
Abstract
Background
Meat tenderness is the consumer’s most preferred sensory attribute. This trait is affected by a number of factors, including genotype, age, animal sex, and pre- and post-slaughter management. In view of the high percentage of Zebu genes in the Brazilian cattle population, mainly Nellore cattle, the improvement of meat tenderness is important since the increasing proportion of Zebu genes in the population reduces meat tenderness. However, the measurement of this trait is difficult once it can only be made after animal slaughtering. New technologies such as RNA-Seq have been used to increase our understanding of the genetic processes regulating quantitative traits phenotypes. The objective of this study was to identify differentially expressed genes related to meat tenderness, in Nellore cattle in order to elucidate the genetic factors associated with meat quality. Samples were collected 24 h postmortem and the meat was not aged.
Results
We found 40 differentially expressed genes related to meat tenderness, 17 with known functions. Fourteen genes were up-regulated and 3 were down-regulated in the tender meat group. Genes related to ubiquitin metabolism, transport of molecules such as calcium and oxygen, acid-base balance, collagen production, actin, myosin, and fat were identified. The PCP4L1 (Purkinje cell protein 4 like 1) and BoLA-DQB (major histocompatibility complex, class II, DQ beta) genes were validated by qRT-PCR. The results showed relative expression values similar to those obtained by RNA-Seq, with the same direction of expression (i.e., the two techniques revealed higher expression of PCP4L1 in tender meat samples and of BoLA-DQB in tough meat samples).
Conclusions
This study revealed the differential expression of genes and functions in Nellore cattle muscle tissue, which may contain potential biomarkers involved in meat tenderness.
Similar content being viewed by others

Background
Meat quality traits in Brazilian animal breeding programs have not been fully explored because of the late expression of these attributes and the complex evaluation that can only be made after slaughter. Furthermore, on the domestic market, producers are generally not paid for meat quality, a fact that diminishes interest in improving meat quality traits and has hindered their inclusion in traditional selection objectives. In contrast, on international markets, meat tenderness is one of the most valued traits [39], a fact that highlights the importance towards improving this trait since Brazil is one of the world’s largest beef exporters.
Meat tenderness is the preferred sensory attribute of consumers [7]. According to Scollan et al. [46], the European food industry has sought to improve this trait to gain market share over other types of food. In Brazil, about 80% of the cattle herd consists of Zebu animals or their crossbreeds [1]. In this respect, the improvement of meat tenderness becomes important since Ferguson et al. [17] have shown that the higher the proportion of Zebu genes in a population, mainly Nellore cattle, the less tender the meat. Meat tenderness can only be measured after slaughter making this trait more complex to select animals. Thus, alternative tools are useful to include meat tenderness in animal breeding programs [9].
Modern recently developed large-scale RNA sequencing technologies (RNA-Seq) have been useful in understanding the genetic and physiological processes that regulate the phenotype of quantitative traits in a certain situation [34]. RNA-Seq permits analysis of the transcriptional profiles of cells, tissues or organs in a certain situation and the discovery of known and unknown genes involved in a given cellular process [57]. This new technique can be used to identify novel potential molecular markers that permit more accurate and early genetic predictions [51], with a consequent reduction in the generation interval that would contribute to the improvement of difficult-to-measure traits such as meat tenderness.
RNA-Seq has been widely used in recent studies to investigate differentially expressed genes related to meat tenderness in different species. For example, genes related to the degradation of filamins, lipogenesis and collagen synthesis have been identified in a study on meat tenderness in broiler chickens [40]. Gonçalves [20] found genes related to metabolic pathways involved in apoptosis, calcium transport, proteolysis and ribosome synthesis in castrated Nellore cattle, classified as extreme for tenderness based on estimated breeding values for shear force measured after 14 days of aging. Bongiorni et al. [8], who studied gene expression in longissimus dorsi muscle of animals of two Italian beef breeds (Maremmana and Chianina) representing the extremes for meat tenderness, detected differentially expressed genes related to growth and sodium-potassium pumps, among others.
Despite the above-mentioned publications, studies investigating differentially expressed genes related to meat tenderness in cattle are rare. In this respect, the better understanding and identification of the transcripts and biological processes, associated with this complex and economically important trait, will permit to highlight genes that could contain potential biomarkers involved in meat tenderness.
The objective of this study was to identify genes differentially expressed in muscle tissue (longissimus dorsi) of Nellore cattle with divergent meat tenderness using RNA-Seq in order to obtain data that increase our understanding of the genetic and metabolic mechanisms underlying this trait.
Results
RNA sequencing, alignment, and assembly of the transcripts
The TopHat2 program identified a total of 942 million reads (2 × 100 bp) and the sequencing coverage was 63X (coverage for all transcripts of all samples). An average of almost 24 million reads were obtained per sample and 88.3% of the reads were mapped. For tender meat group, an average of 24,928,506 (89%) million reads were mapped, while for tough meat group, an average of 22,170,021 million reads were mapped (89%) (Additional file 1: Table S1).
We found transcript for 28,059 genes and 103,309 potential new isoforms.
To evaluate the quality of sequencing, the expression profiles of the Glucuronidase Beta (GUSB), erythrocyte hydroxymethylbilane synthase (HMBS), Hypoxanthine Phosphoribosyltransferase 1 (HPRT1), phosphoglycerate kinase 1 (PGK1) and TATA-Box Binding Protein (TBP) genes were analyzed, which exhibited a similar expression profile in the tender and tough meat groups (Additional file 2: Figure S1).
A box plot (Additional file 3: Figure S2) containing the transformed FPKM values (log10) for each group and the plot of principal component analysis (PCA) (Additional file 4: Figure S3) were constructed using the cummerRbund package. As can be seen in the box plot, the distribution of quartiles was consistent between groups, indicating high quality of the data. In addition, the medians were similar in the two groups and close to −1, indicating that the level of sequencing coverage permitted the identification of low-expressed genes [11, 51]. PCA showed the formation of different groups (tender and tough meat), indicating differences in the expression of genes between the tender and tought meat groups’.
Analysis of differentially expressed genes
Analysis of differential expression in the tender and tough meat groups revealed 40 differentially expressed genes (q-value <0.05) (Table 1). Seventeen of these genes have a known function. The log2 signal (fold change) was used was used to partition the DE genes into up- and down-regulated groups. In this analysis, 35 genes were found to be up-regulated and 5 were down-regulated in relation to the tough meat group. Among the genes with known function, 14 were up-regulated and 3 were down-regulated.
Combined functional annotation using all differentially expressed (up- and down-regulated) genes for meat tenderness was performed with the DAVID v6.7 database using Bos taurus as a reference. This analysis permitted the identification of seven functional groups (annotation clusters; Additional file 5: Table S2). These genes were classified according to their function: cell fraction (GO: 000267), cell junction (GO: 0030054), intrinsic component of membrane (GO: 0031224), regulation of cell communication (GO: 0010646), catalytic activity (GO: 0003824), organelles (GO: 0043226), and binding (GO: 0005488), among others.
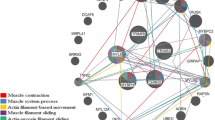
Using the ClueGO plug-in, the differentially expressed transcripts HMOX1, AT2, CLDN19, CLEC4G, CLEC12A, PNP and SYP were found to be inter-related through biological processes (cell communication, regulation of response to stimuli), molecular function (binding proteins), or cell components (integral membrane component) (Fig. 1). The gene HMOX1 was expressed more in tough meat group, while the other five genes were expressed more in tender meat group. The proteins encoded by these transcripts are involved in the transport of molecules such as sodium, potassium, calcium, and oxygen [15, 29, 36, 43].

Enrichment analysis of the HMOX1, CLDN19, CLEC4G, CLEC12A, PNP and SYP genes using the ClueGO plug-in of the Cytoscape program. Note the interrelationships between these genes, which are related to the transport of molecules
Using the same programs, the DMGDH gene (dimethylglycine dehydrogenase) was identified as a member of the “glycine, serine and threonine metabolism” pathway (Fig. 2). Glycine makes up about one-third of the helical polypeptide chains of collagen [30]. On the other hand, according to Bailey [2], collagen is degraded by serine proteases, with serine also being part of the glycine metabolic pathway, and by cysteine proteases whose metabolic pathway (“cysteine and methionine metabolism”) is associated with the DMGDH pathway. In the present study, the transcript of this gene was expressed more in tender meat.

Enrichment analysis of the DMGDH gene with the ClueGO plug-in. The yellow circlels highlight the biological processes and serine and glycine metabolic pathway in which this gene is involved
Figure 3 illustrates the interrelationships between the TCF7L1, EXOSC2, DMGDH and ASAH1 transcripts obtained by enrichment analysis. This analysis shows that the main link between these genes is the cell component called “intracellular membrane-bound organelle”. This category refers to structures found inside the cell such as the nucleus and mitochondria [10]. Gene expression analysis in Angus cattle also showed a relationship between meat tenderness and this cell component category [59]. The genes identified in this study are related to actin-myosin assembly, collagen synthesis, lipid accumulation, and serine and glycine metabolic pathways [2, 22, 30, 38].

Enrichment analysis of the TCF71, EXOSC2, DMGDH and ASAH1 genes with the ClueGO plug-in
Validation of differentially expressed genes
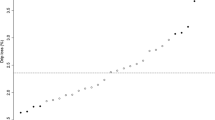
The relative expression values (log2) of the transcripts were similar for the two techniques used, RNA-Seq and qRT-PCR, with values of 2.12 and 2.03 (standard deviation = 0.89) for PCP4L1 and of −0.84 and −0.644 (standard deviation = 0.44) for BoLA-BQD, respectively (Fig. 4). Similar to the RNA-Seq analysis, higher expression of the PCP4L1 and BoLA-BQD genes was observed in the tender and tough meat groups, respectively. Thus, these transcripts showed similar patterns of mRNA abundance in the RNA-Seq and qRT-PCR analyses, with the same direction of expression (i.e., up-regulated and down-regulated, respectively, in relation to the tender meat group).

Comparison of the relative expression values of two differentially expressed transcripts obtained by RNA-Seq and qRT-PCR
Discussion
A higher proportion of Zebu genes in cattle herds considerably reduces meat tenderness when compared to taurine breeds. In Brazil, the herd mostly consists in Zebu cattle, mainly Nellore, then improve meat tenderness is very important, because for the beef export market, in which Brazil plays an important role, tenderness is paramount in determining the value of the product.
Gene expression studies have been used as a tool to identify gene candidates and metabolic pathways related to traits of economic interest. In the present study, the USP32 (ubiquitin specific peptidase 32) transcript was expressed more in tender meat. Members of the ubiquitin-proteasome system are important during the transformation of muscle into meat. These proteins are involved in proteolysis, causing the degradation of myofibrillar proteins in muscle cells [47].
In a genome-wide association study (GWAS) of Nellore cattle using different meat tenderness measures, Tizioto et al. [52] identified genes of the USP family, including USP32. Another study on cattle also associated genes of the USP family with meat tenderness. In Wagiu cattle, the USP2 gene was strongly associated with meat tenderness [12] and gene expression analysis in Nellore cattle showed that the USP2 gene was expressed more in tender meat samples [20].
The functional categories cell junction, regulation of cell communication and intrinsic component of membrane are related to the binding, communication and transport of molecules between cells [10]. Among the transcripts related to these categories, CTNNB1 (catenin - cadherin-associated protein beta 1), which was expressed more in tender meat, is involved in the same metabolic pathway as actin and myosin. Actin and myosin are the proteins found in thin and thick myofilaments, respectively, which form the myofibril that is responsible for muscle contraction. These proteins are the most abundant in the mechanism of muscle contraction, accounting for 52 to 56% of all muscle proteins [48].
Each actin filament binds to the plasma membrane of the cell through a structure, called focal contact. This structure consists of binding proteins and of a transmembrane protein that are products of the “focal adhesion” pathway to which the CTNNB1 and TCF7L1 (transcription factor 7 like 1) genes belong. On the outer side of the cell, in the extracellular matrix, the transmembrane protein binds to a collagen fiber [14, 23]. According to Bailey [2], a direct association exists between collagen content and the toughening of meat. However, in the present study, the CTNNB1 and TCF7L1 transcripts were expressed more in tender meat.
The SYP (synaptophysin) transcript, which was expressed more in tender meat, encodes an integral membrane protein found in small synaptic vesicles. In a study on rats, [44] showed that the phosphorylation of synaptophysin is calcium dependent. The authors observed a four-fold increase in serine phosphorylation of synaptophysin in the presence of the calmodulin-calcium complex. According to Bailey et al. [2], serine proteases are responsible for the degradation of collagen, which, in turn, directly influences meat tenderness. In addition, calcium is essential for muscle contraction by acting as a catalyst of enzymatic proteolytic activity, which is directly related to the process of meat tenderization [37].
The AT2 transcript, which encodes angiotensin II, was expressed more in tender meat. This protein is involved in vasoconstriction and regulates the secretion of aldosterone, which, in turn, stimulates the reabsorption of sodium by the kidneys. In this respect, after slaughter and during bleeding, angiotensin is activated to restore blood pressure. The result of these stimuli is the depolarization of the cell membrane, altering the distribution of sodium and potassium, in addition to permitting the flow of calcium ions [43]. In a study on crossbred cattle (Luxi-Simmental), Zhong-Liang et al. [60] observed a decline in shear force after the injection of angiotensin II into the carcass for 7 days after slaughter. Bongiorni et al. [8], studying gene expression in longissimus dorsi muscle of Italian Maremmana and Chianina breeds, also found the differential expression of genes to be related to sodium and potassium flow.
The functional category “catalytic activity” is related to increases in the velocity of a biochemical reaction at physiological temperatures [10]. Some reactions that occur during the postmortem period depend on calcium and cellular pH, which decrease in the first 24 h after slaughter [25]. A member of this functional category is ASAH1 (N-acylsphingosine amidohydrolase (acid ceramidase) 1), which belongs to a family of hydrolases that catalyze the synthesis and degradation of ceramide into sphingolipid and free fatty acid and are acid pH dependent [32]. A genetic deficiency in ASAH1 that reduces its catalytic activity causes a lysosomal sphingolipid storage disorder characterized by the accumulation of lipids in cells and tissues throughout the organism [38]. ASAH1 also belongs to the “sphingolipid signaling pathway” and “sphingolipid metabolism” categories in which serine is also involved, with serine protease degrading collagen [2]. Thus, ASAH1, which was expressed more in tender meat, may be related to the process of meat tenderization.
Another member of the “catalytic activity” category is HMOX1 (heme oxygenase 1), which was expressed more in tough meat. This gene encodes a protein involved in the metabolism of porphyrins, molecules whose catalytic activity is activated by iron [35]. Porphyrins are precursors of hemes, the main components of hemoglobin, myoglobin and cytochromes which are responsible for the transport of oxygen and electrons in tissues [36].
The C-type lectin (CLEC) family comprises calcium-dependent carbohydrate-binding protein domains that are involved in cell-cell adhesion [15]. In the present study, the CLEC4G and CLEC12A transcripts were expressed more in tender meat. GWAS in Nellore cattle demonstrated an association of the CLEC12A gene with different meat tenderness measures [52].
The IQCG transcript (IQ motif containing G), which was expressed more in tender meat, encodes a protein that functions as a binding site for different proteins, including myosin light chains and calmodulins. Calmodulin phosphorylates myosin, a process that permits the sliding of fibers and muscle contraction. In this case, calcium present in the reaction, binds to calmodulin, attached to IQ motif, and stimulates the ATPase activity of myosin [42]. According to Duston [16], in addition to factors such as collagen content, the structure and state of contraction of myofibrils (which mainly consists of myosin and actin) directly affect meat tenderness.
The protein encoded by the PNP transcript (purine nucleoside phosphorylase), which was expressed more in tough meat, plays a role in nicotinate and nicotinamide metabolism. Nicotinate (niacin or vitamin B3) is a precursor of NAD+ and NADP+ coenzymes, which are essential for the production of ATP in the cell [28].
Numerous structural changes and biochemical events occur in the first 24 h after slaughter of the animal, which are responsible for the conversion of muscle into meat [25]. In the early postmortem stages, ATP levels are maintained constant by the conversion of ADP plus phosphocreatine into ATP and oxygen supply ceases because of the cessation of blood circulation. At this stage, slow production of lactate is observed and the onset of rigor mortis occurs (slow phase). The decrease in phosphocreatine levels characterizes the rapid phase, which consists of a rapid decline in available ATP that is used as an energy reserve after the consumption of glycogen and other carbohydrates and is therefore hydrolyzed again to ADP. The scarcity of ATP during this phase is accompanied by the release of calcium ions into the myofibrillar space, which causes muscle shortening with a direct influence on meat tenderness [5].
Another event that occurs during this phase is the anaerobic conversion of glycogen into glucose, producing lactate and reducing the pH of the medium. In addition, the transport of sodium and potassium across the cell membrane, which uses the energy released by the hydrolysis of ATP into ADP, is impaired because it occurs against the concentration gradient. The protons generated during the hydrolysis of ATP into ADP cause a significant decline in intracellular pH [3]. According to Darrel et al. [13], this drop in pH directly influences the final tenderness of meat, especially during the process of aging.
According to Koohmaraie et al. [26], calcium is responsible for the activation of calpains and calpastatins (calcium-dependent cysteine proteases) and calpain I has been shown to be the main enzyme responsible for postmortem tenderization of meat by degrading cytoskeletal proteins that confer the structural integrity of the myofibrillar matrix. Nevertheless, in the present study, the calpain and calpastatin genes were not differentially expressed in the tender and tough meat groups. This finding might be explained by the fact that the amount of calpastatin in cells is higher 24 h after slaughter [43] and in this study the samples were collected immediately after cleaning the carcasses. Other GWAS and gene expression studies of muscle tissue in Nellore cattle also found no relationship between meat tenderness and calpain or calpastatin [20, 52].
The EXOSC2 transcript, which encodes exosome component 2, was expressed more in tender meat. According to Jong et al. [22], this gene is related with collagen activity in humans. This found could indicated a relationship between this genes and collagen activity in bovines, because there is a direct association exists between collagen content and the toughening of meat [2].
The ZKSCAN2 transcript (zinc finger with KRAB and SCAN domains 2), which was expressed more in tender meat, is vertebrate specific and synthesizes zinc finger proteins that bind through an N-terminus to the SCAN domain (dimerization motif). The function of this gene is not well known, but zinc finger proteins have been associated with the regulation of growth factor transcription and lipid metabolism [45].
In cattle, the main histocompatibility complex class II is called BoLA-DQB (bovine leukocyte antigen) [24]. In the present study, the BoLA-DQB transcript was expressed more in tough meat. We found no studies investigating the association of this gene with meat tenderness. However, this gene has been associated with growth traits in Holstein and beef cattle (Angus, Charolais, Hereford, Limousin, Simmental); [4, 49] and, according to Koohmaraie et al. [27], animals with higher growth rates have more palatable and more tender meat.
When we compared this study with a GWAS study for meat tenderness using the same Nellore population, we do not found common genes between them, but there were some shared functions related to phosphorylation and catalytic activity [33]. These functions are related with oxygen and calcium transport, and collagen degradation, important processes for the the toughening of meat, especially after slaughter. In a GWAS study using another Nellore cattle population, Tizioto et al. [52] identified regions that influence tenderness at three different time points (24 h and 7 and 14 days after slaughter). Some of the genes reported by these authors were also identified in the present study, such as CLDN19, CLEC12A and USP32. In addition to these genes, the authors reported an association of genes belonging to the family of BoLA-BQD, CTNNB1, EXOSC2 and IQCG transcripts and meat tenderness.
Conclusions
Global gene expression analysis in animals phenotypically divergent for meat tenderness identified genes related to ubiquitin metabolism, transport of molecules such as calcium and oxygen, acid-base balance, collagen synthesis, actin and myosin, and fat accumulation. These results contribute to the understanding of the molecular mechanisms involved in the meat tenderization process, at the time of slaughter, and to the development of strategies to select animals with more tender meat.
Methods
Animals and sample collection
Meat samples were collected from 132 intact male (non castrated) Nellore animals belonging to the same contemporary group (i.e., animals that remained together from birth to slaughter). The animals were from the Capivara Farm that participates in the Qualitas Nelore Breeding Program. All animals were finished in feedlots for, approximately, 90 days and slaughtered at an average age of 731 ± 81 days on the same day and under the same conditions.
The slaughter occurred in a commercial plant, under usual process in Brazilian beef industry: the animals are slaughtered and the half-carcasses are refrigerated by 24 h. After that, the carcass is deboned, frozen and commercialized. All samples were frozen and none of them was aged.
For RNA, muscle tissue (longissimus dorsi) samples were collected immediately after slaughtering and stored in 15-mL Falcon tubes containing 5 mL RNA holder (BioAgency, São Paulo, SP, Brazil) at −80 °C until the time for total RNA extraction. Additionally, for shear force measurements, a longissimus muscle sample was removed during deboning, after 24 h in a cold chamber, between the 12th and 13th rib of each left half-carcass.
Transcriptome studies show the genes expressed in a specific time for a specific cell, i.e. it shows which genes are been expressed at the moment of the sample collection. So, we have chosen to study the gene expression related with tenderness using the phenotype measured closest to the sample collection for RNA extraction, that is, after 24 h postmortem.
Analysis of shear force
Longissimus dorsi samples measuring 2.54 cm in thickness were obtained for analysis of tenderness. The standardized procedure proposed by Wheeler et al. [58] was used for shear force determination in a mechanical Salter Warner-Bratzler Shear Force device. The samples analyzed were not submitted to any type of aging process. From this analysis (n = 132), 40 samples derived from animals extreme for meat tenderness (20 with tender meat and 20 with tough meat) were selected. The Student t-test implemented in the R environment [41] was applied to verify differences between the tender and tough meat groups (Table 2).
RNA sequencing
Total RNA was extracted from the samples obtained from the extreme animals selected (n = 40). Muscle tissue (longissimus dorsi) samples that were collected immediately after slaughter and stored in 15-mL Falcon tubes containing 5 mL RNA holder (BioAgency, São Paulo, SP, Brazil) at −80 °C were used to extract total RNA. An average of 50 mg of the muscle tissue previously stored in RNA holder (BioAgency, São Paulo, SP, Brazil) was used for extraction with the RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA, USA) according to manufacturer recommendations. The purity of the extracted RNA was determined by reading absorbance in a NanoDrop 1000 spectrophotometer (Thermo Fisher Scientific, Santa Clara, CA, USA, 2007). The quality of the total RNA extracted was evaluated in an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA, 2009) and its concentration and contamination with genomic DNA were measured in a Qubit® 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA, 2010).
Sequencing (RNA-Seq) was performed on an Illumina HiSeq 2500 System. Messenger RNA was obtained from the total RNA extracted and libraries containing 200 bp fragments were constructed and pooled to perform multiplexing sequencing. The reads obtained were paired-end of 2 × 100 bp.
Sequence processing and alignment
The sequence data generated with the Illumina HiSeq 2500 System were converted into FastQ format, using the Casava software (https://support.illumina.com/downloads/casava_18_changes.html). The computational analyses were performed on CyVerse platform [19].
First, sequenced fragments (reads) of low quality were trimmed using the Sickle program (github.com/najoshi/sickle). The TopHat2 v2.0.9 program [54] was then used to map the fragments and to align them with the bovine reference genome (UMD3.1) available in the NCBI database (http://www.ncbi.nlm.nih.gov/genome/?term=bos+taurus). For each library, a .bam file containing the aligned reads in relation to the reference genome was generated.
Assembly and quantification of the transcripts
The Cufflinks2 v2.1.1 program [55] was used to assemble the aligned reads of each sample and to estimate the number of transcripts, expressed as fragments per kilobase of transcript per million reads mapped (FPKM). The Cufflinks2 result per sample was concatenated in a single file with the Cuffmerge2 v2.1.1 program, which was used as a reference in the differential gene expression analysis.
Differential gene expression analysis
Using the Cuffdiff2 v 2.1.1 program [53, 55], the sequence alignment files generated (.bam) were divided into two contrasting groups according to meat tenderness. The FPKM values of each transcript were calculated for each sample. The Cuffdiff2 program uses a t-test for the calculation of p-values. False discovery rates (FDR) were controlled by the Benjamini-Hochberg procedure considering a q-value of less than 5%.
The CummeRbund package [55], implemented in the R environment [41], was used for exploration and visualization of the data obtained and generate PCA and boxplot graphics.
Annotation of differentially expressed genes
The Database for Annotation, Visualization, and Integrated Discovery (DAVID) v6.7, which consists of an integrated system of biological databases and analytical tools designed to systematically extract the biological meaning from a large list of genes and/or proteins [21], was used to annotate and interpret the lists of differentially expressed genes. The Functional Annotation Tool was used for this purpose, which determines the most relevant Gene Ontology (GO) terms for each list of genes. The Functional Annotation Clustering algorithm was applied to generate annotations of functional groups. DAVID pathway mapping was used to identify metabolic pathways in which the differentially expressed genes are involved.
The ClueGo plug-in of the Cytoscape program was used to visualize non-redundant biological terms for genes in functionally grouped networks [6].
Validation of differentially expressed genes
Real-time quantitative PCR (qRT-PCR) was used to validate the differential expression of the genes identified by RNA-Seq analysis. All the 40 RNA samples used in the RNA-Seq analyses was used to validate the data by qRT-PCR. Two differentially expressed genes were chosen randomly for this purpose: bovine leukocyte antigen (BoLA-DQB) and Purkinje cell protein 4-like 1 (PCP4L1). In addition to these genes, three reference genes were chosen and quantified by qRT-PCR, as proposed by Vandesompele et al. [56], to normalize the data. The RNA-Seq technique detected no differences in the expression of the beta-glucuronidase (GUSB), hypoxanthine phosphoribosyltransferase 1 (HPRT1) and TATA box binding protein (TBP) genes between the groups studied and these genes were therefore chosen as housekeeping genes and were tested by qRT-PCR.
The method (conditions and equipment) described by Fonseca et al. [18] was used for validation of the differentially expressed genes by qRT-PCR: One μg total RNA was used to synthetize the first complementary DNA (cDNA) strand using SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (Invitrogen). To design the primers (Table 3), the Primer Express 3.0 software (Applied Biosystems, 2004) was used and the GenBank database (http://www.ncbi.nlm.nih.gov) was accessed to obtain the mRNA nucleotide sequences. The primers specificity was tested by NCBI BLAST algorithm (https://blast.ncbi.nlm.nih.gov/Blast.cgi). Genorm (https://genorm.cmgg.be/) and Expression Suite softwares v1.0 (Applied Biosystems, Foster, CA, USA, 2012) were used to test the expression stability of the housekeeping genes.
All qRT-PCR reactions were done with 7500 Real-Time PCR (Applied Biosystems, 2009). For these reactions we used: 0.1 μg cDNA; 1X SYBR Green Master Mix and forward and reverse primers. Primers concentrations were determined by titration: 600 nanoMolar (nM) forward and reverse primers (600/600) for BoLA-BQD and GUSB; 300 nanoMolar (nM) forward and reverse primers (300/300) for HPRT1 and 100 nanoMolar (nM) forward and reverse primers (100/100) for PCP4L1 and TBP. The analyses were performed in triplicate. For each gene (target and housekeeping), we included a negative and a positive control in every reaction.
Serial dilutions of cDNA (1:5) were used to build a standard curve and to calculate the qRT-PCR efficiency for each gene. Only PCR primers showing an efficiency of 90–110% were used [31].
The amplification conditions were: 40 cycles at 50 °C for 2 min, 95 °C for 10 min, and 60 °C for 1 min. Dissociation analyzes were performed to monitor the reactions specificity.
For the housekeeping genes, the geometric means of Ct values were calculated [56]. For the analysis of relative expression, a mixed linear model was fitted [50]:
where: Ygikr is the Ct obtained from the thermocycler software for gene g, in the rth well of the plate (referring to the technical replicate) in a sample obtained from animal k of treatment i (low or high meat tenderness group). Tig is the group of animals effect i (low or high meat tenderness group) on the expression of gene g; Dik is a random sampling specific effect which captures differences between samples shared by genes, particularly those affecting RNA concentration such as different extraction and amplification efficiency, and egikr is a residual effect.
Abbreviations
- ADP:
-
Adenosine Diphosphate
- ASAH1:
-
N-Acylsphingosine Amidohydrolase (Acid Ceramidase) 1
- AT2:
-
Angiotensin II Receptor Type 2
- ATP:
-
Adenosine Triphosphate
- BoLA-DQB:
-
Major Histocompatibility Complex, Class II, DQ Beta
- cDNA:
-
Complementary DNA
- CLDN19:
-
Claudin 19
- CLEC:
-
C-Type Lectin Family
- CLEC12A:
-
C-Type Lectin Domain Family 12 Member A
- CLEC4G:
-
C-Type Lectin Domain Family 4 Member G
- Ct:
-
Threshold Cycle
- CTNNB1:
-
Catenin - Cadherin-Associated Protein Beta 1
- DMGDH:
-
Dimethylglycine Dehydrogenase
- EXOSC2:
-
Exosome Component 2
- FDR:
-
False Discovery Rates
- FPKM:
-
Fragments Per Kilobase Of Transcript Per Million Reads Mapped
- GO:
-
Gene Ontology
- GUSB:
-
Glucuronidase Beta
- GWAS:
-
Genome-Wide Association Study
- HMBS:
-
Erythrocyte Hydroxymethylbilane Synthase
- HMOX1:
-
Heme Oxygenase 1
- HPRT1:
-
Hypoxanthine Phosphoribosyltransferase 1
- IQCG:
-
IQ Motif Containing G
- NAD+ :
-
Nicotinamide Adenine Dinucleotide
- NADP+ :
-
Nicotinamide Adenine Dinucleotide Phosphate
- nM:
-
nanoMolar
- PCA:
-
Principal Component Analysis
- PCP4L1:
-
Purkinje Cell Protein 4 Like 1
- PGK1:
-
Phosphoglycerate Kinase 1
- PNP:
-
Purine Nucleoside Phosphorylase
- qRT-PCR:
-
Quantitative Real Time Polymerase Chain Reaction
- RNA-Seq:
-
RNA Sequencing
- SYP:
-
Synaptophysin
- TBP:
-
TATA-Box Binding Protein
- TCF7L1:
-
Transcription Factor 7 Like 1
- USP:
-
Ubiquitin Specific Peptidase Family
- USP2:
-
Ubiquitin Specific Peptidase 2
- USP32:
-
Ubiquitin Specific Peptidase 32
- ZKSCAN2:
-
Zinc Finger With KRAB And SCAN Domains 2
References
ABIEC, Associação Brasileira das Indústrias Exportadoras de Carne, 2016. http://www.abiec.com.br/. Accessed 8 Feb 2016.
Bailey AJ, Paul RG, Knott L. Knott. Mechanisms of maturation and aging of collagen. Mech Ageing Dev.1998, 106:1–56.
Bate-Smith EC, Bendall JR. Factors determining the time course of rigor mortis. J Physiol 1949, 110:47–65.
Batra TR, Lee AJ, Gavora JS, Stear MJ. CLASS I alleles of the bovine major histocompatibility system and their association with economic traits. J Dairy Sci. 1989;72:2115–2124.
Bendall JR. Posmortem changes in muscle. In: GH BOURNE, editor. The structure and function of muscle, vol. 2. New York: Academic Press; 1973. p. 244–309.
Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. J Bioinformatics. 2009;25(8):1091–3.
Boleman SJ, Boleman SL, Miller RK, Taylor JF, Cross HR, Wheeler TL, Koohmaraie M, Shackelford SD, Miller MF, West RL, Johnson DD, Savell JW. Consumer evaluation of beef of known categories of tenderness. J Anim Sci. 1997;75:1521–4.
Bongiorni S, Gruber CEM, Bueno S, Chillemi G, Ferr F, Failla S, Moioli B, Valentini A. Transcriptomic investigation of meat tenderness in two Italian cattle breeds. Anim Genet. 2016. doi:10.1111/age.12418.
Campo MM, Sañudo C, Panea B, Alberti P, Santolaria P. Breed type and aging time effects on sensory characteristics of beef strip loin steaks. Meat Sci. 1999;51:383–91.
Carbon S, Ireland A, Mungall CJ, Shu S, Marshall B, Lewis S. AmiGO: online access to ontology and annotation data. Bioinformatics. 2009;2009(25):288–9.
Chapple RH, Tizioto PC, Wells KD. Characterization of the rat developmental liver transcriptome. Physiol Genomics. 2013;45:301–11.
CRC: cooperative Resersh Centre for Beef Genetic Technologies. Annual Report of CRC for Beef Genetics Tecnologies. High Quality beef for Global Consumers. Armidale, Australia, 2008.
Darrel E, Goll DE, Valery F, Thompson VF, Li H. The calpain system. Physiol Rev. 2003;83:731–801.
De Robertis E. Bases da Biologia Celular e Molecular. Rio de Janeiro: Editora Guanabara Koogan; 2010.
Drickamer K. C-Type lectin-like domains. Curr Opin Struct Biol. 1999;5:585–90.
Duston TR, Hostetler RL, Carpenter ZL. Effect of collagen levels and sarcomere shortening on muscle tenderness. J Food Sci. 1976;41:863–6.
Ferguson DM, et al. Effect of electrical stimulation on protease activity and tenderness of M. Longissimus from cattle with different proportions of Bos Indicus content. Meat Sci. 2000;55:265–72.
Fonseca LFS, Gimenez DF, Mercadante ME, Bonilha SF, Ferro JA, Baldi F, Souza FRP, Albuquerque LG. Expression of genes related to mitochondrial function in Nellore cattle divergently ranked on residual feed intake. Mol Biol Rep. 2015;42:559–65.
Goff SA, Vaughn M, Mckay S. The iPlant collaborative: cyberinfrastructure for plant biology. Front Plant Sci. 2011;2:34. doi:10.3389/fpls.2011.00034.
Gonçalves TM. Dissertation. In: differential expression of genes related with meat tenderness in Nellore cattle. ESALQ, USP; 2015. p. 97. http://www.teses.usp.br/teses/disponiveis/11/11139/tde-12052015-165345/pt-br.php. Accessed 16 Mar 2016.
Huang W, Sherman BT, Lempick RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57.
Jong OG, Balkom BWM, Gremmels H, Verhaar MC. Exosomes from hypoxic endothelial cells have increased collagen crosslinking activity through up-regulation of lysyl oxidase-like 2. J Cell Mol Med. 2015;XX:1–9.
Junqueira LCU, Carneiro J. Biologia Celular e Molecular. 8th ed. Guanabara; 2005.
Klein J, Bontrop RE, Dawkins RL, Erlich HA, Gyllensten UB, Heise ER, Jones PP, Parham P, Wakeland EK, Watkins DI. Nomenclature for the major histocompatibility complexes of different species: a proposal. Immunogenetics. 1990;4:217–9.
Koohmaraie M. The biological basis of meat tenderness and potential genetic approaches for its control and prediction. Proc Recip Meat Conf. 1995;48:69–75.
Koohmaraie M. Biochemical factors regulating the toughening and tenderization process of meat. Meat Sci. 1996;43:S193–201.
Koohmaraie M, Kent MP, Shackelford SD, Veiseth E, Wheeler TL. Meat tenderness and muscle growth: is there any relationship? Meat Sci. 2002;62:345–52.
LAMP: Library of Apicomplexan Metabolic Pathways. Nicotinate and nicotinamide metabolism. http://www.llamp.net/?q=Nicotinate%20metabolism. Accessed 29 Dec 2015.
Lee NP, Tong MK, Leung PP. Kidney claudin-19: localization in distal tubules and collecting ducts and dysregulation in polycystic renal disease. FEBS Lett. 2006;580:923–31.
Lehninger AL, Nelson LD, Cox MM. Princípios de bioquímica. 3rd ed. São Paulo: SARVIER; 2002. p. 1009.
Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–8.
Lucki NC, Sewer MB. Genistein stimulates MCF-7 breast cancer cell growth by inducing acid ceramidase (ASAH1) gene expression. JBC. 2011;286:19399–409.
Magalhães AFB, de Camargo GMF, Junior FGA, Gordo DGM, Tonussi RL, et al. Genome-wide association study of meat quality traits in Nellore cattle. PLoS One. 2016;11(6):e0157845. doi:10.1371/journal.pone.0157845
Malone JH, Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011;9:34.
Manso CMCP, Neri CR, Vidoto EA, Sacco HC, Ciuffi KJ, Iwamoto LS, Iamamoto Y, Nascimento OR, Serra OA. Characterization of iron(III) porphyrin-hidroxo complexes in organic media through Uv-Vis and EPR spectroscopies. J. Inorg. Biochem. 1999;73:85–93.
Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol. 2003;24:449–55.
Ouali A, Gagaoua M, Boudida Y, Becila S, Boudjellal A, Herrera-Mendez CH, Sentandreu MA. Biomarkers of meat tenderness: presente knowledge and perspectives in regards to our current understanding of the mechanisms involved. Meat Sci. 2013;95:854–70.
Park JH, Schuchman EH. Acid ceramidase and human disease. Biochim Biophys Acta. 2006;1758:2133–8.
Paz CCP, Luchiari Filho A. Melhoramento genético e diferenças de raças com relação à qualidade da carne bovina. Pecuária de corte. 2000;101:58–63.
Piorkowska K, Żukowski K, Nowak J, Połtowicz K, Ropka-Molik K, Gurgul A. Genome-wide RNA-Seq analysis of breast muscles of two broiler chicken groups differing in shear force. Anim Genet. 2015;47(1):68–80.
R Core Team. R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2015. https://www.R-project.org/.
Rhoads AR, Friedberg F. Sequence motifs for calmodulin recognition. FASEB J Official Publ Federation of American Societies for Exp Biol. 1997;11:331–40.
Rubensam JM, Transformações Post Mortem E Qualidade Da Carne Suína. 1ª Conferência Internacional Virtual sobre Qualidade de Carne Suína. 2000. http://www.cnpsa.embrapa.br/sgc/sgc_publicacoes/anais00cv_jane_pt.pdf. Accessed 30 Dec 2015.
Rubenstein JL, Greengard P, Czernik AJ. Calcium-dependent serine phosphorylation of synaptophysin. Synapse. 1993;13:161–72.
Sander TL, Stringer KF, Maki JL, Szauter P, Stone JR, Collins T. The SCAN domain defines a large family of zinc finger transcription factors. Gene. 2003;310:29–38.
Scollan N, Hocquette J, Nuernberg K, Dannenberger D, Ian R, Moloney A. Innovations in beef production systems that enhance the nutritional and health value of beef lipids and their relationship with meat quality. Meat Sci. 2006;74:17–33.
Sekikawa M, Seno K, Mikami M. Degradation of ubiquitin in beef during storage. Meat Sci. 1998;48:201–4.
Sgarbieri VC. Proteínas em alimentos protéicos. São Paulo: Varela; 1996. p. 517.
Stear MJ, Pokorny TS, Muggli NE, Stone RT. The relationships of birth weight, preweaning gain and postweaning gain with the bovine major histocompatibility system. J Anim Sci. 1989;67:641–9.
Steibel JP, Poletto R, Coussens PM, Rosa JMG. A powerful and flexible linear mixed model framework for the analysis of relative quantification RT-PCR data. Genomics. 2009;94:146–52.
Tizioto PC, Coutinho LL, Decker JE, Schnabel RD, Rosa KO, Oliveira PSN, Souza MM, Mourão GB, Tullio RR, Chaves AS, Lannad PD, Zerlotini-Neto A, Mudadu MA, Taylor JF, Regitano LCA. Global liver gene expression differences in Nelore steers with divergent residual feed intake phenotypes. BMC Genomics. 2015;16:216.
Tizioto PC, Decker JE, Taylor JF, Schnabel RD, Mudadu MA. Genome scan for meat quality traits in Nelore beef cattle. Physiol Genomics. 2013;45:1012–20.
Trapnell C, Hendrickson DG, Sauvageau M, Goff L, RInn JL, Pacther L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and cufflinks. Nat Protoc. 2012;7(3):562–78.
Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7):RESEARCH0034.
Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Advance Online Plublication: Nat. Rev. Genet; 2008.
Wheeler TL, Koohmaraie M, Shackelford SD. Standardized Warner-Bratzler shear force procedures for meat tenderness measurement. Clay Center: Roman L. Hruska U. S. MARC. USDA, 1995.
Zhao C, Tian F, Yu Y, Liu G, Zan L, Scott M, Song J. miRNA-dysregulation associated with tenderness variation induced by acute stress in Angus cattle. J Anim Sci Biotechnol. 2012;3:12.
Zhong-liang HU, Xing JL, Xiao-feng YE. Effects of Angiotensin II on Beef Quality. Acta Agric Jiangxi. 2009:11.
Acknowledgments
We thank the Qualitas Nelore breeding program company for providing the tissue samples and database used in this study.
Funding
The RNA sequencing was fund by the project "Genomic tools for the genetic improvement of traits of direct economic importance in Nelore cattle". This was finance also by the São Paulo Research Foundation – FAPESP. (FAPESP grant # 2009/16118–5). The LFSF scholarship was fund by the São Paulo Research Foundation – FAPESP (FAPESP grant # 2013/09190–7).
Availability of data and materials
The dataset utilized in this study belongs to a Qualitas Nelore breeding program company, and could be available on request. The author do not have authorization to share the data.
Author information
Authors and Affiliations
Contributions
LFSF, DFJG, LGA, JAF and FB conceived and designed the experiment; LFSF and DFJG performed the experiments; LFSF, LGA, DBSS and RB analyzed and interpreted the results; LFSF, LGA, JAP and RB drafted and revised the manuscript. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval
All experimental procedures were approved by Ethics Committee of the Faculty of Agrarian and Veterinary Sciences of Sao Paulo State, Jaboticabal, São Paulo (protocol number 18,340/16). The animals were provideded by Qualitas Nelore breeding program company and they were slaughtered in commercial slaughterhouses. These slaughterhouses have animal welfare departments staffed by professionals trained by WAG (World Animal Protection) to ensure that the animals are killed humanely using a captive bolt pistol for stunning.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
Samples number (N), classification of the sample, shear force (kgf/cm2), number of transcripts aligned in pairs (N reads), and percentage of transcripts aligned in pairs (% reads). (DOCX 16 kb)
Additional file 2: Figure S1.
Expression profile of reference genes in the experimental groups (tender and tough meat). (TIFF 176 kb)
Additional file 3: Figure S2.
Box plot of expression values (log10 FPKM) obtained for the groups studied (tender and tough meat). (TIFF 162 kb)
Additional file 4: Figure S3.
Principal component analysis (PCA) of the transcripts found in the tender (red) and tough (blue) meat groups. (TIFF 215 kb)
Additional file 5: Table S2.
Enriched GO terms obtained with the DAVID software for differentially expressed genes. (XLSX 14 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fonseca, L.F.S., Gimenez, D.F.J., dos Santos Silva, D.B. et al. Differences in global gene expression in muscle tissue of Nellore cattle with divergent meat tenderness. BMC Genomics 18, 945 (2017). https://doi.org/10.1186/s12864-017-4323-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-4323-0