) and Thermotogae (OGT: 75 °C) and found that the proportion of glutamic acid, isoleucine, tyrosine and valine increased in thermophilic strains while the proportion of alanine, glutamine, histidine and threonine decreased. Generally, an increase in the content of hydrophobic, charged and aromatic residues and a decrease in the content of polar uncharged residues occurs in thermophile proteins [32,33,34]. However, the types of amino acid substitutions have been observed to vary in different thermophilic taxa [32,33,34]. Therefore, studying the amino acid substitution types of various thermophiles, including their genera, will broaden our understanding of their thermophilic mechanisms.
Although some comparative genomic studies concerning amino acid substitutions in thermophile proteins have been performed [32,33,34], few studies have been focused on the class Alphaproteobacteria, especially the family Erythrobacteraceae. To interpret the thermophilic mechanisms in this taxon, here, we sequenced the genomes of five Porphyrobacter type strains using a NGS platform and performed comparative genomic and statistical analysis among eight genomes of the genus Porphyrobacter type strains.
Results and discussion
Range and optimum growth temperature of type strains belonging to the genus Porphyrobacter.
Except for P. cryptus DSM 12079T and P. tepidarius DSM 10594T, other Porphyrobacter strains, whose optimal temperature for growth was in the range of 30–35 °C, could not grow at 45 °C (Fig. 1). The optimal growth temperatures for P. cryptus DSM 12079T and P. tepidarius DSM 10594T were 50 and 45 °C, respectively (Fig. 1). Furthermore, the ranges for the growth of P. cryptus DSM 12079T and P. tepidarius DSM 10594T were 40–60 and 35–50 °C, respectively (Fig. 1); the upper limits of these ranges were higher than those of other Porphyrobacter species.

Ranges and optimum growth temperatures for seven Porphyrobacter type strains
Although the optimal temperatures for the growth of P. cryptus DSM 12079T and P. tepidarius DSM 10594T were higher than those of other Porphyrobacter strains, P. cryptus DSM 12079T was the only thermophile within the genus Porphyrobacter based on the definition of a thermophile, which has an optimal temperature that exceeds 45 °C [14]. Notably, P. cryptus DSM 12079T could grow above 50 °C, whereas P. tepidarius DSM 10594T could not.
Comparative genomic and phylogenomic analyses
The genome size, number of coding sequences (CDSs), and G + C content of the genus Porphyrobacter were 2.9–4.3 Mbp, 2839–4042 and 63.6–67.9 mol%, respectively (Table 1). Compared with other Porphyrobacter strains, P. cryptus DSM 12079T had the smallest genome size and the second lowest number of CDSs (Table 1). However, the p values of significant tests on genome size and the number of CDSs between P. cryptus DSM 12079T and other Porphyrobacter strains were 0.056 and 0.100, respectively, which showed that P. cryptus was not significantly different from other Porphyrobacter species in terms of genome size and the number of CDSs. Therefore, the small genomic size and reduced number of CDSs were not related to the thermophily of P. cryptus DSM 12079T.
The number of unique OCs harbored by each Porphyrobacter strain varied. P. mercurialis CoronadoT had the most unique OCs, and P. cryptus DSM 12079T, being a thermophile, harbored the fewest unique OCs (Fig. 2). It was found that over 80% of the unique OCs held by P. cryptus DSM 12079T were shared by the closest relatives belonging to the orders Rhizobiales, Rhodobacterales, Rhodospirillales and Sphingomonadales, which do not include typical thermophiles.

Pan-genomic curve (a) showing the relationship between OC number and genome number. Schematic view (b) of the core and unique OCs of Porphyrobacter species. Central number indicates the shared OCs of the genus Porphyrobacter, whereas the ambient number represents the unique OCs harboured by each Porphyrobacter species
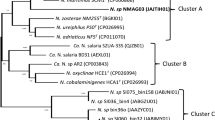
Analysis of the genus Porphyrobacter showed that their pan-genome was open (Fig. 2), which suggests that the genus Porphyrobacter colonizes multiple environments [35], matching the isolation sources of the genus Porphyrobacter (Table 1). It was found that 1434 OCs were shared by all eight Porphyrobacter strains (Fig. 2). When screening the single-copy shared OCs of eight Porphyrobacter strains and Altererythrobacter epoxidivorans CGMCC 1.7731T, the number of shared OCs decreased to 1322. A maximum-likelihood phylogenetic tree reconstructed based on 1322 shared protein sequences revealed that P. cryptus DSM 12079T formed a distinct and independent clade from other Porphyrobacter strains (Fig. 3). The phylogenetic distance from other members of the same genus may be the result of amino acid substitutions that have been selected for to allow the organism to adapt to thermal environments.

Maximum-likelihood tree based on the contatenatation of 1322 protein sequences showing the phylogenetic relationship of type strains belonging to the genus Porphyrobacter. Bootstrap values are based on 100 replicates. Bar, 0.1 substitutions per amino acid position. Red branch shows the clade within P. cryptus DSM 12079T. Altererythrobacter epoxidivorans CGMCC 1.7731T was used as an outgroup (not shown)
Analysis of amino acid substitutions
There were 274 amino acid substitutions in P. cryptus DSM 12079T, and the total number that varied amongst all strains was 5698 (Fig. 4, Additional files 1 and 2). Among the whole amino acid substitutions, the major variations (> 3.0%) were changes from valine to isoleucine, asparatic acid to glutamic acid, lysine to arginine, serine to alanine, and glycine to alanine. When compared with proteins from other thermophiles, where the common amino substitutions reported were changes from lysine to arginine and serine to alanine, the number of variations from valine to isoleucine, asparatic acid to glutamic acid, and glycine to alanine were higher here than in other studies [32,33,34]. Due to bidirectional substitutions, the substituted/substituting ratio analysis indicated that amino acids were most frequently changed to alanine, which resulted in a net substitutional number that was higher for alanine (a ratio above one) than for other amino acids (Fig. 5, Additional file 3).

Hot spot map of amino acid variations of Porphyrobacter cryptus. Amino acids located on the vertical axis represent substituting amino acids, whereas amino acids lying on the horizontal axis are substituted amino acids. A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, lysine; M, methionine; N, asparagine; P, proline; Q, glutamine; R, arginine; S, serine; T, threonine; V, valine; W tryptophan; Y tyrosine

Net substitutional variations of each Porphyrobacter cryptus DSM 12079T amino acid based on the alignments of proteins encoded by single-copy genes of the genus Porphyrobacter. The dashed line shows the position at which the net variation equals zero. Red indicates hydrophobic amino acids, while blue represents hydrophilic amino acids. A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, lysine; M, methionine; N, asparagine; P, proline; Q, glutamine; R, arginine; S, serine; T, threonine; V, valine; W tryptophan; Y tyrosine
In view of amino acid substitutions causing variations in amino acid frequencies, principal component analysis (PCA) of the amino acid frequency revealed that P. cryptus DSM 12079T was distinct from other Porphyrobacter strains (Fig. 6, Additional file 4), which indicated that the amino acid frequencies of P. cryptus DSM 12079T were also different from other strains with PC1 and PC2 accounting for 43 and 40% of the total variances, respectively. It was observed that the frequencies of asparagine, aspartic acid, glutamine, methionine, serine, threonine, tyrosine and valine were the lowest in P. cryptus DSM 12079T proteins, whereas the frequencies of alanine, arginine, leucine and proline were the highest in this strain (Fig. 7). The t-tests performed on the frequency of these amino acids showed that the p values were all lower than 1.0 × 10− 4, indicating that these differences were significant.

Principal component analysis (PCA) of amino acid frequencies among eight Porphyrobacter type strains. PC1 and PC2 account for 43 and 40% of the total variances. Red indicates P. cryptus DSM 12079T, whereas blue represents seven other Porphyrobacter type strains

Comparisons of amino acid frequencies among shared OCs in eight Porphyrobacter type strains. A, alanine; C, cysteine; D, aspartic acid; E, glutamic acid; F, phenylalanine; G, glycine; H, histidine; I, isoleucine; K, lysine; L, lysine; M, methionine; N, asparagine; P, proline; Q, glutamine; R, arginine; S, serine; T, threonine; V, valine; W tryptophan; Y tyrosine. OTG of P. donghaensis DSM 16220T and P. sanguineus JCM 20691T is 30 °C; OTG of P. colympi JCM 18338T, P. dokdonensis DSM 17193T and P. neustonensis DSM 9434T is 35 °C; OTG of P. tepidarius DSM 10594T is 45 °C; OTG of P. cryptus DSM 12079T is 50 °C
The results showed that the major amino acids with net gains were alanine, arginine, glutamic acid, isoleucine and serine based on the substituted/substituting residues and amino acid frequency analyses (Figs. 5 and 7). Substitutions of these five amino acids occurred in 1039 proteins out of all 1405 shared and orthologous proteins in the genus Porphyrobacter.
Except for 12 overly long proteins that failed to be analyzed in secondary structure predictions, the percentages of α-helices, β-sheets, random coils and turns were identical in the 291 original and substituted protein pairs. However, the percentage of α-helices were clearly lower when the substitutional proteins were compared with their corresponding original proteins in P. cryptus DSM 12079T (Additional file 5). Furthermore, 37 conserved proteins were selected through a comparative genomic study of selected Alphaproteobacteria genomic data. Except for five proteins not showing amino acid substitutions, the percentage of α-helices decreased in 18 proteins (Additional file 6). Previous studies showed that net increases in α-helix content could enhance the thermal stability of proteins [36, 37]. It is speculated that a net increase in α-helix content plays a key role in raising protein thermostability in the genus Porphyrobacter.
Further analysis of substitutional proteins with decreased ratios of α-helix regions indicated that 68.3% (348/509), 41.1% (209/509) and 39.8% (203/509) of these proteins had increased percentages of β-sheets, random coils and turns, respectively (Fig. 8). It was found that alanine was the most commonly substituted amino acid (43.9%, 739/1682) among the five major substituted amino acids in substitutional proteins, resulting in fewer amino acids forming α-helix, and the most common replacement amino acids were glycine (20.8%, 154/739) and serine (21.4%, 158/739). In addition, P. cryptus DSM 12079T had a distinctly higher frequency (p = 4.75 × 10− 6) of alanine than the other Porphyrobacter species (Fig. 7). Comparisons of secondary structures of P. cryptus DSM 12079T proteins in which alanine was replaced by glycine/serine indicated that variations of secondary structures occurred in 57.1% (88/154) and 42.4% (67/158) of alanine to glycine/serine sites, respectively. It was investigated whether α-helices significantly changed into other secondary structures, and secondary structure changes occurred in 89.8% (79/88) of alanine to glycine sites and 94.0% (63/67) of alanine to serine sites. Previous studies showed that alanine was the best helix-forming amino acid [27] and that it was found at a higher frequency in thermophiles than in other organisms [38]. The side chain of alanine is methyl (-CH3), which promotes the interactions of neighboring residues in proteins and contributes to packing in protein structures [39]. In addition, hydrophobic residues located in the protein interior enhance conformational stability in the inner part of proteins [31]. Glycine and serine are both hydrophilic amino acids, and they can decrease the core hydrophobicity of proteins. Moreover, glycine tends to increase molecular flexibility by weakening protein stability at higher temperatures [40]; and serine can be released from proteins at higher temperatures, which results in unstable protein structures [41, 42]. Hence, alanine substituting for glycine or serine might promote the formation of α-helices in proteins, which could increase protein thermostability within P. cryptus DSM 12079T.

Variational percentages of protein secondary structures of Porphyrobacter cryptus DSM 12079T original proteins to substituting proteins, according to sites where alanine was replaced by other amino acids. C, E, H and T stand for random coil, β-sheet, α-helix and turn, respectively. Red and green indicate decreases or increases in this kind of secondary structure, respectively, and yellow shows no changes
The analysis of neighboring amino acids of the substituted alanine indicated that hydrophobic amino acids occurred mostly in the N-terminus (41.1%, 58/141) and C-terminus (42.6%, 60/141), and this occurrence was higher than for other amino acid types. Furthermore, alanine, isoleucine and leucine were frequently located in the N-terminus, whereas alanine, leucine and proline were always found in the C-terminus among the neighboring hydrophobic amino acids of the substituted alanine. In addition, it was also often found that arginine and glycine were located in neighboring N- and C-terminal ends.
Conclusions
Comparative genomics analysis of the thermophile Porphyrobacter cryptus DSM 12079T with seven non-thermophilic Porphyrobacter strains broadened our knowledge of bacterial thermophilic mechanisms, especially in the class Alphaproteobacteria. Phylogenomic analysis of the genus Porphyrobacter and alignments of proteins encoded by shared single-copy genes revealed that amino acid substitutions play an important role in the thermophily of P. cryptus DSM 12079T. As a whole, substitutions from glycine/serine to alanine in P. cryptus DSM 12079T proteins increased the frequency of α-helices in these proteins, which could promote protein thermostability and ensure the growth of P. cryptus DSM 12079T at high temperatures. It is hoped that the findings of this study will help in the design of thermostable proteins using techniques such as site-directed mutagenesis.
Methods
Range and optimum of growth temperature
Seven type strains of the Porphyrobacter species were purchased from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (P. cryptus DSM 12079T, P. dokdonensis DSM 17193T, P. neustonensis DSM 9434T and P. tepidarius DSM 10594T) and Japan Collection of Microorganisms (P. colympi JCM 18338T and P. sanguineus JCM 20691T). All type strains were cultured in appropriate media [2,3,4,5,6, 8, 9] and maintained in glycerol suspensions (30%, v/v) at − 80 °C. The temperature range for growth (4, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55 and 60 °C) was inspected in the appropriate medium of each strain. The absorbance at a wavelength of 600 nm for Porphyrobacter cultures was measured using an ultraviolet and visible spectrophotometer (TU-1810, Persee).
Whole-genomic sequencing, assembly and annotation
Except for the genomic sequences of P. cryptus DSM 12079T, P. mercurialis CoronadoT and P. neustonensis DSM 9434T were acquired under NCBI GenBank accession numbers AUHC00000000, JTDN00000000 [43] and CP016033 [44], respectively. Genomic DNA of the five Porphyrobacter type strains was extracted according to the protocol of AxyPrep™ Bacterial Genomic DNA Miniprep Kit (Axygen®, Corning) and detected through 0.6% (w/v) agarose gel electrophoresis with λ-HindIII digest DNA (Takara, Dalian). Whole-genomic sequencing was carried out using Solexa paired-end sequencing technology (HiSeq2000 system, Illumina, USA) based on a whole-genome shotgun strategy in Anoroad Gene Technology Co. Ltd. (Beijing). Raw reads containing ≥9 bp of N were removed and trimmed by removing adaptor sequences. Clean reads were assembled using ABySS 1.9.0 [45]. The ORFs were predicted by Glimmer v.3.0 [46] based on assembled genomes and then annotated by using the online Rapid Annotation using Subsystems Technology (RAST) server [47]. Clusters of Orthologous Groups (COGs) protein annotations were carried out by the online WebMGA server [48, 49].
Comparative-genomic and phylogenomic analyses
The predicted proteins of eight Porphyrobacter species as well as Altererythrobacter epoxidivorans CGMCC 1.7731T (GenBank accession number: CP012669) were compared through OrthoMCL version 2.0 based on OrthoMCL algorithm [50, 51] to detect orthologous clusters (OCs) among the genomes of nine species. Pan- and core-genomic analyses were performed using in-house shell and R scripts (Additional file 7). The BLASTp searches [52] of unique OCs harbored by each Porphyrobacter species against a nr database were used to detect the closest relatives with thresholds of the identity above 70% and coverage not less than the half-length of query annotated proteins. Moreover, conserved proteins were selected by comparative genomic analysis with other Alphaproteobacteria genomes (Additional file 8) using OrthoMCL version 2.0 [50, 51].
Single-copy shared OCs of eight Porphyrobacter species and Altererythrobacter epoxidivorans were screened by in-house shell scripts (Additional file 7). Each single-copy shared OC was aligned by using MAFFT version 7 [53]. Aligned sequences refined by trimAL version 1.4.1 [54] were concatenated manually. Then, a phylogenetic tree was constructed by maximum-likelihood [55] algorithms with IQ-TREE 1.6.1 software [56] based on concatenated aligned single-copy OCs with bootstraps analysis set to 100 replicates, with the best amino acid substitutional model set as LG + F + R3 as proposed by IQ-TREE 1.6.1 software [56]. The maximum-likelihood phylogenetic tree was visualized using MEGA 7 software [57]. The t-tests of genomic size and the number of CDSs were calculated by using the MASS package within R programming language (https://cran.r-project.org/web/packages/MASS/index.html).
Amino acid substitutional analyses
Single-copy shared OCs of eight Porphyrobacter strains were screened and aligned according to the methods described above. Each site of the aligned protein sequences was detected through MEGA7 software [57] to determine a mutational site, which was defined as cases where P. cryptus DSM 12079T held a different amino acid and other Porphyrobacter species had another identical amino acid. The amino acid content of each Porphyrobacter species was summarized, and principal components analysis (PCA) was carried out by using the psych package within R programming language (https://cran.r-project.org/web/packages/psych/index.html). Proteins in P. cryptus DSM 12079T involved in major types of amino acid substitutions with net positive variation were screened using R scripts. Substitutional proteins were created manually by replacing the substituting amino acids with the substituted ones. Original and substitutional proteins were submitted to Chou & Fasman Secondary Structure Prediction Server (http://www.biogem.org/tool/chou-fasman/) to predict their secondary structures based on the Chou & Fasman algorithm [58, 59]. Variations about the percentages of α-helices, β-sheets, random coils and turns in the original and substitutional proteins were summarized to check intrinsic mechanisms leading to the thermophily of P. cryptus DSM 12079T.
Abbreviations
- CDSs:
-
Coding sequences
- NGS:
-
Next-generation sequencing
- OTG:
-
Optimal temperature for growth
- UTLG:
-
Upper temperature limit for growth
References
Coil DA, Flanagan JC, Stump A, Alexiev A, Lang JM, Eisen JA. Porphyrobacter mercurialis sp. nov., isolated from a stadium seat and emended description of the genus Porphyrobacter. PeerJ. 2015;3:e1400.
Fuerst JA, Hawkins JA, Holmes A, Sly LI, Moore CJ, Stackebrandt E. Porphyrobacter neustonensis gen. Nov., sp. nov., an aerobic bacteriochlorophyll-synthesizing budding bacterium from fresh water. Int J Syst Bacteriol. 1993;43:125–34.
Furuhata K, Edagawa A, Miyamoto H, Kawakami Y, Fukuyama M. Porphyrobacter colymbi sp. nov. isolated from swimming pool water in Tokyo, Japan. J Gen Appl Microbiol. 2013;59:245–50.
Hanada S, Kawase Y, Hiraishi A, Takaichi S, Matsuura K, Shimada K, Nagashima KV. Porphyrobacter tepidarius sp. nov., a moderately thermophilic aerobic photosynthetic bacterium isolated from a hot spring. Int J Syst Bacteriol. 1997;47:408–13.
Hiraishi A, Yonemitsu Y, Matsushita M, Shin YK, Kuraishi H, Kawahara K. Characterization of Porphyrobacter sanguineus sp. nov., an aerobic bacteriochlorophyll-containing bacterium capable of degrading biphenyl and dibenzofuran. Arch Microbiol. 2002;178:45–52.
Rainey FA, Silva J, Nobre MF, Silva MT, da Costa MS. Porphyrobacter cryptus sp. nov., a novel slightly thermophilic, aerobic, bacteriochlorophyll a-containing species. Int J Syst Evol Microbiol. 2003;53:35–41.
Rathgeber C, Yurkova N, Stackebrandt E, Schumann P, Humphrey E, Beatty JT, et al. Porphyrobacter meromictius sp. nov., an appendaged bacterium, that produces bacteriochlorophyll a. Curr Microbiol. 2007;55:356–61.
Yoon JH, Kang SJ, Lee MH, Oh HW, Oh TK. Porphyrobacter dokdonensis sp. nov., isolated from sea water. Int J Syst Evol Microbiol. 2006;56:1079–83.
Yoon JH, Lee MH, Oh TK. Porphyrobacter donghaensis sp. nov., isolated from sea water of the East Sea in Korea. Int J Syst Evol Microbiol. 2004;54:2231–5.
Parte AC. LPSN-list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2014;42:D613–6.
Al-Mailem DM, Kansour MK, Radwan SS. Capabilities and limitations of DGGE for the analysis of hydrocarbonoclastic prokaryotic communities directly in environmental samples. Microbiol Open. 2017;6(5)
Kristyanto S, Lee SD, Kim J. Porphyrobacter algicida sp. nov., an algalytic bacterium isolated from seawater. Int J Syst Evol Microbiol. 2017;67:4526–33.
Ramanathan T, Ting YP. Alkaline bioleaching of municipal solid waste incineration fly ash by autochthonous extremophiles. Chemosphere. 2016;160:54–61.
Madigan MT, Clark DP, Stahl D, Martinko JM. Brock biology of microorganisms. 13th ed. San Francisco: Benjamin Cummings; 2010.
Brock TD. Thermophilic microorganisms and life at high temperatures. 1st ed. New York: Springer Science & Business Media; 2012.
McMullan G, Christie JM, Rahman TJ, Banat IM, Ternan NG, Marchant R. Habitat, applications and genomics of the aerobic, thermophilic genus Geobacillus. Biochem Soc Trans. 2004;32:214–7.
Busscher HJ, van der KuijlBooij M, van der Mei HC. Biosurfactants from thermophilic dairy streptococci and their potential role in the fouling control of heat exchanger plates. J Ind Microbiol. 1996;16:15–21.
Ibrahim MHA, Lebbe L, Willems A, Steinbüchel A. Chelatococcus thermostellatus sp nov., a new thermophile for bioplastic synthesis: comparative phylogenetic and physiological study. AMB Express. 2016;6:1–9.
Kristjánsson JK, Hjörleifsdóttir S, Marteinsson VT, Alfredsson GA. Thermus scotoductus, sp. nov., a pigment-producing thermophilic bacterium from hot tap water in Iceland and including Thermus sp. X-1. Syst Appl Microbiol. 1994;17:44–50.
Jannasch HW, Wirsen CO, Molyneaux SJ, Langworthy TA. Extremely thermophilic fermentative archaebacteria of the genus Desulfurococcus from deep-sea hydrothermal vents. Appl Environ Microbiol. 1988;54:1203–9.
Reysenbach AL, Liu YT, Banta AB, Beveridge TJ, Kirshtein JD, Schouten S, et al. A ubiquitous thermoacidophilic archaeon from deep-sea hydrothermal vents. Nature. 2006;442:444–7.
Slobodkina GB, Baslerov RV, Novikov AA, Viryasov MB, Bonch-Osmolovskaya EA, et al. Inmirania thermothiophila gen. Nov., sp nov., a thermophilic, facultatively autotrophic, sulfur-oxidizing gammaproteobacterium isolated from a shallow-sea hydrothermal vent. Int J Syst Evol Microbiol. 2016;66:701–6.
Al-Dhabi NA, Esmail GA, Duraipandiyan V, Valan Arasu M, Salem-Bekhit MM. Isolation, identification and screening of antimicrobial thermophilic Streptomyces sp. Al-Dhabi-1 isolated from Tharban hot spring, Saudi Arabia. Extremophiles. 2016;20:79–90.
Anders H, Power JF, MacKenzie AD, Lagutin K, Vyssotski M, Hanssen E, et al. Limisphaera ngatamarikiensis gen. Nov., sp. nov., a thermophilic, pink-pigmented coccus isolated from subaqueous mud of a geothermal hotspring. Int J Syst Evol Microbiol. 2015;65:1114–21.
Hatzenpichler R, Lebedeva EV, Spieck E, Stoecker K, Richter A, Daims H, et al. A moderately thermophilic ammonia-oxidizing crenarchaeote from a hot spring. Proc Natl Acad Sci U S A. 2008;105:2134–9.
Sterner R, Liebl W. Thermophilic adaptation of proteins. Crit Rev Biochem Mol. 2001;36:39–106.
Argos P, Rossman MG, Grau UM, Zuber H, Frank G, Tratschin JD. Thermal stability and protein structure. Biochemistry. 1979;18:5698–703.
Bornscheuer UT, Huisman GW, Kazlauskas RJ, Lutz S, Moore JC, Robins K. Engineering the third wave of biocatalysis. Nature. 2012;485:185–94.
Matthews BW, Nicholson H, Becktel WJ. Enhanced protein thermostability from site-directed mutations that decrease the entropy of unfolding. Proc Natl Acad Sci U S A. 1987;84:6663–7.
Kumar S, Tsai CJ, Nussinov R. Factors enhancing protein thermostability. Protein Eng. 2000;13:179–91.
Creighton TE. Proteins: structures and molecular properties. 2nd ed. New York: Macmillan; 1993.
Nishio Y, Nakamura Y, Kawarabayasi Y, Usuda Y, Kimura E, Sugimoto S, et al. Comparative complete genome sequence analysis of the amino acid replacements responsible for the thermostability of Corynebacterium efficiens. Genome Res. 2003;13:1572–9.
Takami H, Takaki Y, Chee GJ, Nishi S, Shimamura S, Suzuki H, et al. Thermoadaptation trait revealed by the genome sequence of thermophilic Geobacillus kaustophilus. Nucleic Acids Res. 2004;32:6292–303.
Singer GA, Hickey DA. Thermophilic prokaryotes have characteristic patterns of codon usage, amino acid composition and nucleotide content. Gene. 2003;317:39–47.
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R. The microbial pan-genome. Curr Opin Genet Dev. 2005;15:589–94.
Chakravarty S, Varadarajan R. Elucidation of factors responsible for enhanced thermal stability of proteins: a structural genomics based study. Biochemistry. 2002;41:8152–61.
Vogt G, Woell S, Argos P. Protein thermal stability, hydrogen bonds, and ion pairs. J Mol Biol. 1997;269:631–43.
Pack SP, Yoo YJ. Protein thermostability: structure-based difference of amino acid between thermophilic and mesophilic proteins. J Biotechnol. 2004;111:269–77.
Zhou XX, Wang YB, Pan YJ, Li WF. Differences in amino acids composition and coupling patterns between mesophilic and thermophilic proteins. Amino Acids. 2008;34:25–33.
Panasik N, Brenchley JE, Farber GK. Distributions of structural features contributing to thermostability in mesophilic and thermophilic α/β barrel glycosyl hydrolases. BBA Protein Struc. 2000;1543:189–201.
Denisov VP, Venu K, Peters J, Hörlein HD, Halle B. Orientational disorder and entropy of water in protein cavities. J Phys Chem B. 1997;101:9380–9.
Nagendra H, Sukumar N, Vijayan M. Role of water in plasticity, stability, and action of proteins: the crystal structures of lysozyme at very low levels of hydration. Proteins. 1998;32:229–40.
Coil DA, Eisen JA. Draft genome sequence of Porphyrobacter mercurialis (sp. nov.) strain Coronado. Genome Announc. 2015;3:e300856–15.
Liu Q, Wu YH, Cheng H, Xu L, Wang CS, Xu XW. Complete genome sequence of bacteriochlorophyll-synthesizing bacterium Porphyrobacter neustonensis DSM 9434. Stand Genomic Sci. 2017;12:32.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. ABySS: a parallel assembler for short read sequence data. Genome Res. 2009;19:1117–23.
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with glimmer. Bioinformatics. 2007;23:673–9.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 2014;42:D206–14.
Tatusov RL, Natale DA, Garkavtsev IV, Tatusova TA, Shankavaram UT, Rao BS, et al. The COG database: new developments in phylogenetic classification of proteins from complete genomes. Nucleic Acids Res. 2001;29:22–8.
Wu S, Zhu Z, Fu L, Niu B, Li W. WebMGA: a customizable web server for fast metagenomic sequence analysis. BMC Genomics. 2011;12:444.
Fischer S, Brunk BP, Chen F, Gao X, Harb OS, Iodice JB, et al. Using OrthoMCL to assign proteins to OrthoMCL-DB groups or to cluster proteomes into new ortholog groups. Curr Protoc Bioinforma. 2011;12:11–9.
Li L, Stoeckert CJ Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–89.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421.
Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013;30:772–80.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldon T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–76.
Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol. 2015;32:268–74.
Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol Biol Evol. 2016;33:1870–4.
Chou PY, Fasman GD. Prediction of protein conformation. Biochemistry. 1974;13:222–45.
Chou PY, Fasman GD. Conformational parameters for amino acids in helical, β-sheet, and random coil regions calculated from proteins. Biochemistry. 1974;13:211–22.
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 31770004 and No. 41406174), the Scientific Research Fund of the Second Institute of Oceanography, State Oceanic Administration (JG1527), the Natural Science Foundation of Zhejiang Province (LR17D060001) and the National Program for Support of Top-Notch Young Professionals. The funding bodies had no role in the analysis, data interpretation and manuscript writing.
Availability of data and materials
The GenBank accession numbers for Porphyrobacter colymbi JCM 18338T, Porphyrobacter dokdonensis DSM 17193T, Porphyrobacter donghaensis DSM 16220T, Porphyrobacter sanguineus JCM 20691T and Porphyrobacter tepidarius DSM 10594T are MUYK00000000, MUYI00000000, MUYG00000000, MUYH00000000 and MUYJ00000000, respectively.
Author information
Authors and Affiliations
Contributions
X-WX conceived and designed the study. LX and Y-HW carried out the laboratory work. LX, PZ, QL and HC performed bioinformatic analysis. LX wrote the manuscript. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1
Alignment results of protein amino acid sequences encoded by single-copy genes found in eight Porphyrobacter strains. (ZIP 1510 kb)
Additional file 2
Summary of site-directed amino acid variations of Porphyrobacter cryptus DSM 12079T. (XLSX 16 kb)
Additional file 3
Summary of the substituted/substituting amino acid in Porphyrobacter cryptus DSM 12079T. (XLSX 8 kb)
Additional file 4
Frequency of each amino acid in Porphyrobacter type strains. (XLSX 10 kb)
Additional file 5
Summary of protein secondary structure variations in site-directed substitutions. (XLSX 99 kb)
Additional file 6
Summary of protein secondary structure variations in conserved proteins among Alphaproteobacteria bacteria. (XLSX 12 kb)
Additional file 7
In-house shell and R scripts for pan- and core-genomic analyses and screening single-copy shared OCs. (DOCX 13 kb)
Additional file 8
Summary of sources of Alphaproteobacteria strains applied in investigations of conserved proteins. (XLSX 10 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Xu, L., Wu, YH., Zhou, P. et al. Investigation of the thermophilic mechanism in the genus Porphyrobacter by comparative genomic analysis. BMC Genomics 19, 385 (2018). https://doi.org/10.1186/s12864-018-4789-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4789-4